Dynamic Behaviors of Solvent Molecules Restricted in Poly (Acryl Amide) Gels Analyzed by Dielectric and Diffusion NMR Spectroscopy

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
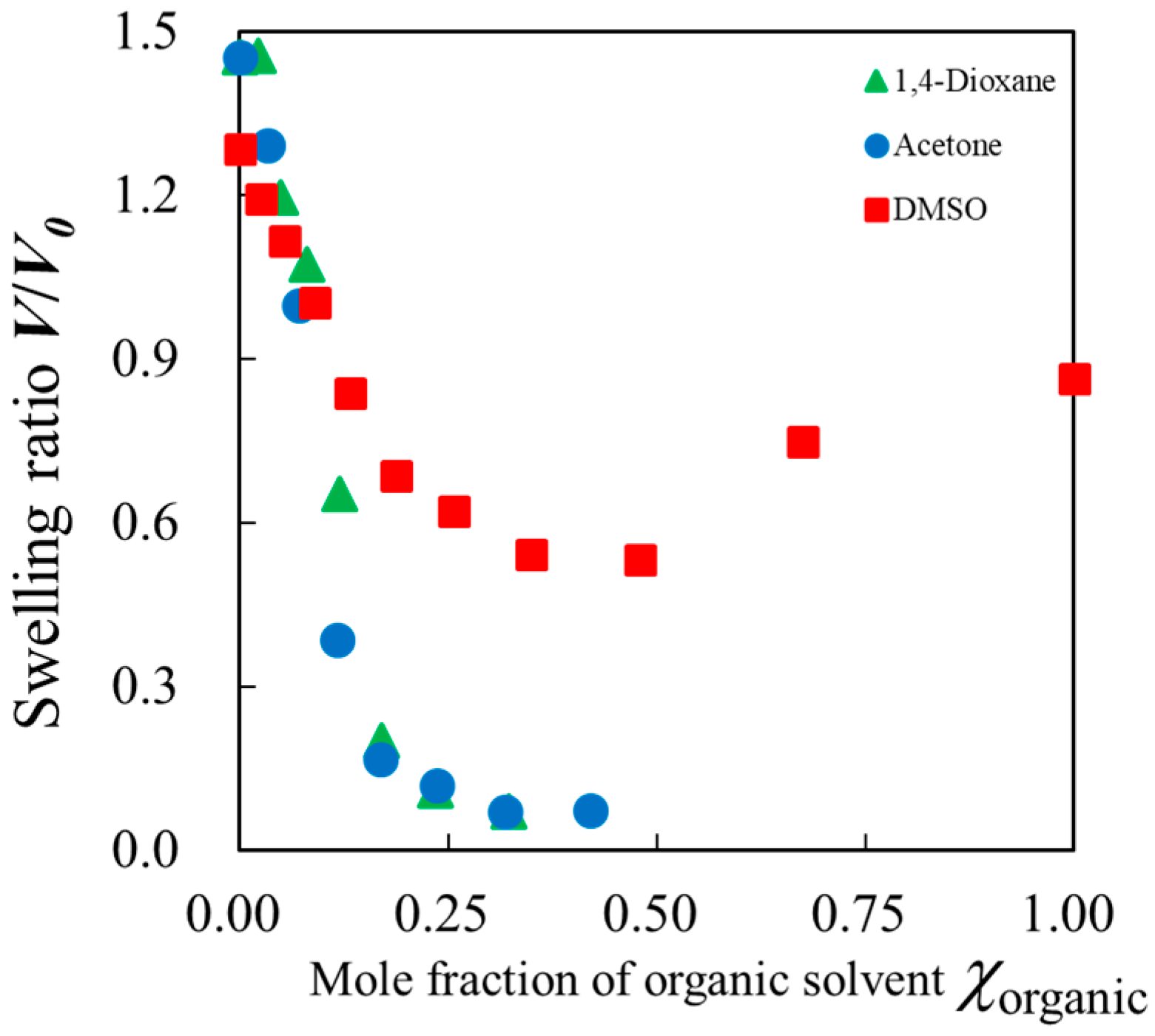
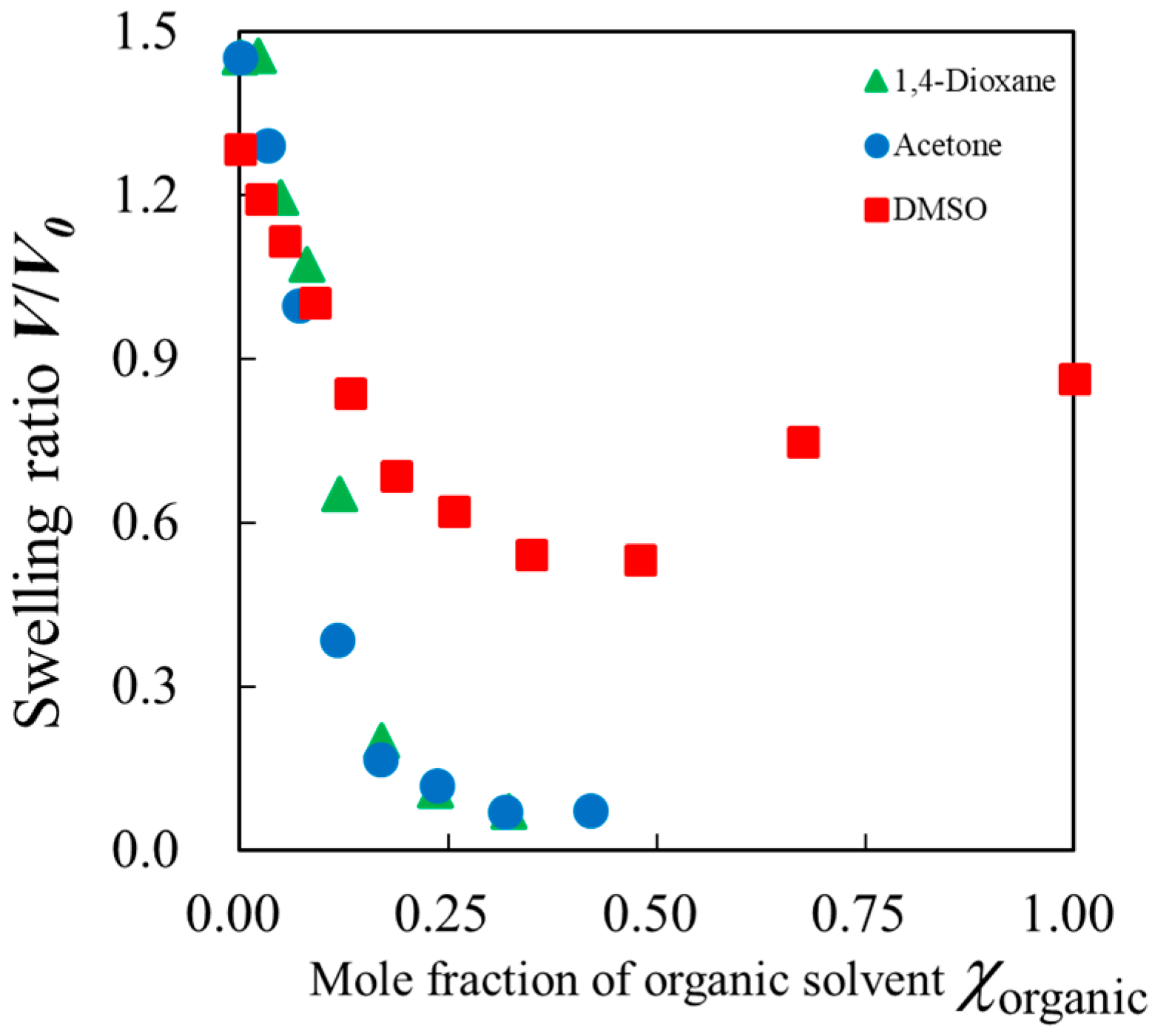
2.1. Swelling Ratio of Gels
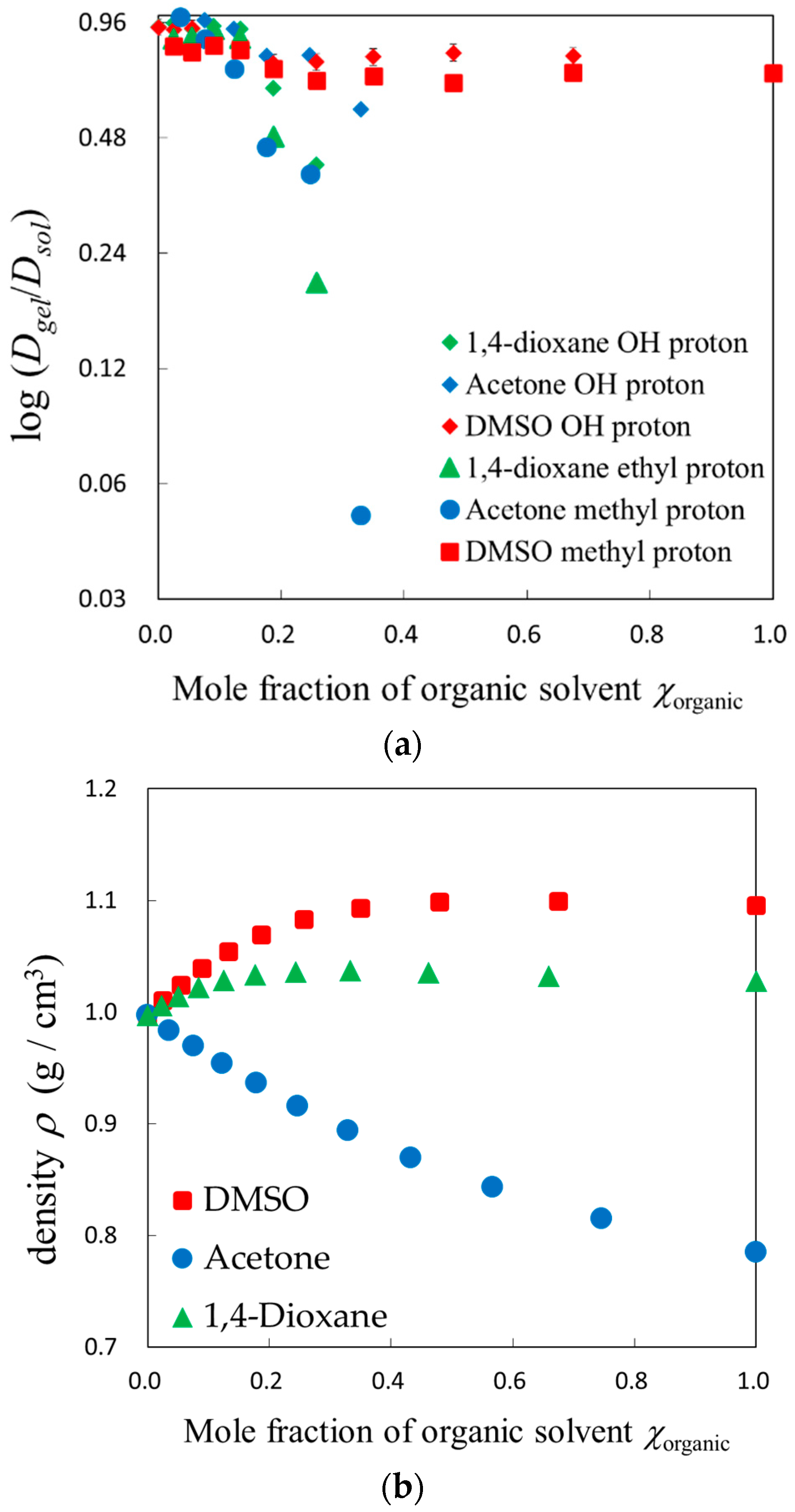
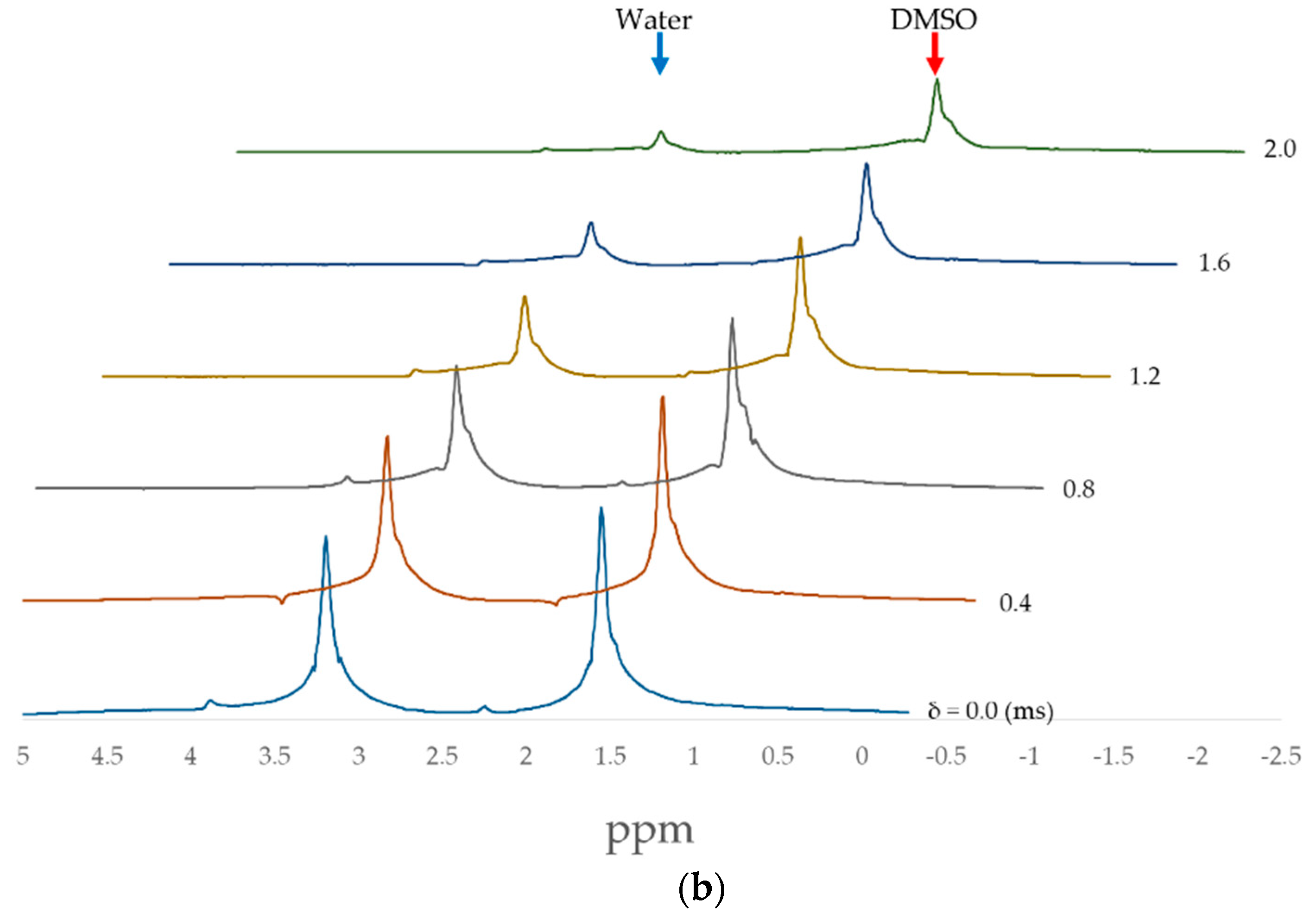
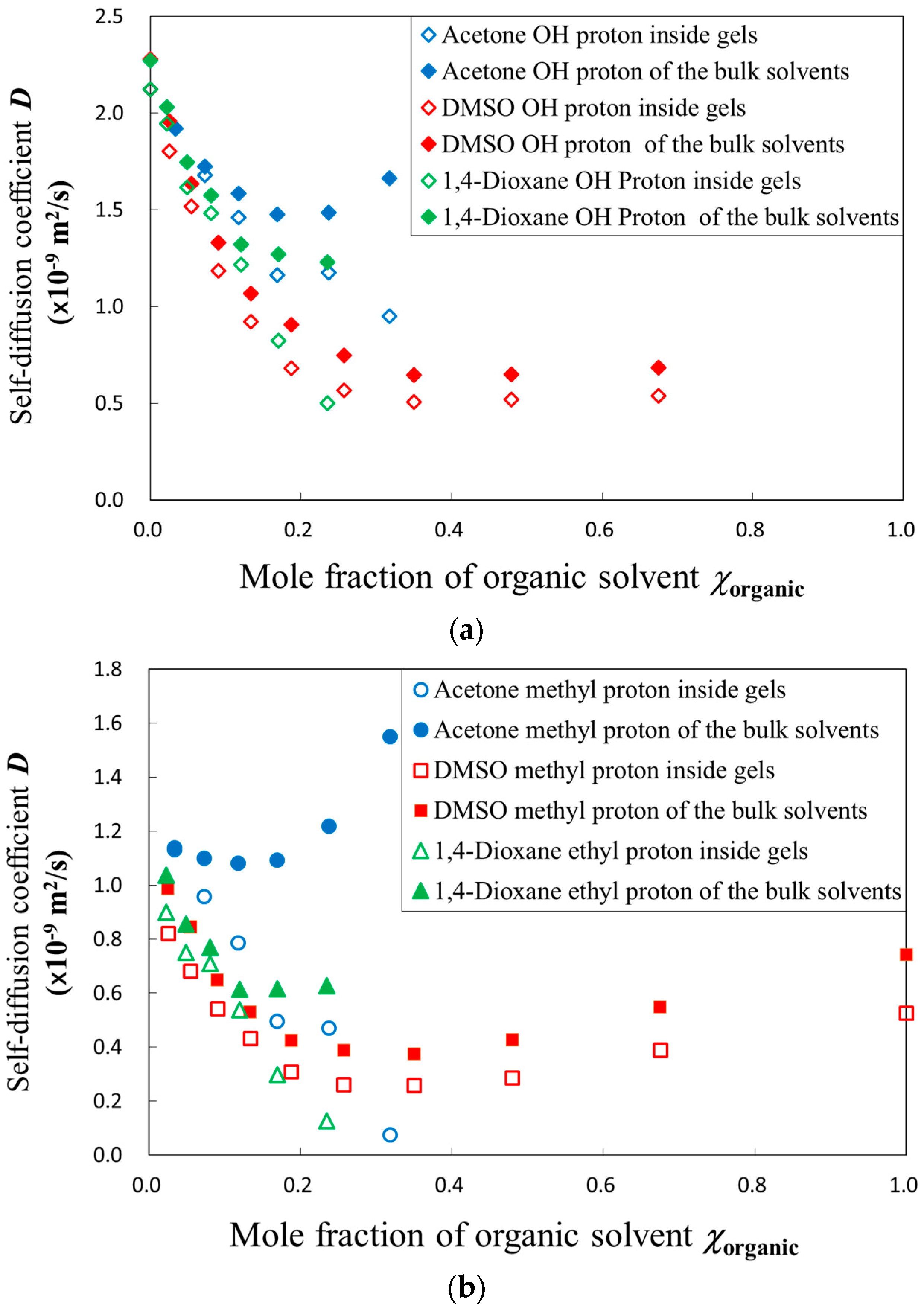
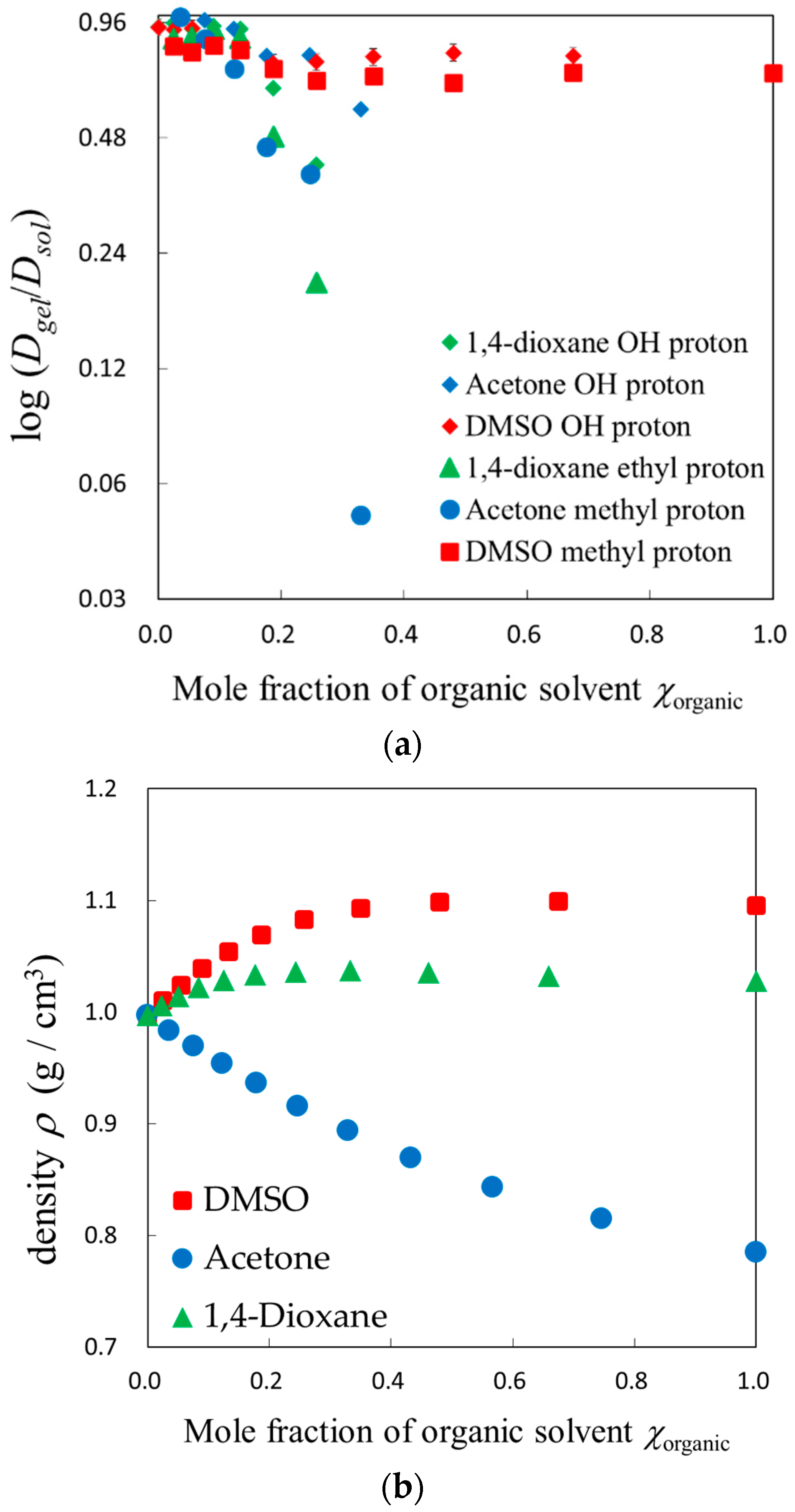
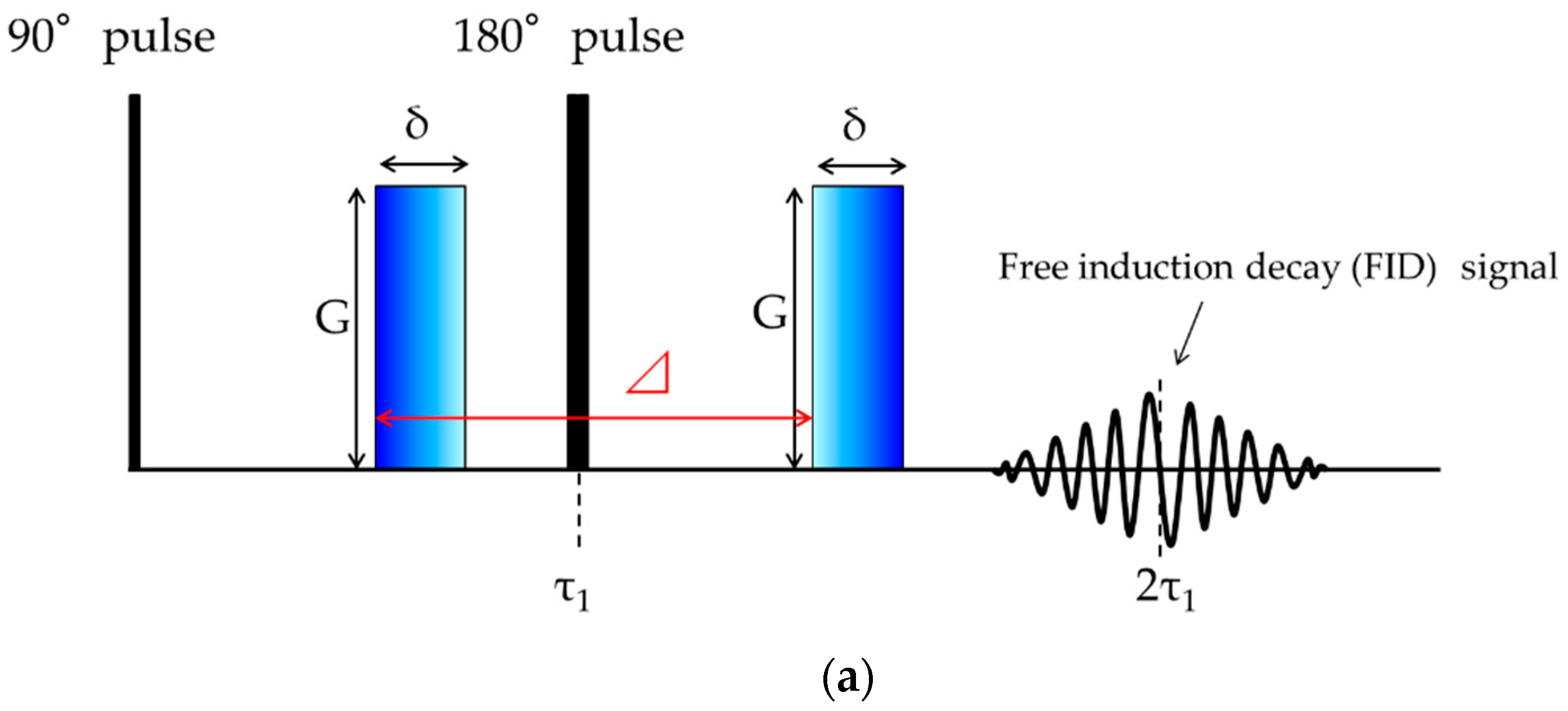
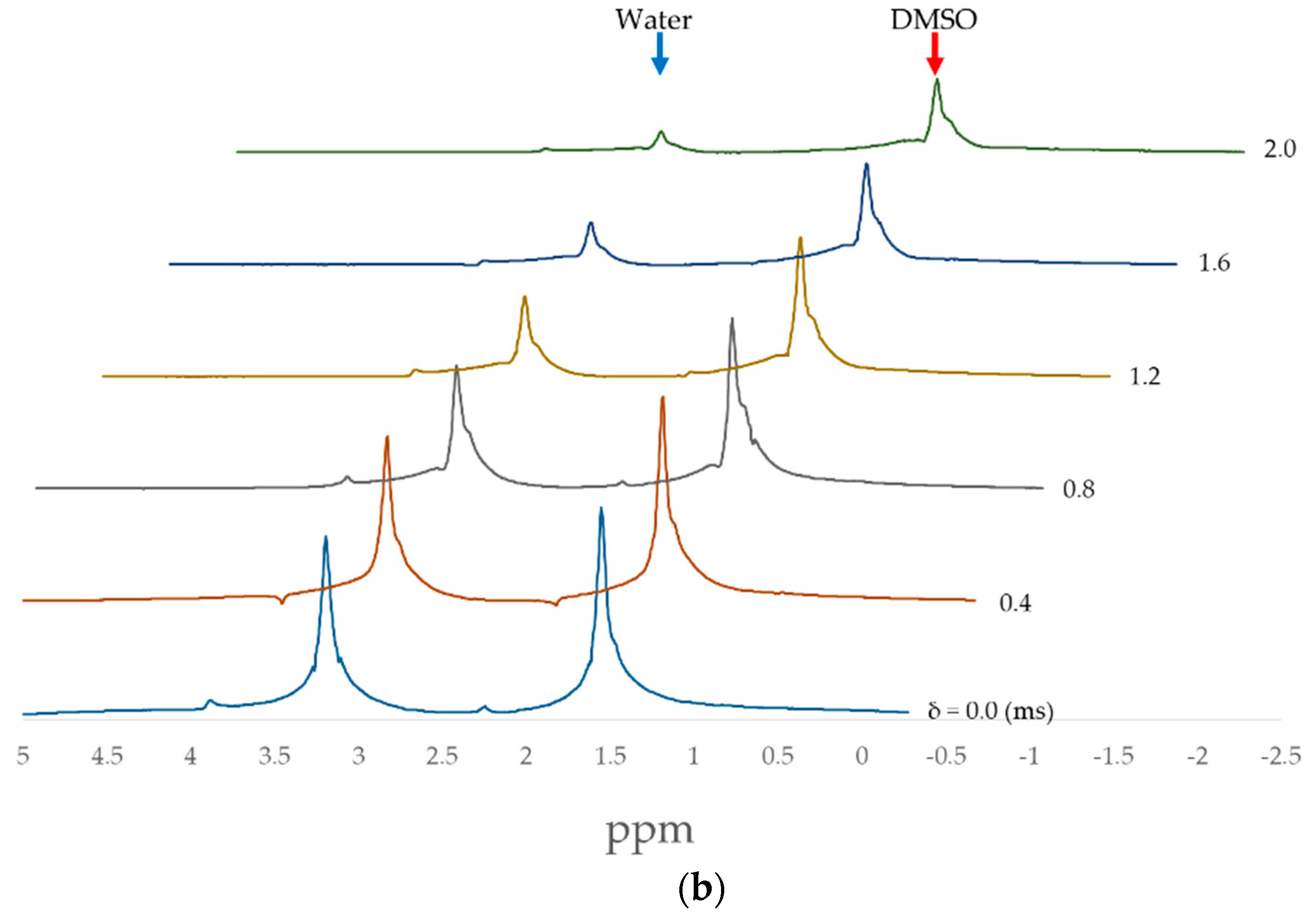
2.2. PFG-NMR Measurements
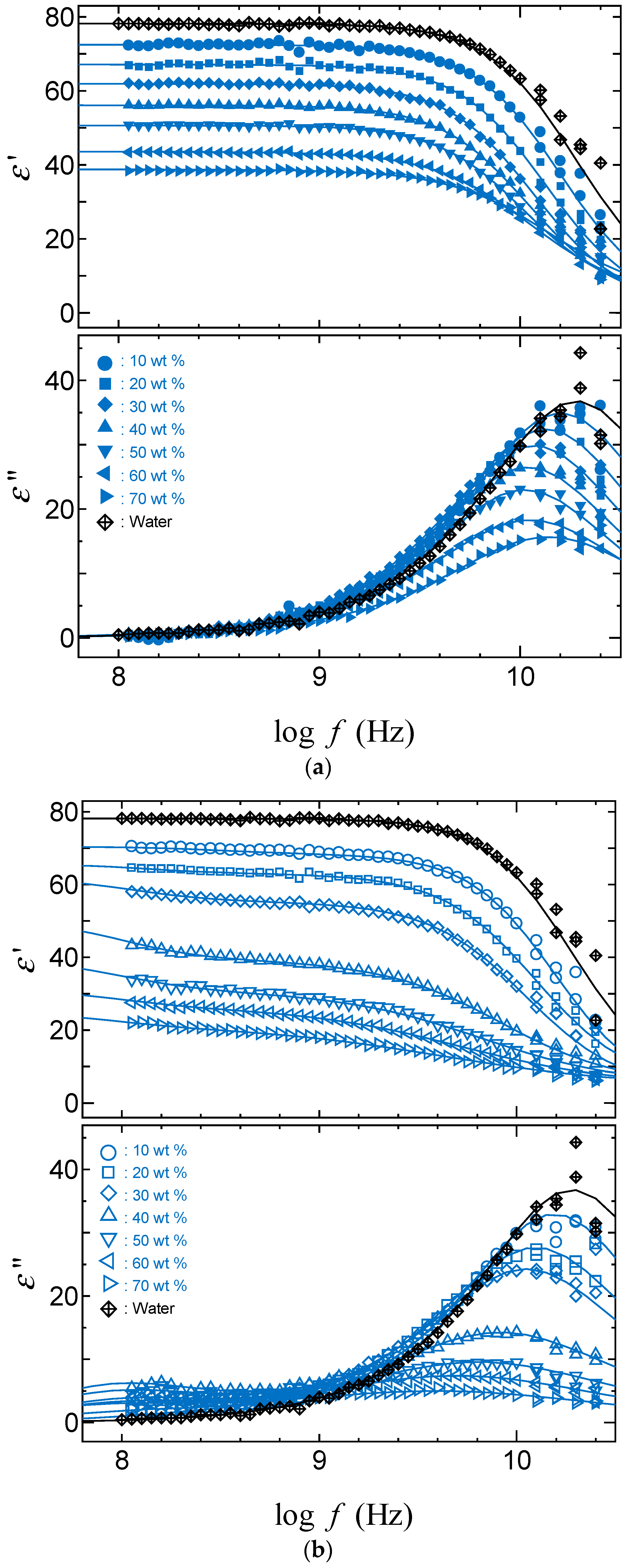
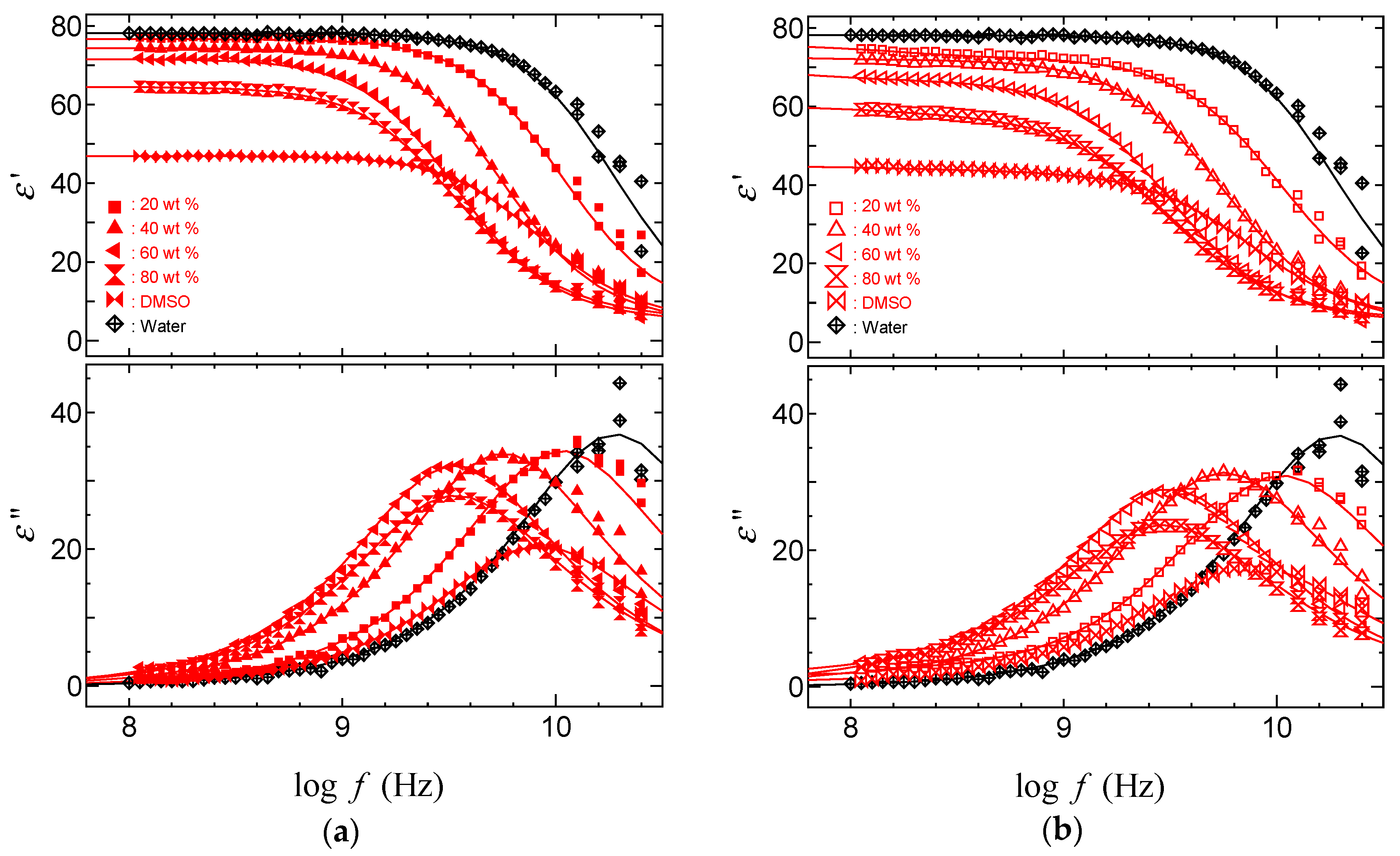
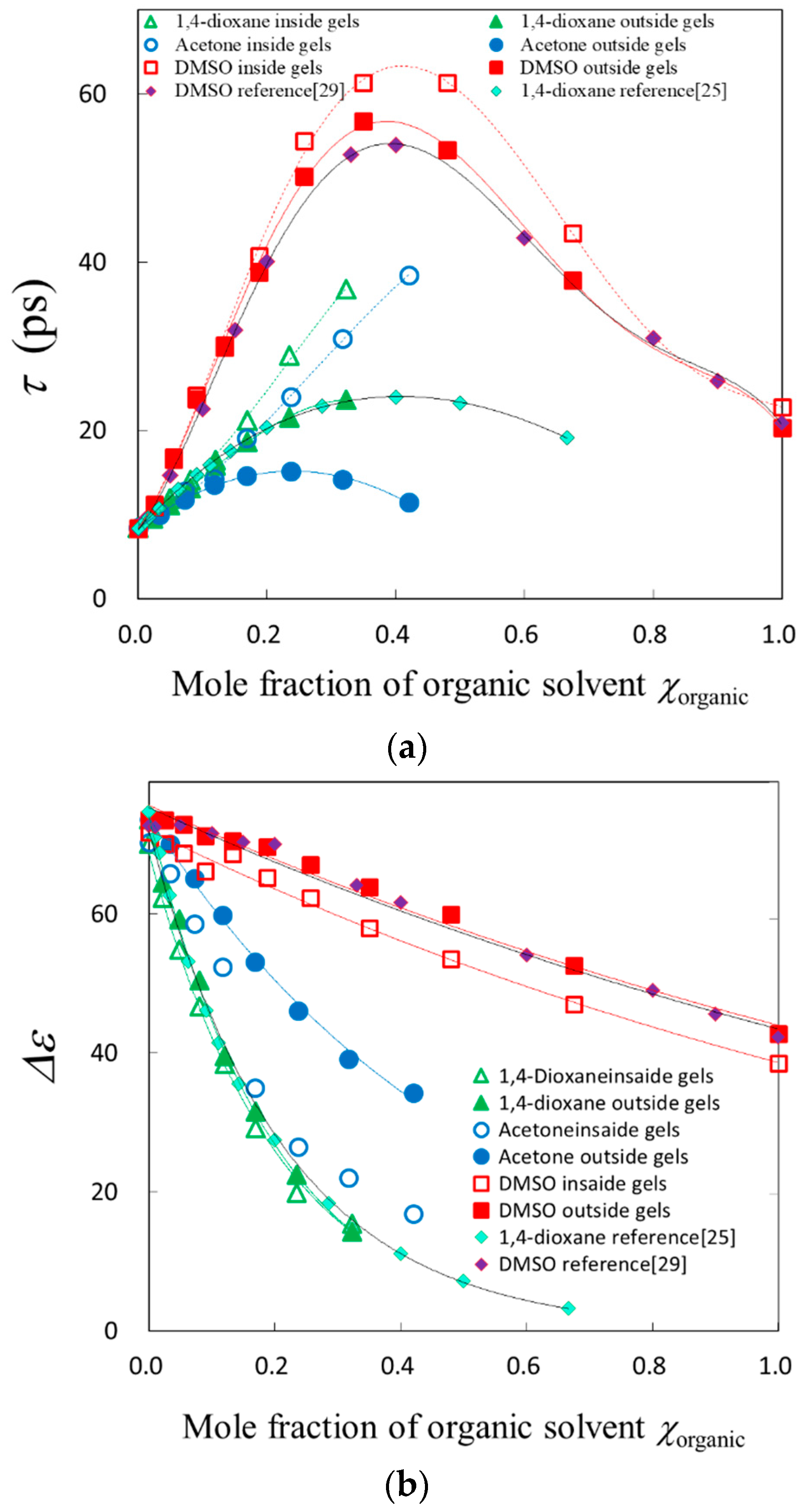
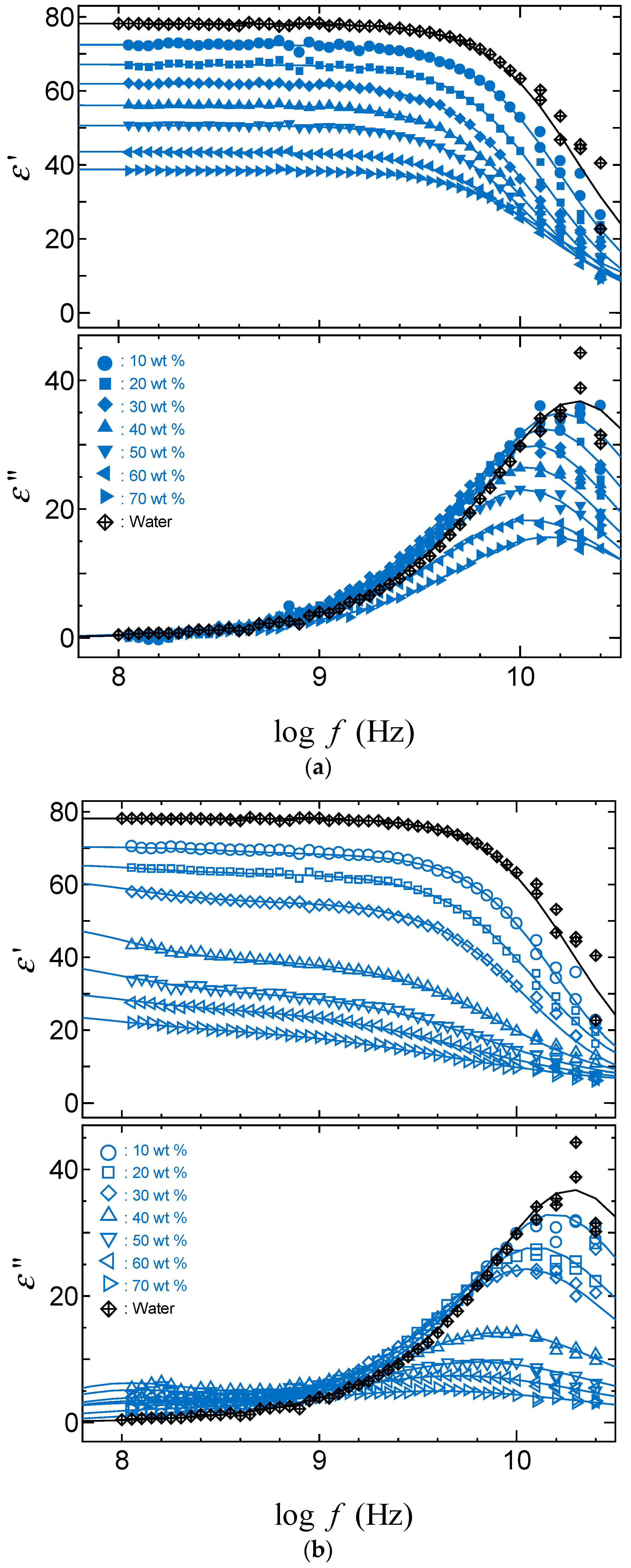
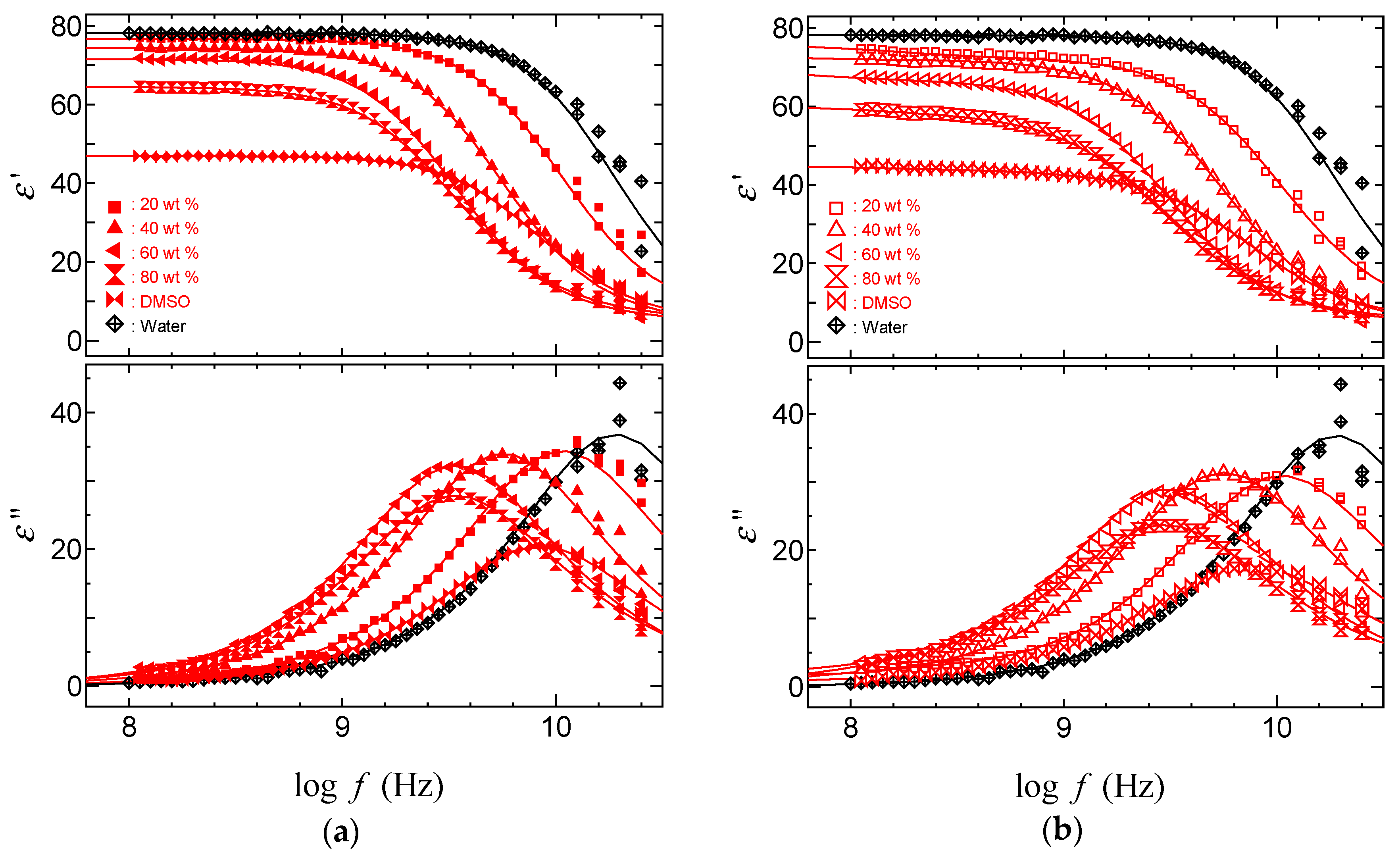
2.3. TDR Meserments
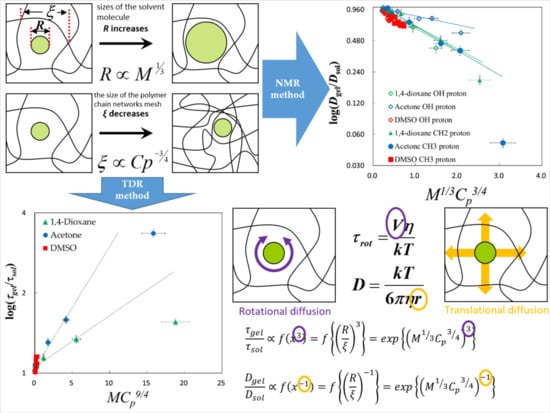
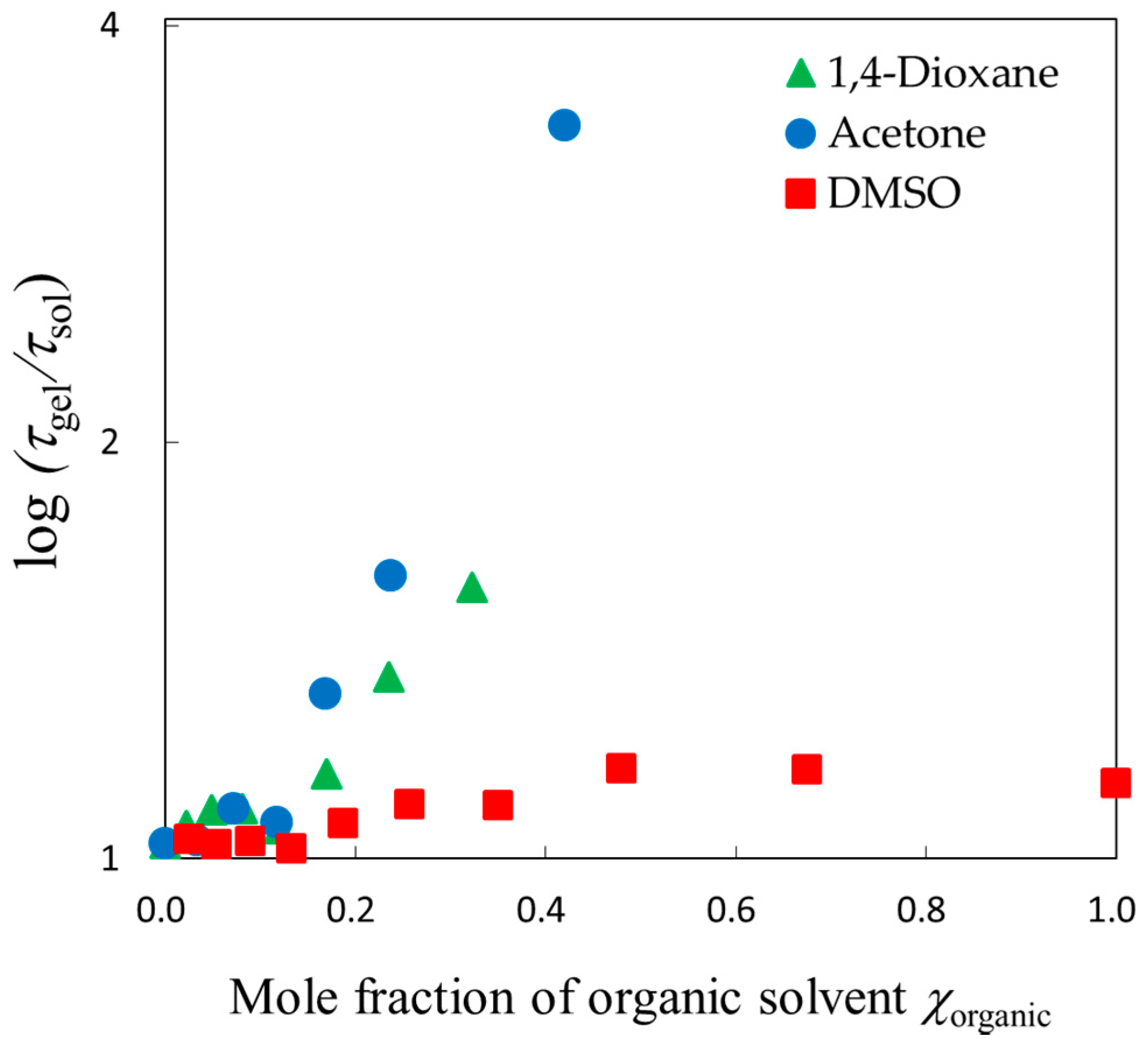
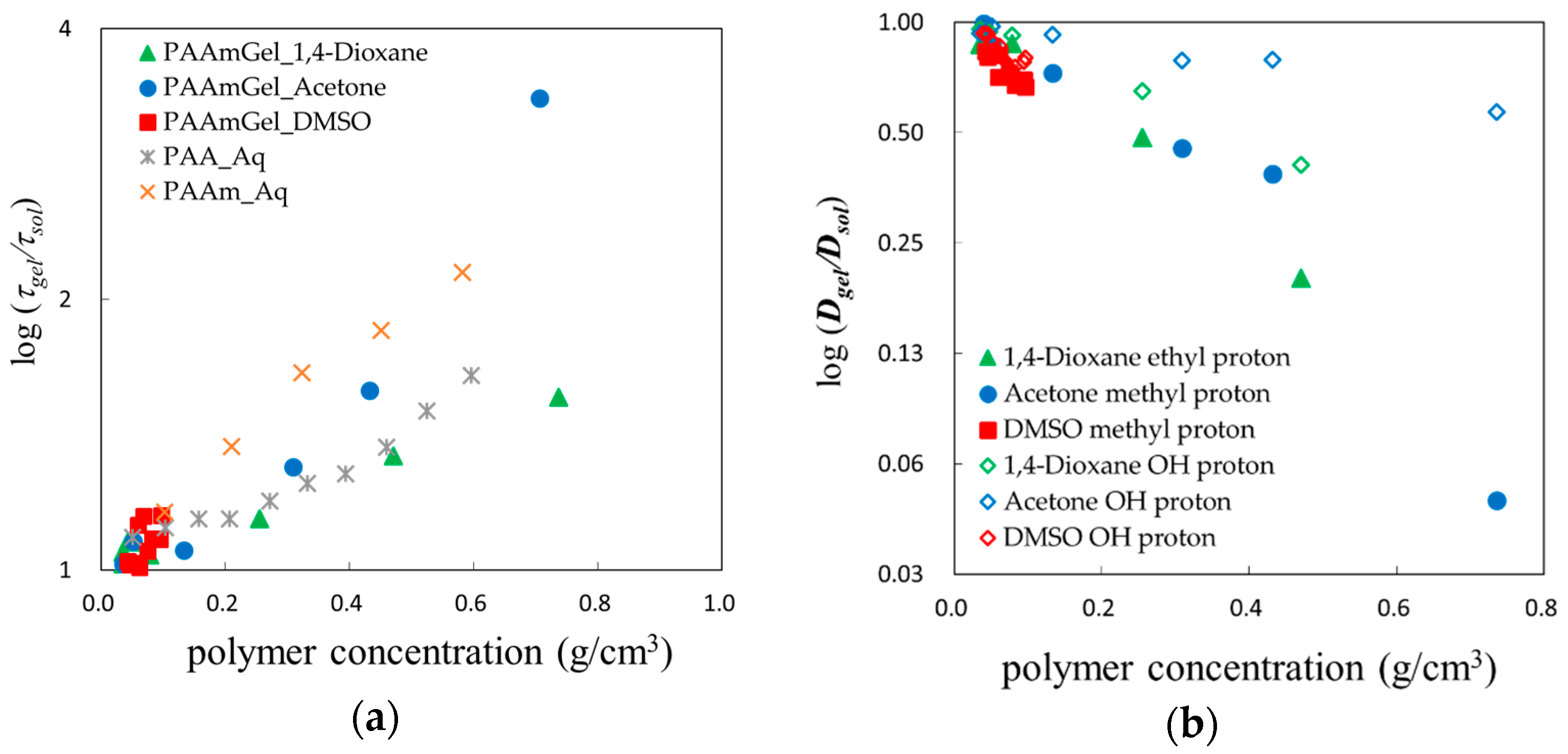
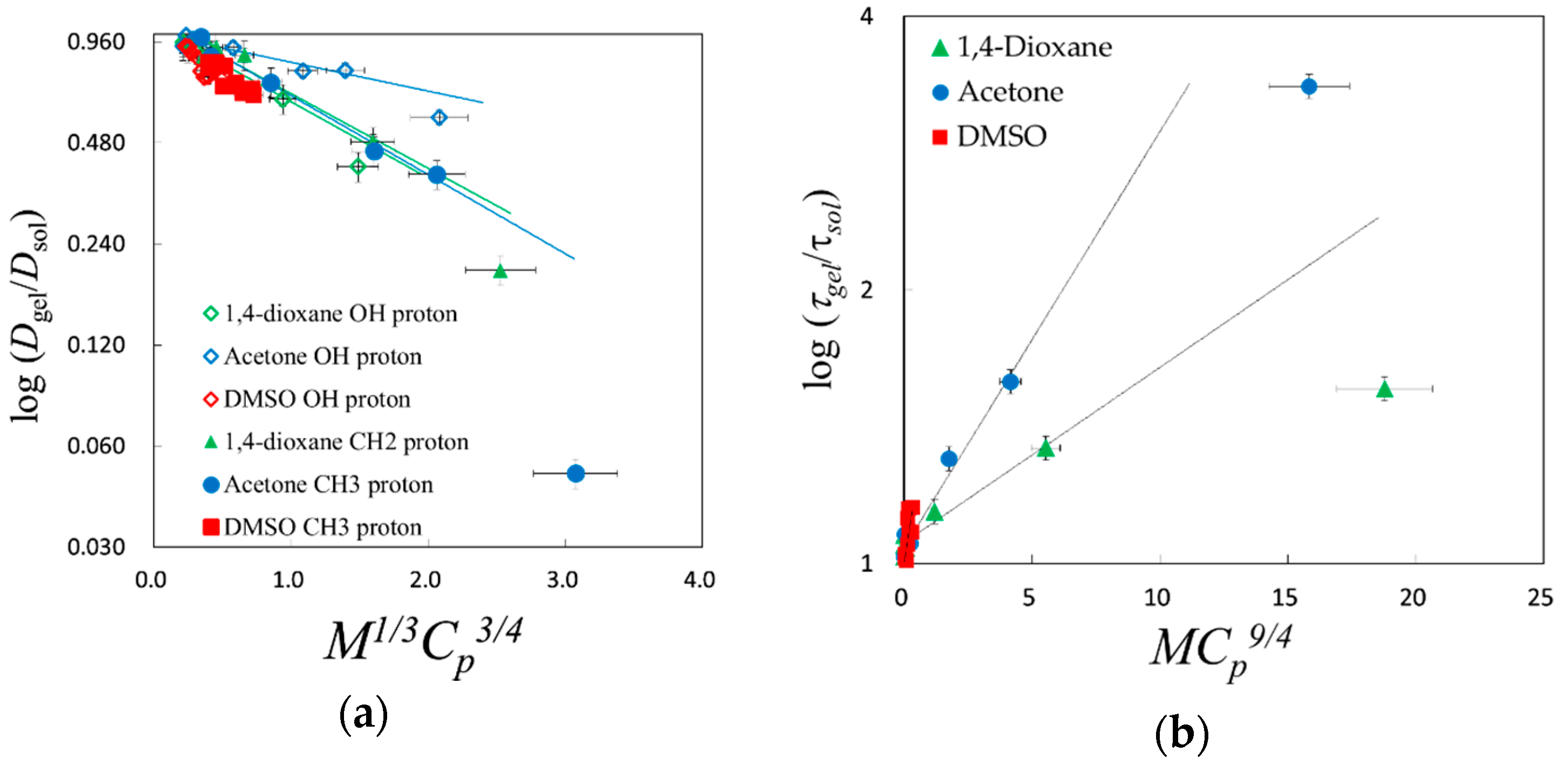
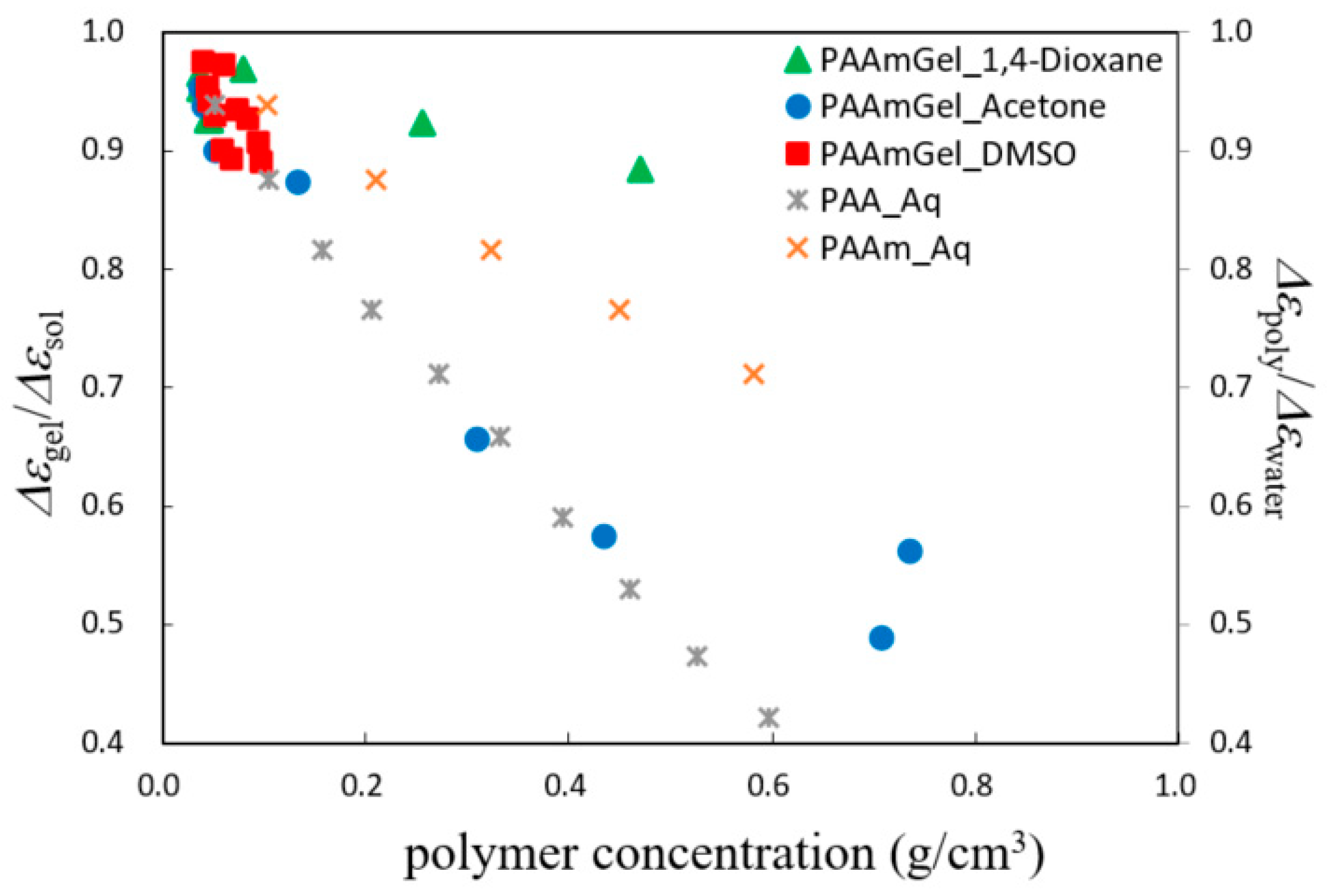
2.4. Scaling Concepts
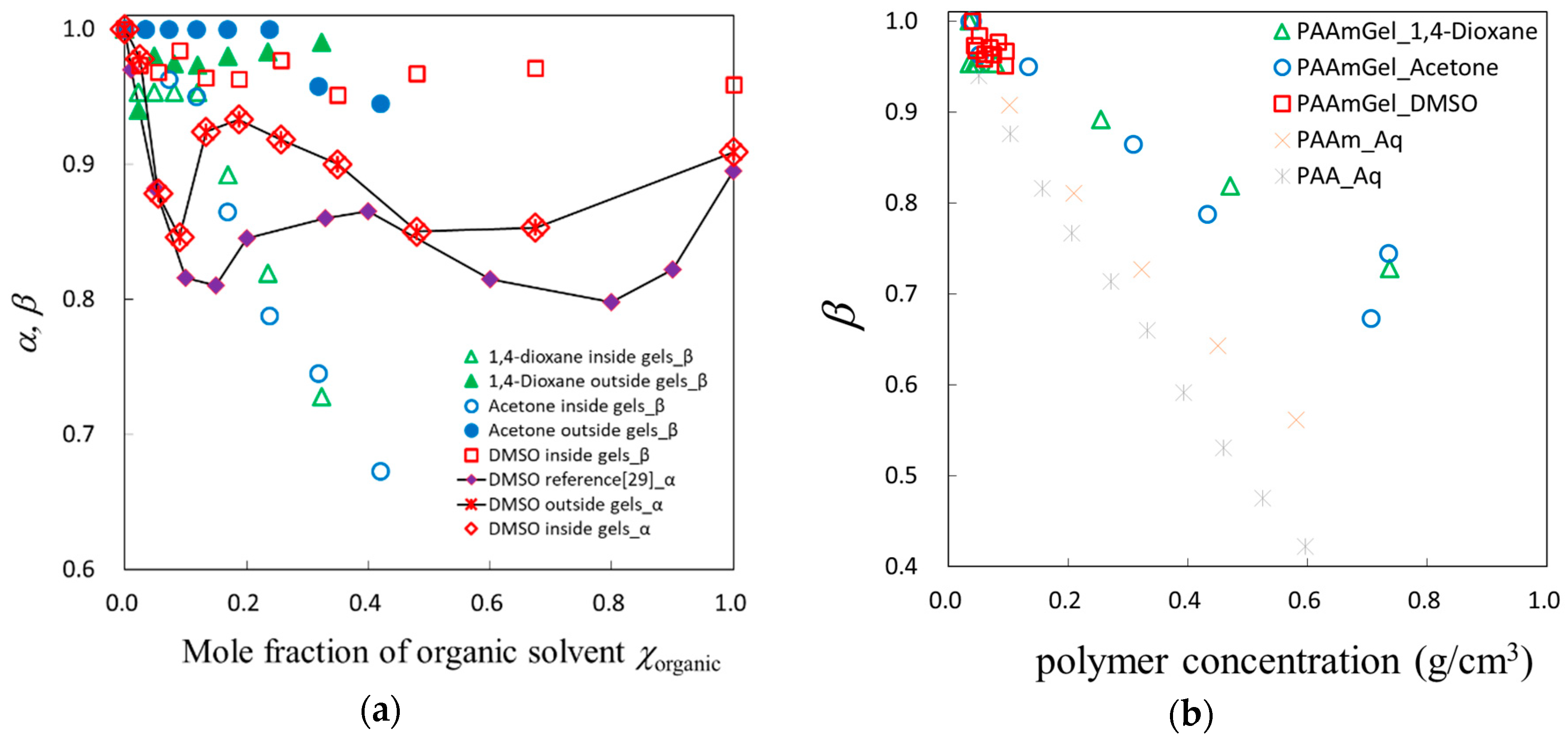
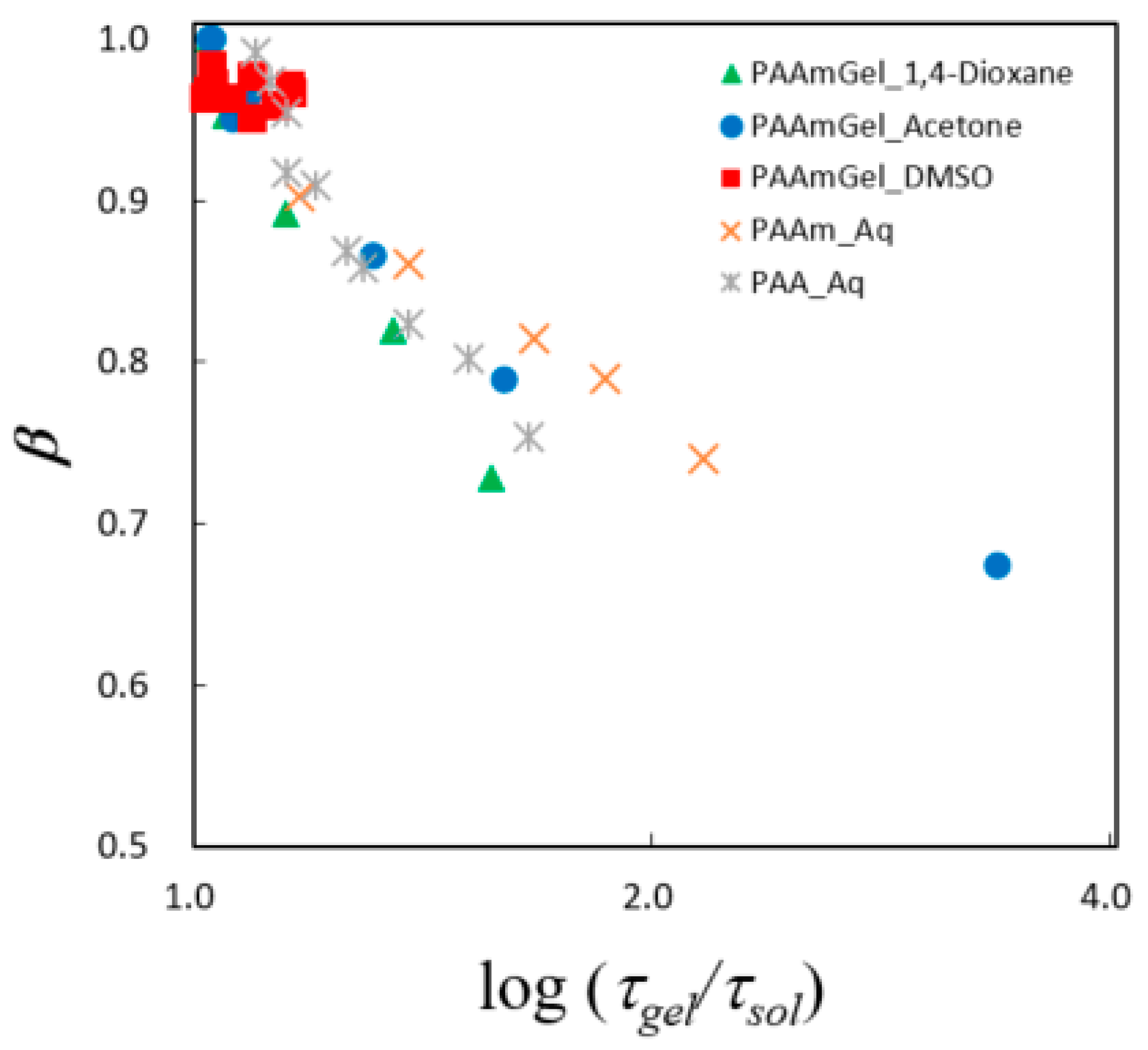
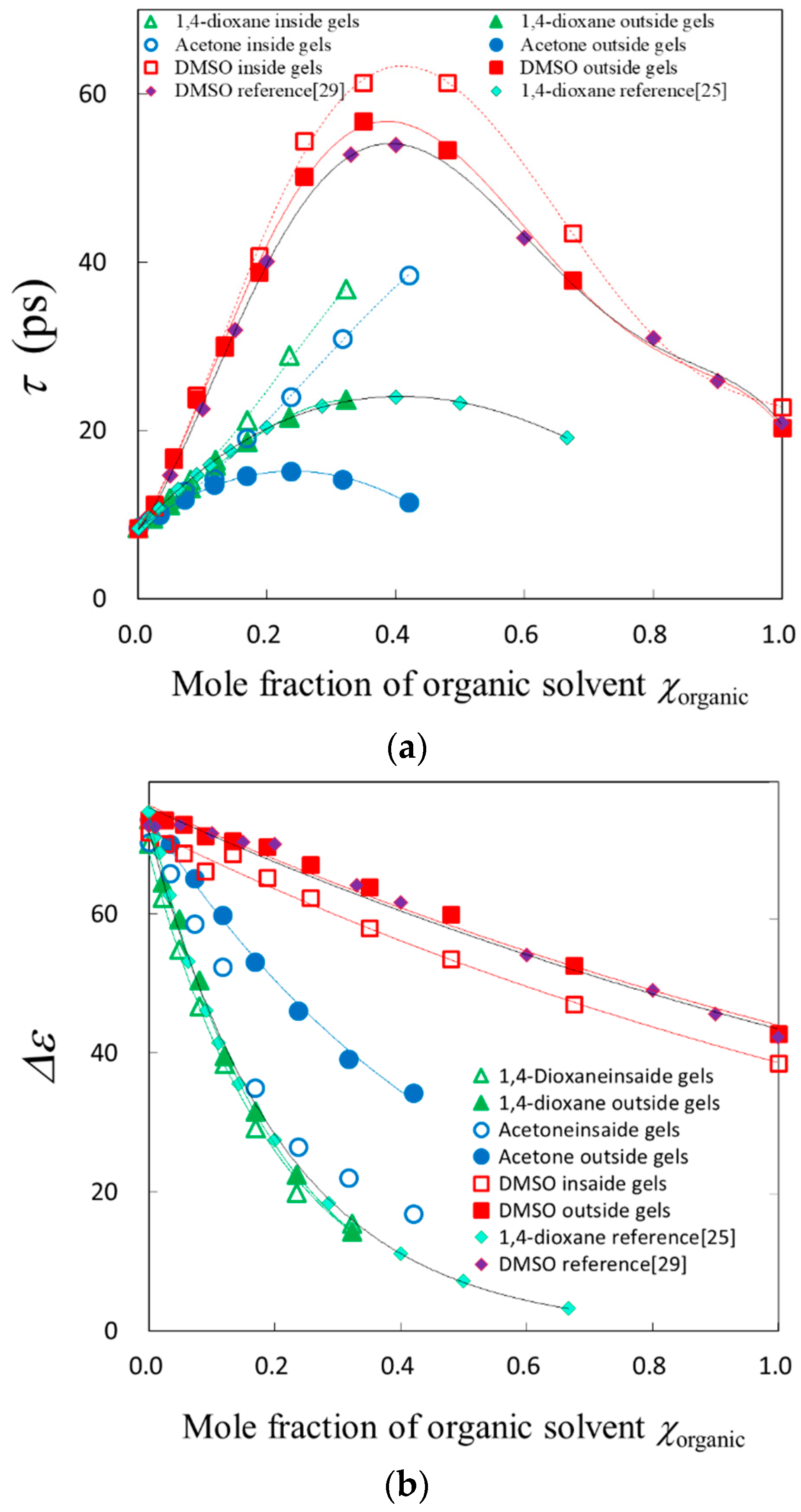
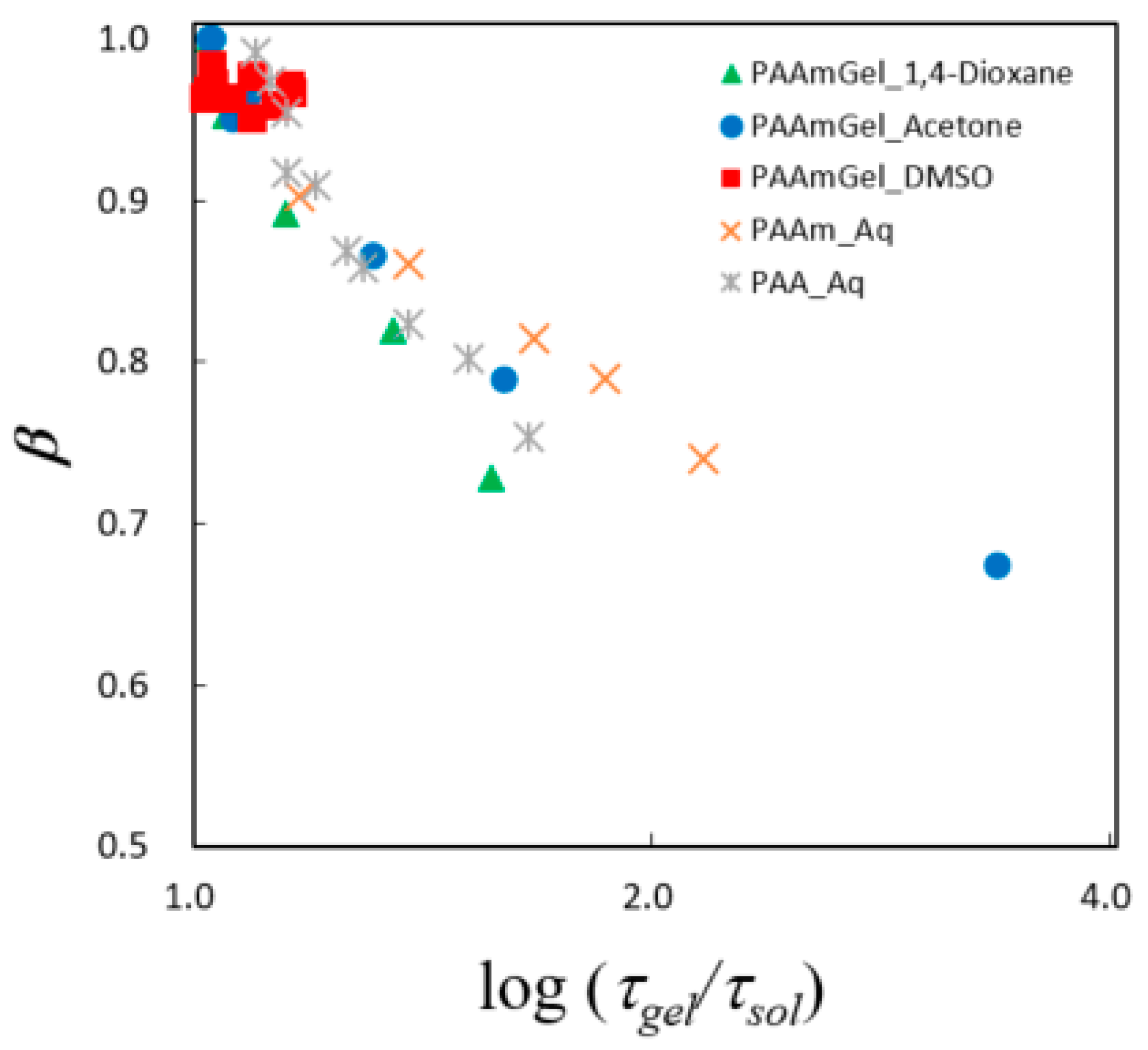
2.5. Fractal Analysis with τ–β Diagram
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Experimental Methods
4.2.1. Volume, Density, and Viscosity
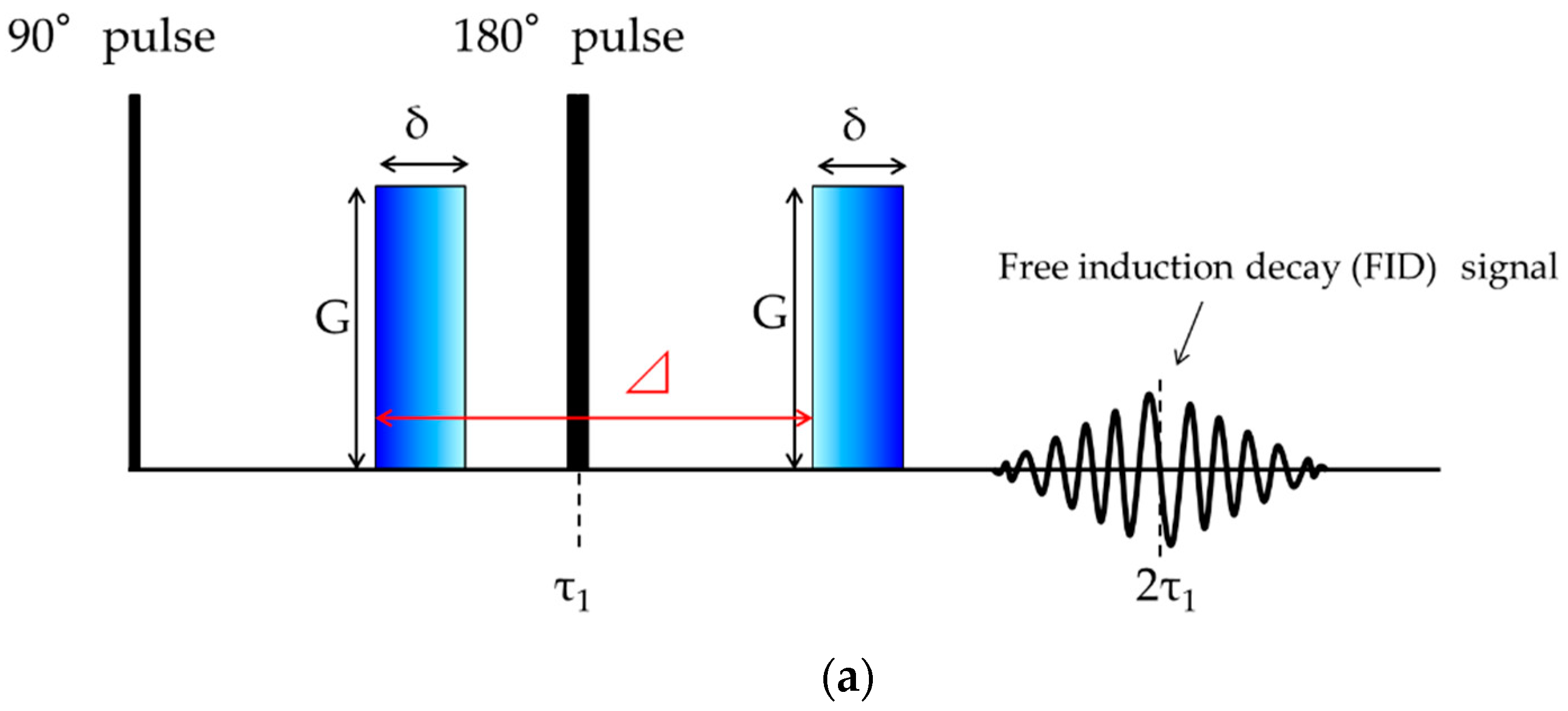
4.2.2. NMR Measurements
4.2.3. Dielectric Measurements
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Tanaka, T. Collapse of gels and the critical endpoint. Phys. Rev. Lett. 1978, 40, 820. [Google Scholar] [CrossRef]
- Tanaka, T.; Fillmore, D.J. Kinetics of swelling of gels. J. Chem. Phys. 1979, 70, 1214–1218. [Google Scholar] [CrossRef]
- Tanaka, T.; Fillmore, D.; Sun, S.T.; Nishio, I.; Swislow, G.; Shah, A. Phase transitions in ionic gels. Phys. Rev. Lett. 1980, 45, 1636. [Google Scholar] [CrossRef]
- Tanaka, T.; Sun, S.T.; Hirokawa, Y.; Katayama, S.; Kucera, J.; Hirose, Y.; Amiya, T. Mechanical instability of gels at the phase transition. Nature 1987, 325, 796. [Google Scholar] [CrossRef]
- Sato, M.E.; Tanaka, T. Kinetics of discontinuous volume—Phase transition of gels. J. Chem. Phys. 1988, 89, 1695–1703. [Google Scholar] [CrossRef]
- Miki, H.; Yagihara, S.; Mukai, S.A.; Tokita, M. Swelling equilibrium of a gel in binary mixed solvents. Prog. Colloid Polym. Sci. 2009, 136, 101–106. [Google Scholar]
- Hahn, E.L. Spin echoes. Phys. Rev. 1950, 80, 580. [Google Scholar] [CrossRef]
- Stejskal, E.O.; Tanner, J.E. Spin diffusion measurements: Spin echoes in the presence of a time-dependent field gradient. J. Chem. Phys. 1965, 42, 288–292. [Google Scholar] [CrossRef]
- Tokita, M.; Miyoshi, T.; Takegoshi, K.; Hikichi, K. Probe diffusion in gels. Phys. Rev. E 1996, 53, 1823. [Google Scholar] [CrossRef]
- Tokita, M. Transport phenomena in gel. Gels 2016, 2, 17. [Google Scholar] [CrossRef]
- Matsukawa, S.; Ando, I. A study of self-diffusion of molecules in polymer gel by pulsed-gradient spin—Echo 1H NMR. Macromolecules 1996, 29, 7136–7140. [Google Scholar] [CrossRef]
- Matsukawa, S.; Yasunaga, H.; Zhao, C.; Kuroki, S.; Kurosu, H.; Ando, I. Diffusion processes in polymer gels as studied by pulsed field-gradient spin-echo NMR spectroscopy. Prog. Polym. Sci. 1999, 24, 995–1044. [Google Scholar] [CrossRef]
- Matsukawa, S.; Sagae, D.; Mogi, A. Molecular diffusion in polysaccharide gel systems as observed by NMR. Progr. Colloid Polym. Sci. 2009, 136, 171–176. [Google Scholar]
- Yagihara, S.; Miura, N.; Hayashi, Y.; Miyairi, H.; Asano, M.; Yamada, G.; Shinyashiki, N.; Mashimo, S.; Umehara, T.; Tokita, M.; et al. Microwave dielectric study on water structure and physical properties of aqueous systems using time domain reflectometry with flat-end cells. Subsurf. Sens. Technol. Appl. 2001, 2, 15–30. [Google Scholar] [CrossRef]
- Yamada, G.; Hashimoto, T.; Morita, T.; Shinyashiki, N.; Yagihara, S.; Tokita, M. Dielectric study on dynamics for volume phase transition of PAAm gel in acetone-water system. Trans. Mater. Res. Soc. Jpn. 2001, 26, 701–704. [Google Scholar]
- Cole, R.H. Evaluation of dielectric behavior by time domain spectroscopy. I. Dielectric response by real time analysis. J. Phys. Chem. 1975, 79, 1459–1469. [Google Scholar] [CrossRef]
- Cole, R.H. Evaluation of dielectric behavior by time domain spectroscopy. II. Complex permittivity. J. Phys. Chem. 1975, 79, 1469–1474. [Google Scholar] [CrossRef]
- Cole, R.H.; Mashimo, S.; Winsor, P., IV. Evaluation of dielectric behavior by time domain spectroscopy. 3. Precision difference methods. J. Phys. Chem. 1980, 84, 786–793. [Google Scholar] [CrossRef]
- Yang, M.; Zhao, K. Influence of the structure on the collapse of poly (N-isopropylacrylamide)-based microgels: An insight by quantitative dielectric analysis. Soft Matter 2016, 12, 4093–4102. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Liu, C.; Zhao, K. Concentration dependent phase behavior and collapse dynamics of PNIPAM microgel by dielectric relaxation. Phys. Chem. Chem. Phys. 2017, 19, 15433–15443. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Liu, C.; Lian, Y.; Zhao, K.; Zhu, D.; Zhou, J. Relaxations and phase transitions during the collapse of a dense PNIPAM microgel suspension—Thorough insight using dielectric spectroscopy. Soft Matter 2017, 13, 2663–2676. [Google Scholar] [CrossRef] [PubMed]
- Yagihara, S.; Asano, M.; Kosuge, M.; Tsubotani, S.; Imoto, D.; Shinyashiki, N. Dynamical behavior of unfreezable molecules restricted in a frozen matrix. J. Non-Cryst. Solids 2005, 351, 2629–2634. [Google Scholar] [CrossRef]
- Shinyashiki, N.; Imoto, D.; Yagihara, S. Broadband dielectric study of dynamics of polymer and solvent in poly (vinyl pyrrolidone)/normal alcohol mixtures. J. Phys. Chem. B 2007, 111, 2181–2187. [Google Scholar] [CrossRef] [PubMed]
- Mashimo, S.; Miura, N.; Umehara, T.; Yagihara, S.; Higasi, K. The structure of water and methanol in p-dioxane as determined by microwave dielectric spectroscopy. J. Chem. Phys. 1992, 96, 6358–6361. [Google Scholar] [CrossRef]
- Schrödle, S.; Hefter, G.; Buchner, R. Dielectric spectroscopy of hydrogen bond dynamics and microheterogenity of water + dioxane mixtures. J. Phys. Chem. B 2007, 111, 5946–5955. [Google Scholar] [CrossRef] [PubMed]
- Cole, R.H. Correlation function theory of dielectric relaxation. J. Chem. Phys. 1965, 42, 637–643. [Google Scholar] [CrossRef]
- Davidson, D.W.; Cole, R.H. Dielectric relaxation in glycerol, propylene glycol, and n-propanol. J. Chem. Phys. 1951, 19, 1484–1490. [Google Scholar] [CrossRef]
- Kaatze, U.; Pottel, R.; Schäfer, M. Dielectric spectrum of dimethyl sulfoxide/water mixtures as a function of composition. J. Phys. Chem. 1989, 93, 5623–5627. [Google Scholar] [CrossRef]
- Lu, Z.; Manias, E.; Macdonald, D.D.; Lanagan, M. Dielectric relaxation in dimethyl sulfoxide/water mixtures studied by microwave dielectric relaxation spectroscopy. J. Phys. Chem. A 2009, 113, 12207–12214. [Google Scholar] [CrossRef] [PubMed]
- Havriliak, S.; Negami, S. A complex plane representation of dielectric and mechanical relaxation processes in some polymers. Polymer 1967, 8, 161–210. [Google Scholar] [CrossRef]
- Yagihara, S.; Nozaki, R.; Takeishi, S.; Mashimo, S. Evaluation of dielectric permittivity by dc transient current. J. Chem. Phys. 1983, 79, 2419–2422. [Google Scholar] [CrossRef]
- Nozaki, R.; Mashimo, S. Dielectric relaxation measurements of poly (vinyl acetate) in glassy state in the frequency range 10−6–106 Hz. J. Chem. Phys. 1987, 87, 2271–2277. [Google Scholar] [CrossRef]
- Kato, S.; Kita, R.; Shinyashiki, N.; Yagihara, S.; Fukuzaki, M. Diffusion phenomenon of molecules in 1, 4-dioxane-water system observed by 1H NMR method. Proc. Sch. Sci. Tokai Univ. 2009, 44, 53–61. [Google Scholar]
- De Gennes, P.G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, USA, 1979. [Google Scholar]
- Yagihara, S.; Oyama, M.; Inoue, A.; Asano, M.; Sudo, S.; Shinyashiki, N. Dielectric relaxation measurement and analysis of restricted water structure in rice kernels. Meas. Sci. Technol. 2007, 18, 983. [Google Scholar] [CrossRef]
- Kundu, S.K.; Yagihara, S.; Yoshida, M.; Shibayama, M. Microwave dielectric study of an oligomeric electrolyte gelator by time domain reflectometry. J. Phys. Chem. B 2009, 113, 10112–10116. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, Y.; Numamoto, Y.; Saito, H.; Kita, R.; Shinyashiki, N.; Yagihara, S.; Fukuzaki, M. Complementary analyses of fractal and dynamic water structures in protein-water mixtures and cheeses water structures. Colloids Surf. A Physicochem. Eng. Asp. 2014, 440, 42–48. [Google Scholar] [CrossRef]
- Ryabov, Y.E.; Feldman, Y.; Shinyashiki, N.; Yagihara, S. The symmetric broadening of the water relaxation peak in polymer–water mixtures and its relationship to the hydrophilic and hydrophobic properties of polymers. J. Chem. Phys. 2002, 116, 8610–8615. [Google Scholar] [CrossRef]
- Feldman, Y.; Puzenko, A.; Ryabov, Y. Non-Debye dielectric relaxation in complex materials. Chem. Phys. 2002, 284, 139–168. [Google Scholar] [CrossRef]
- Yagihara, S.; Asano, M.; Shinyashiki, N. Broadband dielectric spectroscopy study on hydration of cement and some aqueous solution and dispersion systems. Conf. Proc. Int. Soc. Electromagn. Aquametry 2007, 7, 11–18. [Google Scholar]
- Holz, M.; Heil, S.R.; Sacco, A. Temperature-dependent self-diffusion coefficients of water and six selected molecular liquids for calibration in accurate 1H NMR PFG measurements. Phys. Chem. Chem. Phys. 2000, 2, 4740–4742. [Google Scholar] [CrossRef]
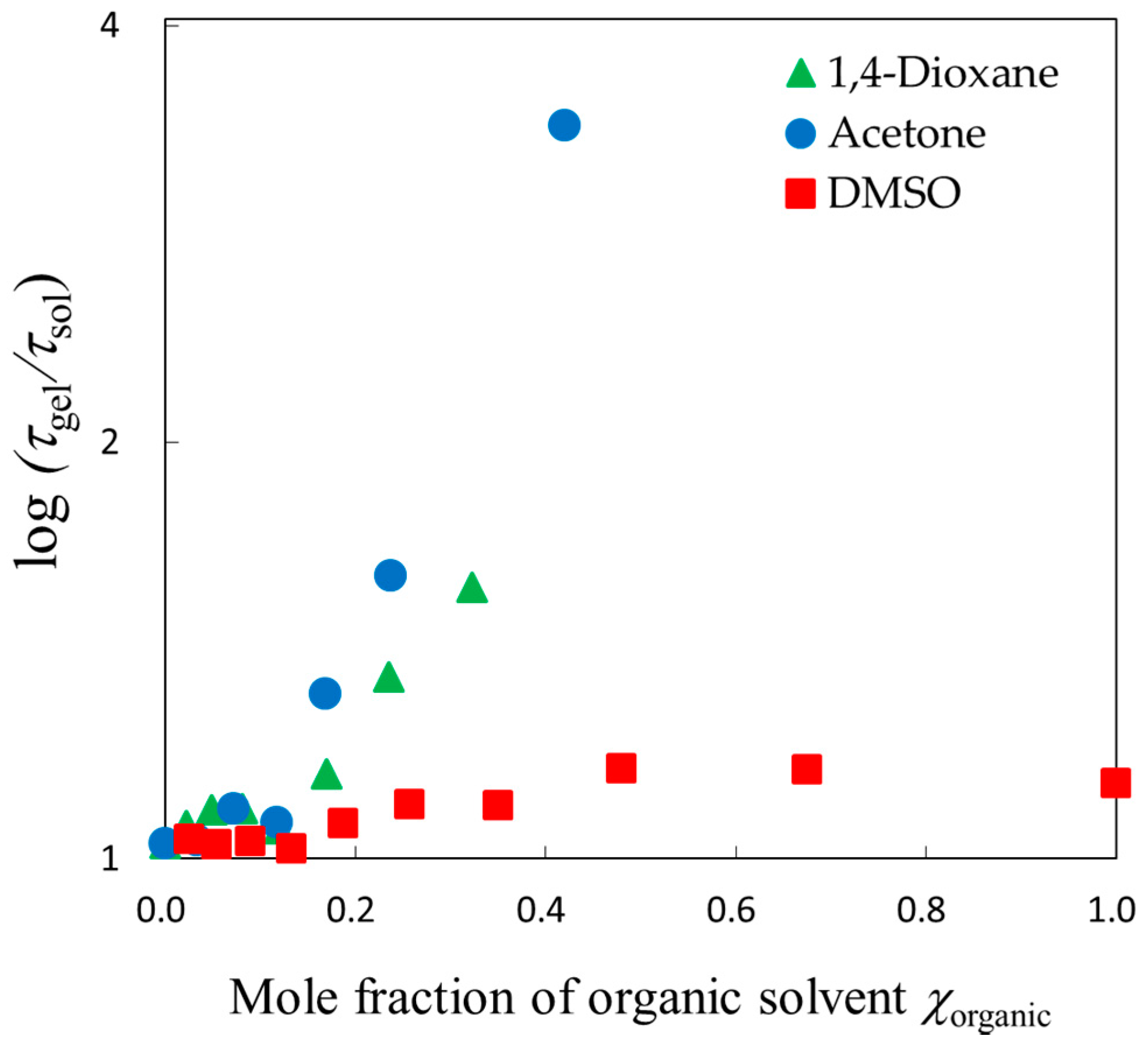
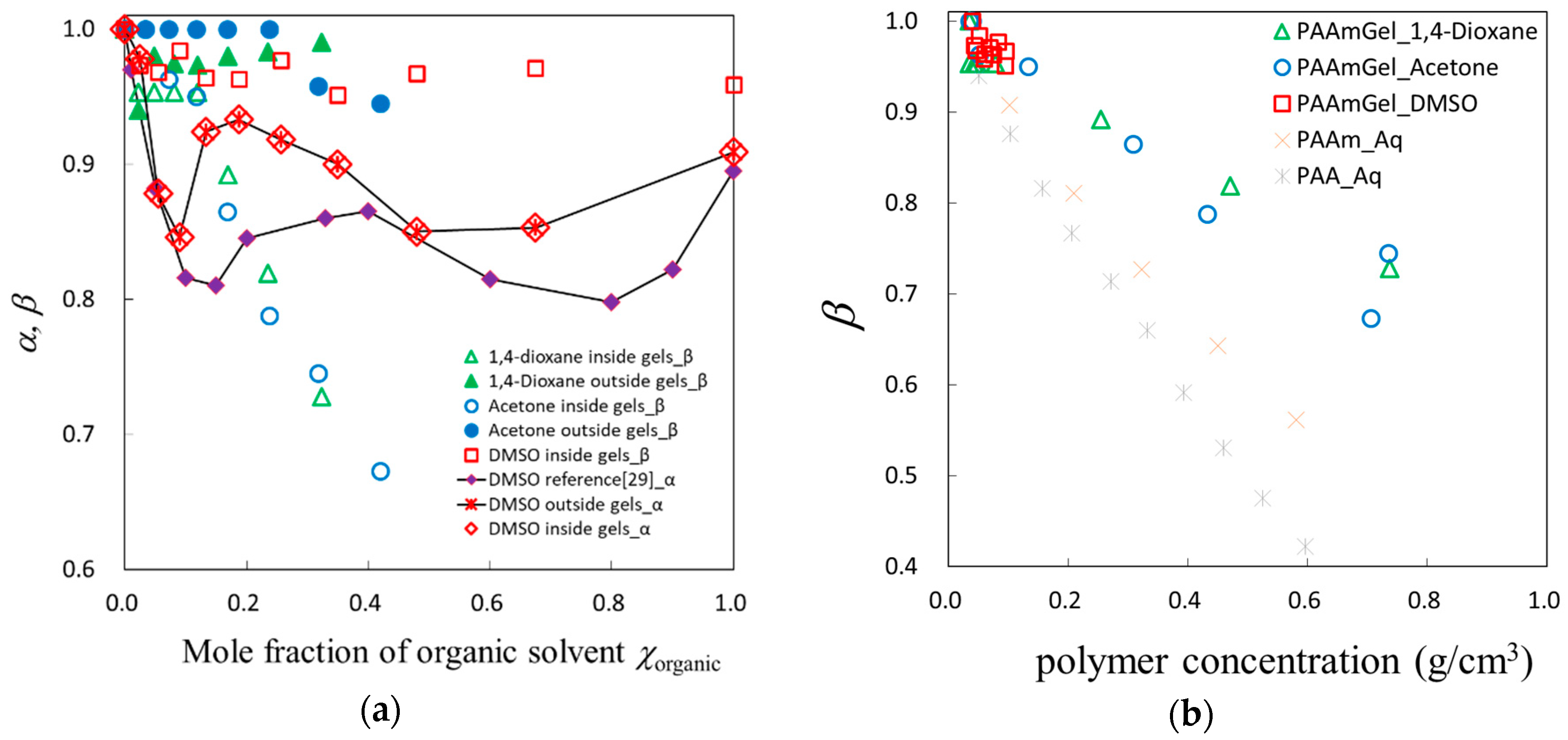
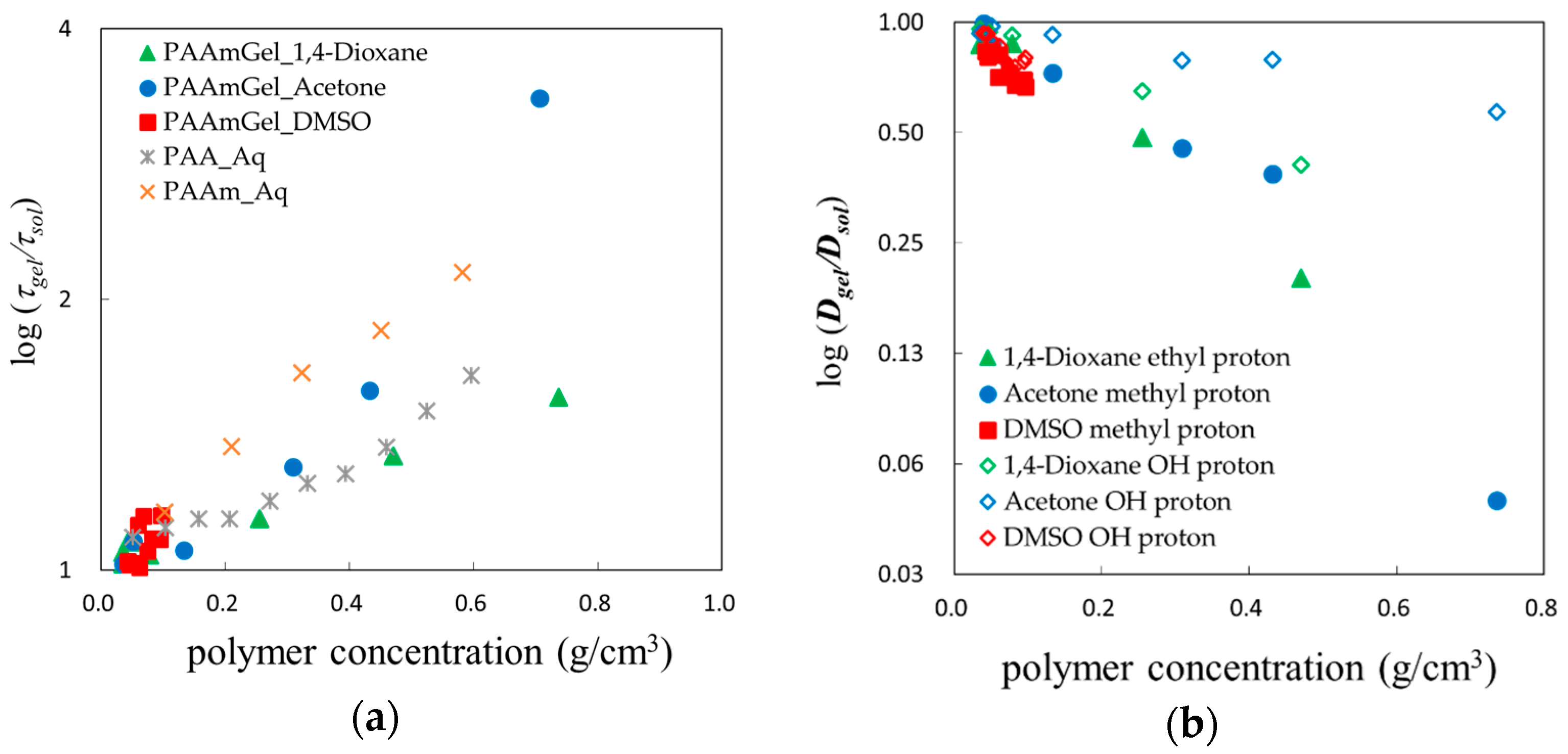
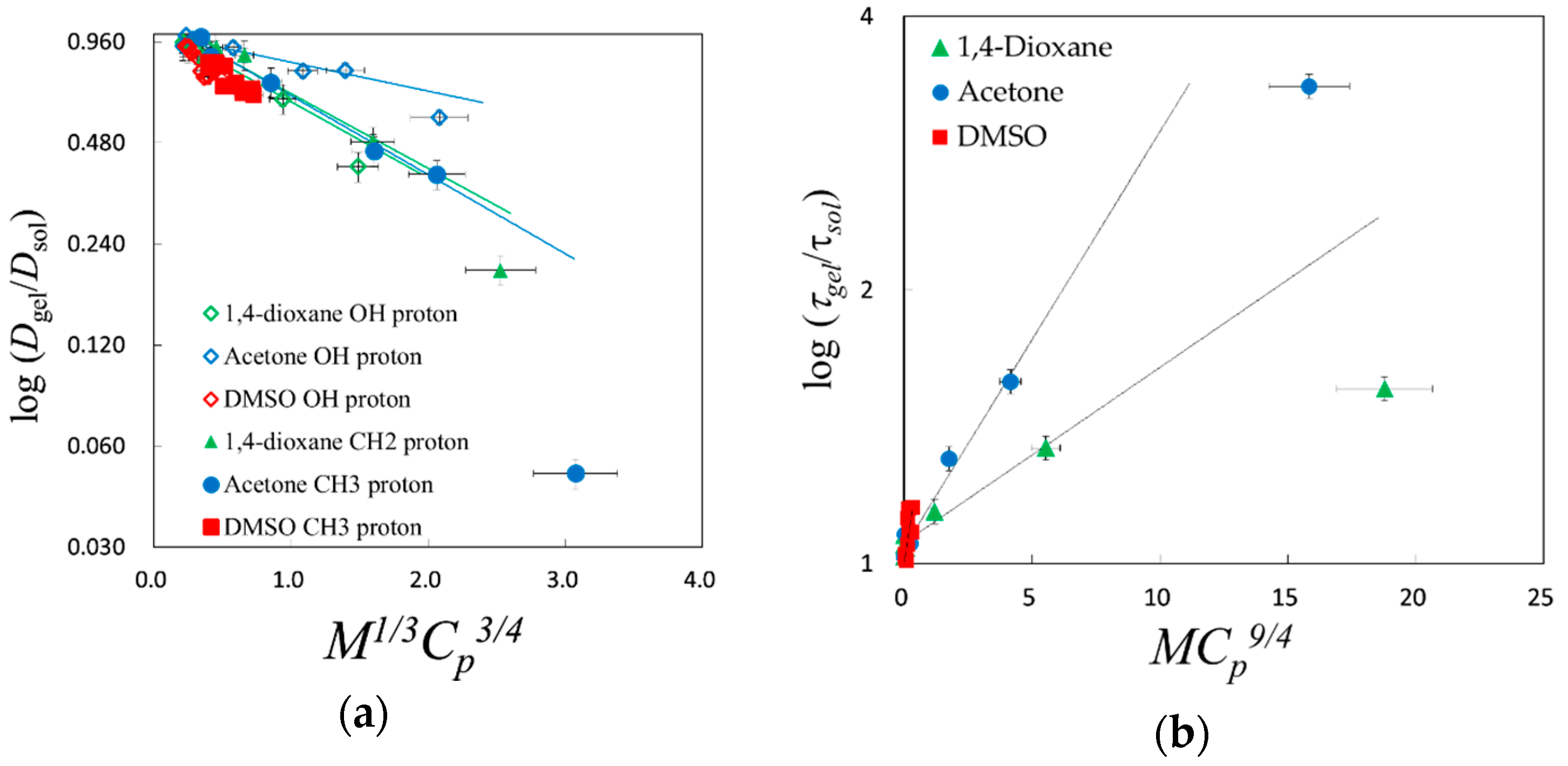
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saito, H.; Kato, S.; Matsumoto, K.; Umino, Y.; Kita, R.; Shinyashiki, N.; Yagihara, S.; Fukuzaki, M.; Tokita, M. Dynamic Behaviors of Solvent Molecules Restricted in Poly (Acryl Amide) Gels Analyzed by Dielectric and Diffusion NMR Spectroscopy. Gels 2018, 4, 56. https://doi.org/10.3390/gels4030056
Saito H, Kato S, Matsumoto K, Umino Y, Kita R, Shinyashiki N, Yagihara S, Fukuzaki M, Tokita M. Dynamic Behaviors of Solvent Molecules Restricted in Poly (Acryl Amide) Gels Analyzed by Dielectric and Diffusion NMR Spectroscopy. Gels. 2018; 4(3):56. https://doi.org/10.3390/gels4030056
Chicago/Turabian StyleSaito, Hironobu, Shunpei Kato, Keisuke Matsumoto, Yuya Umino, Rio Kita, Naoki Shinyashiki, Shin Yagihara, Minoru Fukuzaki, and Masayuki Tokita. 2018. "Dynamic Behaviors of Solvent Molecules Restricted in Poly (Acryl Amide) Gels Analyzed by Dielectric and Diffusion NMR Spectroscopy" Gels 4, no. 3: 56. https://doi.org/10.3390/gels4030056
APA StyleSaito, H., Kato, S., Matsumoto, K., Umino, Y., Kita, R., Shinyashiki, N., Yagihara, S., Fukuzaki, M., & Tokita, M. (2018). Dynamic Behaviors of Solvent Molecules Restricted in Poly (Acryl Amide) Gels Analyzed by Dielectric and Diffusion NMR Spectroscopy. Gels, 4(3), 56. https://doi.org/10.3390/gels4030056