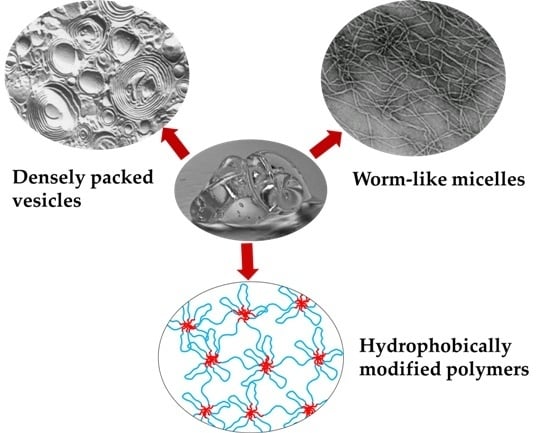
Gels Obtained by Colloidal Self-Assembly of Amphiphilic Molecules



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
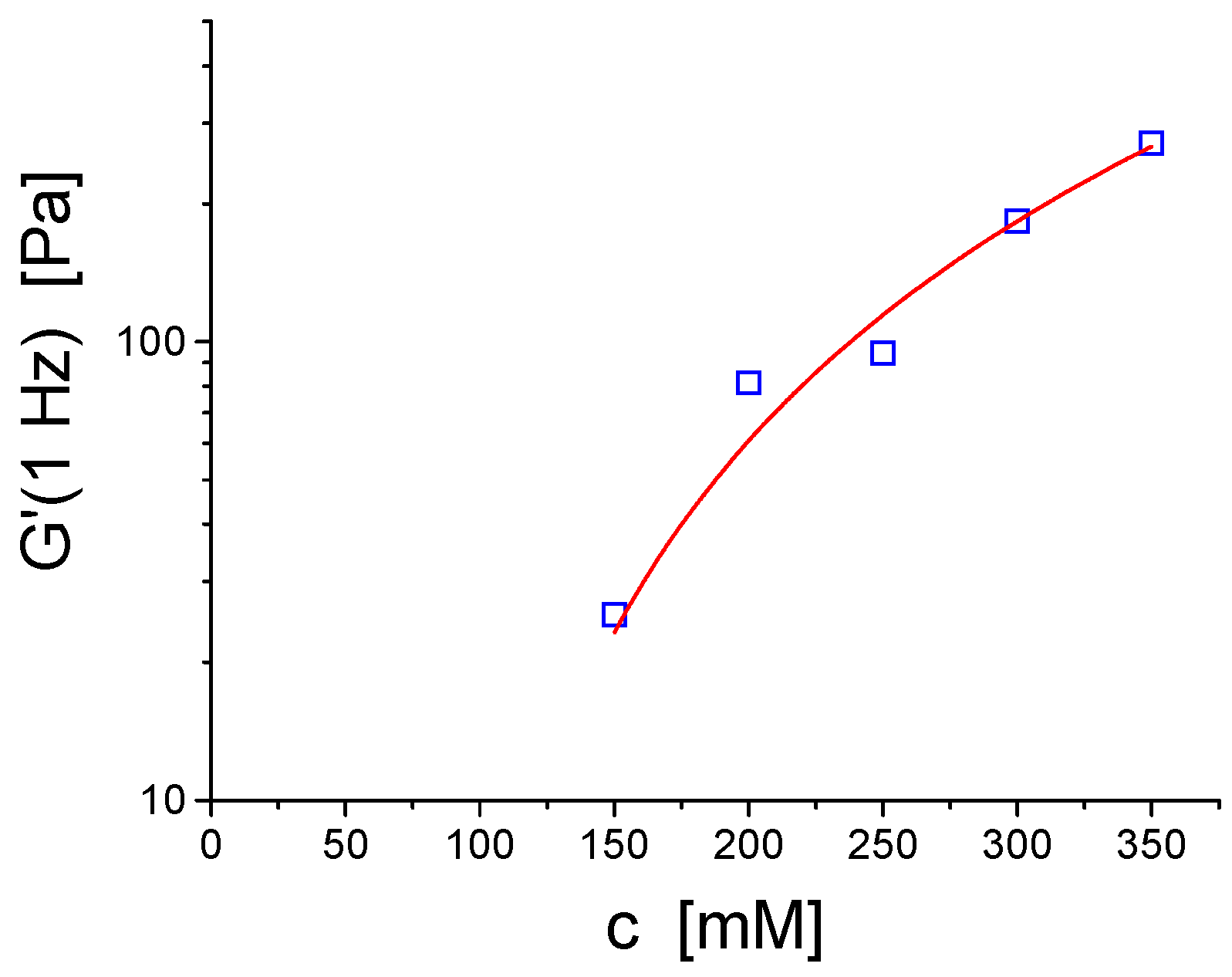
2. Viscoelastic Networks of Wormlike Micelles
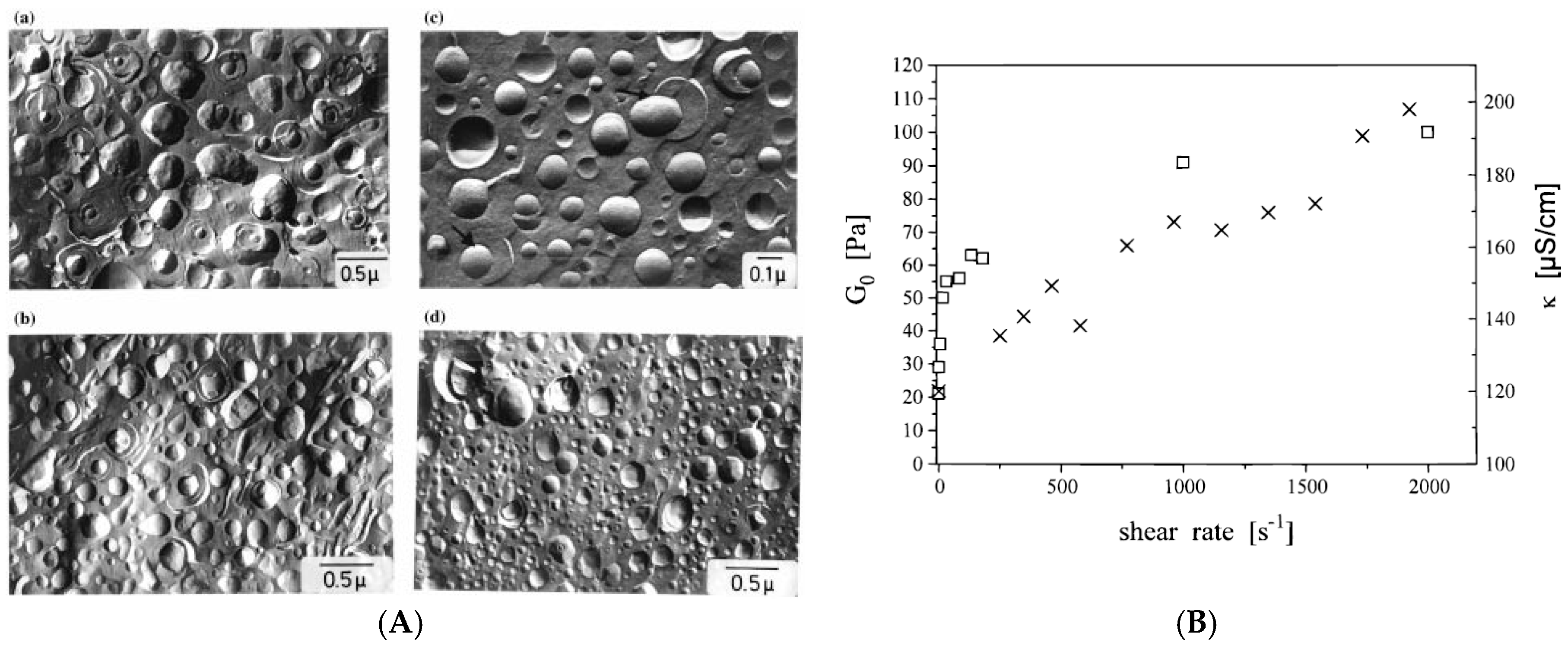
3. Densely Packed Vesicle Gels
3.1. Vesicle Gels Based on Unilamellar Vesicles (ULVs)
3.2. Vesicle Gels Based on Multilamellar Vesicles (MLVs)
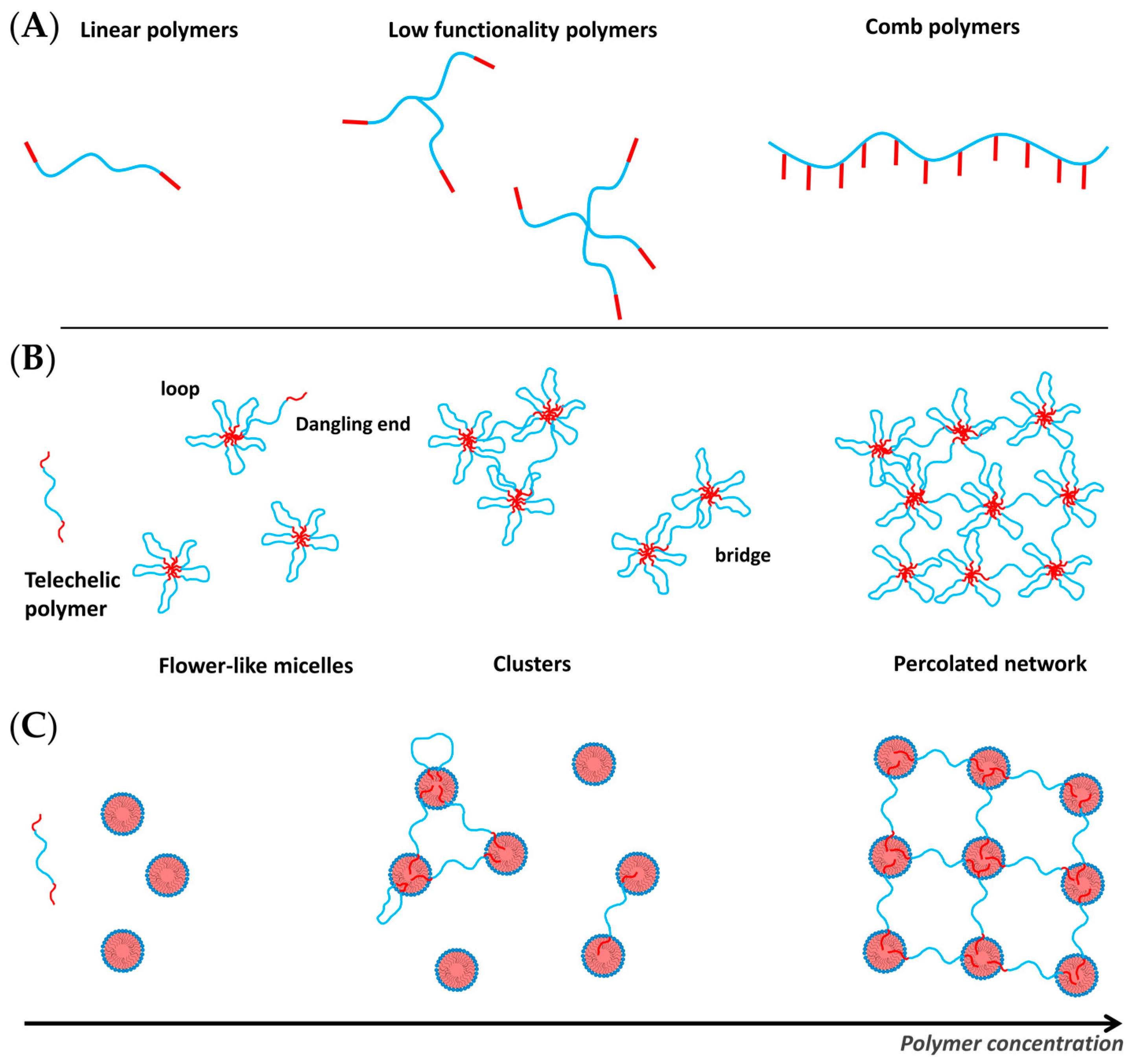
4. Hydrophobically Modified Polymers
- Telechelic polymers, which are linear polymers end-capped with two stickers, alkyl chains or short hydrophobic blocks;
- Low functionality multisticker polymers;
- Multisticker grafted polymer chains with randomly distributed pendant hydrophobes along the hydrophilic chain (comb polymers).
4.1. Telechelic Polymers
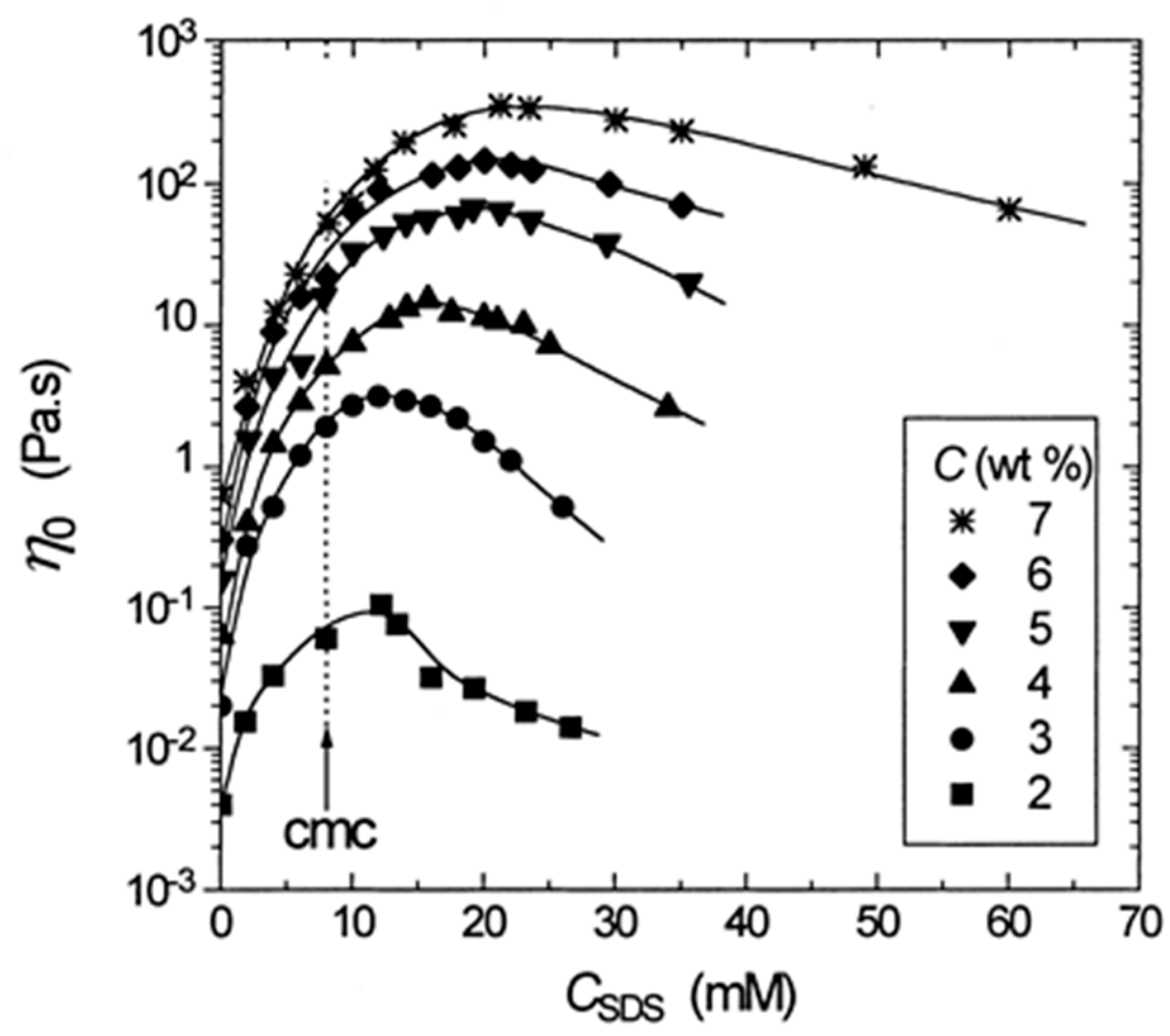
4.2. Amphiphilic Polymers with Multiple Hydrophobic Stickers
4.3. Stimuli Responsive Copolymers
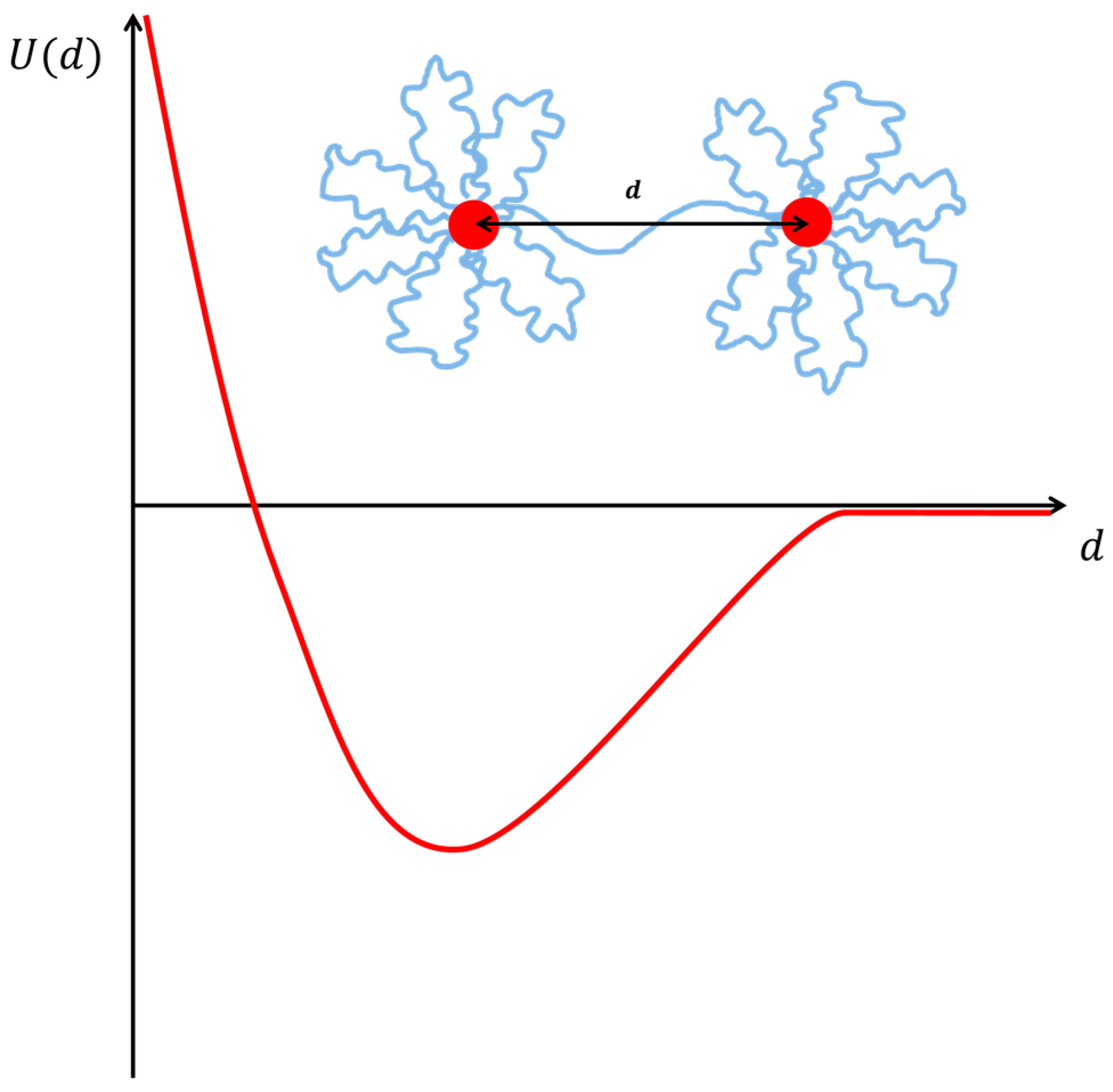
5. Micellar Systems, Microemulsions or Vesicles Cross-linked by Amphiphilic Polymers
5.1. Surfactant Micelles Interacting with Amphiphilic Polymers
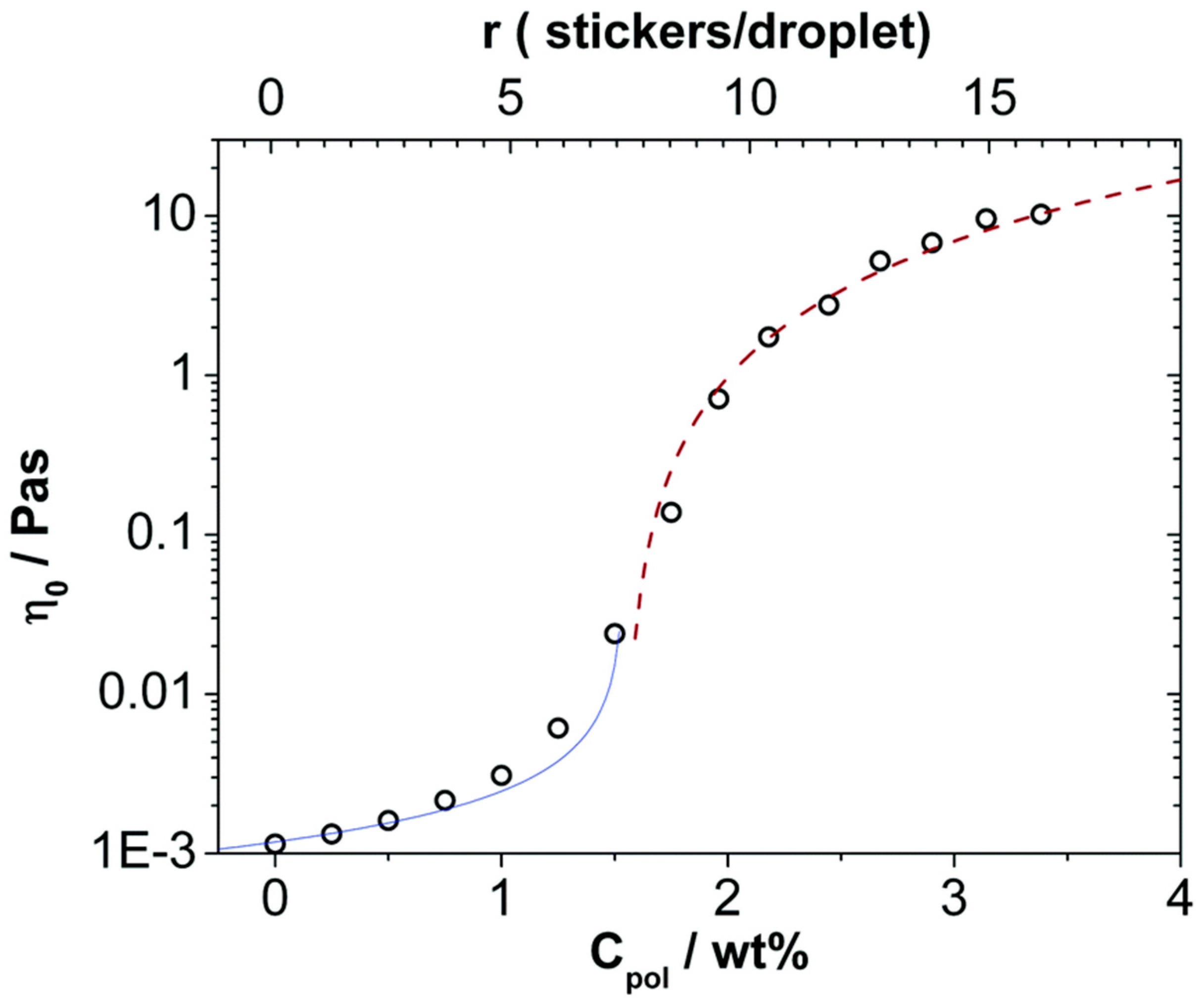
5.2. Microemulsions Interacting with Polymers
- (1)
- (2)
- an entropic attraction induced by the bridging polymer [139];
- (3)
5.3. Vesicles Interacting with Polymers
6. Conclusions
Conflicts of Interest
References
- Evans, F.D.; Wennerstöm, H. The Colloidal Domain: Where Physics, Chemistry, Biology, and Technology Meet (Advances in Interfacial Engineering); Wiley-VCH: Weinheim, Germany, 1996. [Google Scholar]
- Tiddy, G. Surfactant-water liquid crystal phases. Phys. Rep. 1980, 57, 1–46. [Google Scholar] [CrossRef]
- Laughlin, R.G. The Aqueous Phase Behavior of Surfactants; Academic Press: London, UK, 1994; Volume 6. [Google Scholar]
- Estroff, L.A.; Hamilton, A.D. Water Gelation by Small Organic Molecules. Chem. Rev. 2004, 104, 1201–1218. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.; Weissig, V. Liposomes: A Practical Approach; Oxford University Press: Oxford, UK, 2003. [Google Scholar]
- International Union of Pure and Applied Chemistry. Compendium of Chemical Terminology Gold Book, 2nd ed.; International Union of Pure and Applied Chemistry: Research Triangle Park, NC, USA, 2014. [Google Scholar]
- Møller, P.C.F.; Mewis, J.; Bonn, D. Yield stress and thixotropy: On the difficulty of measuring yield stresses in practice. Soft Matter 2006, 2, 274–283. [Google Scholar] [CrossRef]
- Rehage, H.; Hoffmann, H. Viscoelastic surfactant solutions: Model systems for rheological research. Mol. Phys. 1991, 74, 933–973. [Google Scholar] [CrossRef]
- Zana, R.; Kaler, E.W. Giant Micelles: Properties and Applications; CRC Press: Boca Raton, FL, USA, 2007; Volume 140. [Google Scholar]
- Hoffmann, H. Viscoelastic Surfactant Solutions. In ACS Symposium Series, Vol. 578; ACS Publications: Washington, DC, USA, 1994; pp. 2–31. [Google Scholar]
- Cates, M.E. Dynamics of living polymers and flexible surfactant micelles: Scaling laws for dilution. J. Phys. 1988, 49, 1593–1600. [Google Scholar] [CrossRef]
- Raghavan, S.R.; Douglas, J.F. The conundrum of gel formation by molecular nanofibers, wormlike micelles, and filamentous proteins: Gelation without cross-links? Soft Matter 2012, 8, 8539–8546. [Google Scholar] [CrossRef]
- Gradzielski, M. Vesicles and vesicle gels—Structure and dynamics of formation. J. Phys. Condens. Matter 2003, 15, R655–R697. [Google Scholar] [CrossRef]
- Hoffmann, H.; Ulbricht, W. Surfactant gels. Curr. Opin. Colloid Interface Sci. 1996, 1, 726–739. [Google Scholar] [CrossRef]
- Schmolka, I.R. A comparison of block copolymer surfactant gels. J. Am. Oil Chem. Soc. 1991, 68, 206–209. [Google Scholar] [CrossRef]
- Mortensen, K.; Brown, W.; Nordén, B. Inverse melting transition and evidence of three-dimensional cubatic structure in a block-copolymer micellar system. Phys. Rev. Lett. 1992, 68, 2340–2343. [Google Scholar] [CrossRef] [PubMed]
- Hecht, E.; Hoffmann, H. Interaction of ABA block copolymers with ionic surfactants in aqueous solution. Langmuir 1994, 10, 86–91. [Google Scholar] [CrossRef]
- Escobar-Chávez, J.J.; López-Cervantes, M.; Naik, A.; Kalia, Y.; Quintanar-Guerrero, D.; Ganem-Quintanar, A. Applications of thermo-reversible pluronic F-127 gels in pharmaceutical formulations. J. Pharm. Pharm. Sci. 2006, 9, 339–358. [Google Scholar] [PubMed]
- Flory, P.J. Statistical Mechanics of Swelling of Network Structures. J. Chem. Phys. 1950, 18, 108–111. [Google Scholar] [CrossRef]
- Tanford, C. The Hydrophobic Effect: Formation of Micelles and Biological Membranes, 2nd ed.; Wiley: Hoboken, NJ, USA, 1980. [Google Scholar]
- Raghavan, S.R. Distinct character of surfactant gels: A smooth progression from micelles to fibrillar networks. Langmuir 2009, 25, 8382–8385. [Google Scholar] [CrossRef] [PubMed]
- Berret, J.-F. Rheology of wormlike micelles: Equilibrium properties and shear banding transitions. In Molecular Gels; Springer: Berlin/Heidelberg, Germany, 2006; pp. 667–720. [Google Scholar]
- Imae, T.; Kamiya, R.; Ikeda, S. Electron microscopic observation of rod-like micelles of dimethyloleylamine oxide regenerated from its aqueous solutions. J. Colloid Interface Sci. 1984, 99, 300–301. [Google Scholar] [CrossRef]
- Sakaiguchi, Y.; Shikata, T.; Urakami, H.; Tamura, A.; Hirata, H. Electron microscope study of viscoelastic cationic surfactant systems. Colloid Polym. Sci. 1987, 265, 750–753. [Google Scholar] [CrossRef]
- Ziserman, L.; Abezgauz, L.; Ramon, O.; Raghavan, S.R.; Danino, D. Origins of the viscosity peak in wormlike micellar solutions. 1. Mixed catanionic surfactants. A cryo-transmission electron microscopy study. Langmuir 2009, 25, 10483–10489. [Google Scholar] [CrossRef] [PubMed]
- Oelschlaeger, C.; Schopferer, M.; Scheffold, F.; Willenbacher, N. Linear-to-branched micelles transition: A rheometry and diffusing wave spectroscopy (DWS) study. Langmuir 2008, 25, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T.M.; Vinson, P.K.; Minter, J.R.; Davis, H.T.; Talmon, Y.; Miller, W.G. Viscoelastic micellar solutions: Microscopy and rheology. J. Phys. Chem. 1992, 96, 474–484. [Google Scholar] [CrossRef]
- Israelachvili, J.N.; Mitchell, D.J.; Ninham, B.W. Theory of self-assembly of hydrocarbon amphiphiles into micelles and bilayers. J. Chem. Soc. Faraday Trans. 2 Mol. Chem. Phys. 1976, 72, 1525–1568. [Google Scholar] [CrossRef]
- Mu, J.-H.; Li, G.-Z.; Jia, X.-L.; Wang, H.-X.; Zhang, G.-Y. Rheological Properties and Microstructures of Anionic Micellar Solutions in the Presence of Different Inorganic Salts. J. Phys. Chem. 2002, 106, 11685–11693. [Google Scholar] [CrossRef]
- Chu, Z.; Feng, Y. Thermo-switchable surfactant gel. Chem. Commun. 2011, 47, 7191–7193. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.S.; Cates, M.E. Linear viscoelasticity of wormlike micelles: A comparison of micellar reaction kinetics. J. Phys. II 1992, 2, 503–519. [Google Scholar] [CrossRef]
- Cates, M.E.; Candau, S.J. Statics and dynamics of worm-like surfactant micelles. J. Phys. Condens. Matter 1990, 2, 6869–6892. [Google Scholar] [CrossRef]
- Kumars, R.; Kalur, G.C.; Ziserman, L.; Danino, D.; Raghavan, S.R. Wormlike micelles of a C22-tailed zwitterionic betaine surfactant: From viscoelastic solutions to elastic gels. Langmuir 2007, 23, 12849–12856. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Chu, Z. pH-Tunable wormlike micelles based on an ultra-long-chain “pseudo” gemini surfactant. Soft Matter 2015, 11, 4614–4620. [Google Scholar] [CrossRef] [PubMed]
- Bernheim-Groswasser, A.; Zana, R.; Talmon, Y. Sphere-to-cylinder transition in aqueous micellar solution of a dimeric (gemini) surfactant. J. Phys. Chem. 2000, 104, 4005–4009. [Google Scholar] [CrossRef]
- Pei, X.; Zhao, J.; Ye, Y.; You, Y.; Wei, X. Wormlike micelles and gels reinforced by hydrogen bonding in aqueous cationic gemini surfactant systems. Soft Matter 2011, 7, 2953–2960. [Google Scholar] [CrossRef]
- Gradzielski, M. The rheology of vesicle and disk systems—Relations between macroscopic behaviour and microstructure. Curr. Opin. Colloid Interface Sci. 2011, 16, 13–17. [Google Scholar] [CrossRef]
- Fontell, K.; Mandell, L.; Ekwall, P. Some isotropic mesophases in systems containing amphiphilic compounds. Acta Chem. Scand 1968, 22, 3209–3223. [Google Scholar] [CrossRef]
- Gradzielski, M.; Bergmeier, M.; Müller, M.; Hoffmann, H. Novel Gel Phase: A Cubic Phase of Densely Packed Monodisperse, Unilamellar Vesicles. J. Phys. Chem. 1997, 101, 1719–1722. [Google Scholar] [CrossRef]
- Gradzielski, M.; Müller, M.; Bergmeier, M.; Hoffmann, H.; Hoinkis, E. Structural and Macroscopic Characterization of a Gel Phase of Densely Packed Monodisperse, Unilamellar Vesicles. J. Phys. Chem. 1999, 103, 1416–1424. [Google Scholar] [CrossRef]
- Gradzielski, M.; Bergmeier, M.; Hoffmann, H.; Müller, M.; Grillo, I. Vesicle Gel Formed by a Self-Organization Process. J. Phys. Chem. 2000, 104, 11594–11597. [Google Scholar] [CrossRef]
- Gradzielski, M.; Grillo, I.; Narayanan, T. Morphological Transitions in Amphiphilic Systems Probed by Small-Angle Scattering Techniques. In Self-Assembly; Robinson, B.H., Ed.; IOS Press: Amsterdam, The Netherland, 2003; pp. 410–421. [Google Scholar]
- Lasič, S.; Åslund, I.; Oppel, C.; Topgaard, D.; Söderman, O.; Gradzielski, M. Investigations of vesicle gels by pulsed and modulated gradient NMR diffusion techniques. Soft Matter 2011, 7, 3947–3955. [Google Scholar] [CrossRef]
- Jeong, Y.; Uezu, K.; Kobayashi, M.; Sakurai, S.; Masunaga, H.; Inoue, K.; Sasaki, S.; Shimada, N.; Takeda, Y.; Kaneko, K.; et al. Complex made from tetrasodium N, N-bis (carboxylatomethyl) glutamate and sodium oleate that forms a highly ordered lamella in gel phase. Bull. Chem. Soc. Jpn. 2007, 80, 410–417. [Google Scholar] [CrossRef]
- Oppel, C.; Prévost, S.; Noirez, L.; Gradzielski, M. The use of highly ordered vesicle gels as template for the formation of silica gels. Langmuir 2011, 27, 8885–8897. [Google Scholar] [CrossRef] [PubMed]
- Menger, F.M.; Peresypkin, A. V Strings of vesicles: Flow behavior in an unusual type of aqueous gel. J. Am. Chem. Soc. 2003, 125, 5340–5345. [Google Scholar] [CrossRef] [PubMed]
- Seth, M.; Ramachandran, A.; Murch, B.P.; Leal, L.G. Origins of microstructural transformations in charged vesicle suspensions: The crowding hypothesis. Langmuir 2014, 30, 10176–10187. [Google Scholar] [CrossRef] [PubMed]
- Diec, K.H.; Sokolowski, T.; Wittern, K.P.; Schreiber, J.; Meier, W. New liposome gels by self organization of vesicles and intelligent polymers. Cosmet. Toilet. 2002, 117, 55–62. [Google Scholar]
- Hoffmann, H.; Thunig, C.; Schmiedel, P.; Munkert, U. Gels from surfactant solutions with densely packed multilamellar vesicles. Faraday Discuss. 1995, 101, 319–333. [Google Scholar] [CrossRef]
- Hoffmann, H.; Thunig, C.; Schmiedel, P.; Munkert, U. Complex fluids with a yield value; their microstructures and rheological properties. Nuovo Cim. D 1994, 16, 1373–1390. [Google Scholar] [CrossRef]
- Bergmeier, M.; Gradzielski, M.; Hoffmann, H.; Mortensen, K. Behavior of Ionically Charged Lamellar Systems under the Influence of a Shear Field. J. Phys. Chem. 1999, 103, 1605–1617. [Google Scholar] [CrossRef]
- Diat, O.; Roux, D. Preparation of monodisperse multilayer vesicles of controlled size and high encapsulation ratio. J. Phys. 1993, 3, 9–14. [Google Scholar] [CrossRef]
- Long, P.; Hao, J. A gel state from densely packed multilamellar vesicles in the crystalline state. Soft Matter 2010, 6, 4350–4356. [Google Scholar] [CrossRef]
- Hufnagl, A.; Kinzel, S.; Gradzielski, M. Vesicles and Vesicle Gels—Structure and Solubilisation Properties. Tenside Surfactants Deterg. 2007, 44, 110–115. [Google Scholar] [CrossRef]
- Grewe, F.; Ortmeyer, J.; Haase, R.; Schmidt, C. Colloidal Gels Formed by Dilute Aqueous Dispersions of Surfactant and Fatty Alcohol. In Colloid Process Engineering; Springer: Berlin/Heidelberg, Germany, 2015; pp. 21–43. [Google Scholar]
- Cheng, C.-Y.; Wang, T.-Y.; Tung, S.-H. Biological Hydrogels Formed by Swollen Multilamellar Liposomes. Langmuir 2015, 31, 13312–13320. [Google Scholar] [CrossRef] [PubMed]
- Lauger, J.; Linemann, R.; Richtering, W. Shear orientation of a lamellar lyotropic liquid crystal. Rheol. Acta 1995, 34, 132–136. [Google Scholar] [CrossRef]
- Kinzel, S.; Gradzielski, M. Control of phase behavior and properties of vesicle gels by admixing ionic surfactants to the nonionic surfactant brij 30. Langmuir 2008, 24, 10123–10132. [Google Scholar] [CrossRef] [PubMed]
- Zou, A.; Hoffmann, H.; Freiberger, N.; Glatter, O. Influence of ionic charges on the bilayers of lamellar phases. Langmuir 2007, 23, 2977–2984. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Zhong, Z.; Hao, J. Self-assembly of onion-like vesicles induced by charge and rheological properties in anionic–nonionic surfactant solutions. Soft Matter 2012, 8, 7812–7821. [Google Scholar] [CrossRef]
- Dong, R.; Wu, J.; Dong, S.; Song, S.; Tian, F.; Hao, J. Interconvertible Self-Assembly and Rheological Properties of Planar Bilayers and Vesicle Gels in Anionic/Nonionic (CF/CH) Surfactant Solutions. Chem. Asian J. 2013, 8, 1863–1872. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wei, G.; Dong, R.; Hao, J. Multiresponsive viscoelastic vesicle gels of nonionic C12EO4 and anionic AzoNa. Chem. A Eur. J. 2013, 19, 8253–8260. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hao, J. Multiple-stimulus-responsive hydrogels of cationic surfactants and azoic salt mixtures. Colloid Polym. Sci. 2013, 291, 2935–2946. [Google Scholar] [CrossRef]
- Kaler, E.W.; Murthy, A.K.; Rodriguez, B.E.; Zasadzinski, J.A.N. Spontaneous vesicle formation in aqueous mixtures of single-tailed surfactants. Science 1989, 245, 1371–1375. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahem, R.; Hoffmann, H. Novel viscoelastic systems from cationic surfactants and hydrophobic counter-ions: Influence of surfactant chain length. J. Colloid Interface Sci. 2007, 312, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahem, R.; Gradzielski, M.; Hoffmann, H. A novel viscoelastic system from a cationic surfactant and a hydrophobic counterion. J. Colloid Interface Sci. 2005, 288, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Horbaschek, K.; Hoffmann, H.; Thunig, C. Formation and properties of lamellar phases in systems of cationic surfactants and hydroxy-naphthoate. J. Colloid Interface Sci. 1998, 206, 439–456. [Google Scholar] [CrossRef] [PubMed]
- Coldren, B.A.; Warriner, H.; van Zanten, R.; Zasadzinski, J.A.; Sirota, E.B. Lamellar gels and spontaneous vesicles in catanionic surfactant mixtures. Langmuir 2006, 22, 2465–2473. [Google Scholar] [CrossRef] [PubMed]
- Laflèche, F.; Durand, D.; Nicolai, T. Association of Adhesive Spheres Formed by Hydrophobically End-Capped PEO. 1. Influence of the Presence of Single End-Capped PEO. Macromolecules 2003, 36, 1331–1340. [Google Scholar]
- Séréro, Y.; Aznar, R.; Porte, G.; Berret, J.-F.; Calvet, D.; Collet, A.; Viguier, M. Associating Polymers: From “Flowers” to Transient Networks. Phys. Rev. Lett. 1998, 81, 5584–5587. [Google Scholar] [CrossRef]
- Berret, J.-F.; Séréro, Y.; Winkelman, B.; Calvet, D.; Collet, A.; Viguier, M. Nonlinear rheology of telechelic polymer networks. J. Rheol. 2001, 45, 477–492. [Google Scholar] [CrossRef]
- Alami, E.; Almgren, M.; Brown, W.; François, J. Aggregation of Hydrophobically End-Capped Poly(ethylene oxide) in Aqueous Solutions. Fluorescence and Light-Scattering Studies. Macromolecules 1996, 29, 2229–2243. [Google Scholar] [CrossRef]
- Cram, S.L.; Brown, H.R.; Spinks, G.M.; Hourdet, D.; Creton, C. Hydrophobically Modified Dimethylacrylamide Synthesis and Rheol. l Behavior. Macromolecules 2005, 38, 2981–2989. [Google Scholar] [CrossRef]
- Herfurth, C.; Malo de Molina, P.; Wieland, C.; Rogers, S.; Gradzielski, M.; Laschewsky, A. One-step RAFT synthesis of well-defined amphiphilic star polymers and their self-assembly in aqueous solution. Polym. Chem. 2012, 3, 1606–1617. [Google Scholar] [CrossRef]
- Filali, M.; Aznar, R.; Svenson, M.; Porte, G.; Appell, J. Swollen Micelles Plus Hydrophobically Modified Hydrosoluble Polymers in Aqueous Solutions: Decoration versus Bridging. A Small Angle Neutron Scattering Study. J. Phys. Chem. 1999, 103, 7293–7301. [Google Scholar]
- Rufier, C.; Collet, A.; Viguier, M.; Oberdisse, J.; Mora, S. Asymmetric End-Capped Poly(ethylene oxide). Synthesis and Rheological Behavior in Aqueous Solution. Macromolecules 2008, 41, 5854–5862. [Google Scholar]
- Hietala, S.; Mononen, P.; Strandman, S.; Järvi, P.; Torkkeli, M.; Jankova, K.; Hvilsted, S.; Tenhu, H. Synthesis and rheological properties of an associative star polymer in aqueous solutions. Polymer 2007, 48, 4087–4096. [Google Scholar] [CrossRef]
- Taribagil, R.R.; Hillmyer, M.A.; Lodge, T.P. Hydrogels from ABA and ABC triblock polymers. Macromolecules 2010, 43, 5396–5404. [Google Scholar] [CrossRef]
- Chassenieux, C.; Nicolai, T.; Benyahia, L. Rheology of associative polymer solutions. Curr. Opin. Colloid Interface Sci. 2011, 16, 18–26. [Google Scholar] [CrossRef]
- Pham, Q.T.; Russel, W.B. Micellar Solutions of Associative Triblock Copolymers: The Relationship between Structure and Rheology. Langmuir 1999, 5139–5146. [Google Scholar] [CrossRef]
- Yekta, A.; Duhamel, J.; Brochard, P. A fluorescent probe study of micelle-like cluster formation in aqueous solutions of hydrophobically modified poly (ethylene oxide). Macromolecules 1993, 26, 1829–1836. [Google Scholar] [CrossRef]
- Meng, X.X.; Russel, W.B. Structure and size of spherical micelles of telechelic polymers. Macromolecules 2005, 38, 593–600. [Google Scholar] [CrossRef]
- Chassenieux, C.; Nicolai, T.; Durand, D. Association of Hydrophobically End-Capped Poly(ethylene oxide). Macromolecules 1997, 30, 4952–4958. [Google Scholar] [CrossRef]
- Lundberg, D.J.; Brown, R.G.; Glass, J.E.; Eley, R.R. Synthesis, Characterization, and Solution Rheology of Model Hydrophobically-Modified, Water-Soluble Ethoxylated Urethanes. Langmuir 1994, 10, 3027–3034. [Google Scholar] [CrossRef]
- Annable, T. The rheology of solutions of associating polymers: Comparison of experimental behavior with transient network theory. J. Rheol. 1993, 37, 695–726. [Google Scholar] [CrossRef]
- Winnik, M.A.; Yekta, A. Associative polymers in aqueous solution. Curr. Opin. Colloid Interface Sci. 1997, 2, 424–436. [Google Scholar] [CrossRef]
- Zhong, M.; Wang, R.; Kawamoto, K.; Olsen, B.D.; Johnson, J.A. Quantifying the impact of molecular defects on polymer network elasticity. Science 2016, 353, 1264–1268. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.X.; Cooper, S.L. Shear Thickening in Aqueous Solutions of Hydrocarbon End-Capped Poly (ethylene oxide). Macromolecules 2000, 34, 3294–3301. [Google Scholar] [CrossRef]
- Pellens, L.; Gamez Corrales, R.; Mewis, J. General nonlinear rheological behavior of associative polymers. J. Rheol. 2004, 48, 379–393. [Google Scholar] [CrossRef]
- Dozier, W.D.; Huang, J.S.; Fetters, L.J. Colloidal nature of star polymer dilute and semidilute solutions. Macromolecules 1991, 24, 2810–2814. [Google Scholar] [CrossRef]
- Rufier, C.; Collet, A.; Viguier, M.; Oberdisse, J.; Mora, S. Influence of Surfactants on Hydrophobically End-Capped Poly(ethylene oxide) Self-Assembled Aggregates Studied by SANS. Macromolecules 2011, 44, 7451–7459. [Google Scholar] [CrossRef]
- Semenov, A.N.; Joanny, J.F.; Khokhlov, A.R. Associating polymers: Equilibrium and linear viscoelasticity. Macromolecules 1995, 28, 1066–1075. [Google Scholar] [CrossRef]
- Rubinstein, M.; Semenov, A.N. Dynamics of entangled solutions of associating polymers. Macromolecules 2001, 34, 1058–1068. [Google Scholar] [CrossRef]
- Regalado, E.J.; Selb, J.; Candau, F. Viscoelastic behavior of semidilute solutions of multisticker polymer chains. Macromolecules 1999, 32, 8580–8588. [Google Scholar] [CrossRef]
- Lin, H.-H.; Cheng, Y.-L. In-Situ Thermoreversible Gelation of Block and Star Copolymers of Poly(ethylene glycol) and Poly(N-isopropylacrylamide) of Varying Architectures. Macromolecules 2001, 34, 3710–3715. [Google Scholar] [CrossRef]
- Park, S.Y.; Han, D.K.; Kim, S.C. Synthesis and Characterization of Star-Shaped PLLA−PEO Block Copolymers with Temperature-Sensitive Sol−Gel Transition Behavior. Macromolecules 2001, 34, 8821–8824. [Google Scholar] [CrossRef]
- Nagahama, K.; Ouchi, T.; Ohya, Y. Temperature-Induced Hydrogels Through Self-Assembly of Cholesterol-Substituted Star PEG-b-PLLA Copolymers: An Injectable Scaffold for Tissue Engineering. Adv. Funct. Mater. 2008, 18, 1220–1231. [Google Scholar] [CrossRef]
- Burnworth, M.; Tang, L.; Kumpfer, J.R.; Duncan, A.J.; Beyer, F.L.; Fiore, G.L.; Rowan, S.J.; Weder, C. Optically healable supramolecular polymers. Nature 2011, 472, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Holten-Andersen, N.; Harrington, M.J.; Birkedal, H.; Lee, B.P.; Messersmith, P.B.; Lee, K.Y.C.; Waite, J.H. pH-induced metal-ligand cross-links inspired by mussel yield self-healing polymer networks with near-covalent elastic moduli. Proc. Natl. Acad. Sci. USA 2011, 108, 2651–2655. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Habicht, A.; Li, S.; Seiffert, S.; Olsen, B.D. Self-Diffusion of Associating Star-Shaped Polymers. Macromolecules 2016, 49, 5599–5608. [Google Scholar] [CrossRef]
- Guo, M.; Pitet, L.M.; Wyss, H.M.; Vos, M.; Dankers, P.Y.W.; Meijer, E.W. Tough stimuli-responsive supramolecular hydrogels with hydrogen-bonding network junctions. J. Am. Chem. Soc. 2014, 136, 6969–6977. [Google Scholar] [CrossRef] [PubMed]
- Appel, E.A.; Forster, R.A.; Koutsioubas, A.; Toprakcioglu, C.; Scherman, O.A. Activation energies control the macroscopic properties of physically cross-linked materials. Angew. Chem. Int. Ed. 2014, 53, 10038–10043. [Google Scholar] [CrossRef] [PubMed]
- Dürrschmidt, T.; Hoffmann, H. Organogels from ABA triblock copolymers. Colloid Polym. Sci. 2001, 279, 1005–1012. [Google Scholar] [CrossRef]
- Monge, S.; Joly-Duhamel, C.; Boyer, C.; Robin, J.-J. Synthesis and Characterisation of Organogels from ABA Triblock Copolymers. Macromol. Chem. Phys. 2007, 208, 262–270. [Google Scholar] [CrossRef]
- Meier, W.; Falk, A.; Odenwald, M.; Stieber, F. Microemulsion elastomers. Colloid Polym. Sci. 1996, 274, 218–226. [Google Scholar] [CrossRef]
- Blochowicz, T.; Gögelein, C.; Spehr, T.; Müller, M.; Stühn, B. Polymer-induced transient networks in water-in-oil microemulsions studied by small-angle X-ray and dynamic light scattering. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2007, 76, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Schmaljohann, D. Thermo-and pH-responsive polymers in drug delivery. Adv. Drug Deliv. Rev. 2006, 58, 1655–1670. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Tang, Y.; Billingham, N.C.; Armes, S.P.; Lewis, A.L. Synthesis of biocompatible, stimuli-responsive, physical gels based on ABA triblock copolymers. Biomacromolecules 2003, 4, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Tang, Y.; Armes, S.P.; Morris, C.J.; Rose, S.F.; Lloyd, A.W.; Lewis, A.L. Synthesis and characterization of biocompatible thermo-responsive gelators based on ABA triblock copolymers. Biomacromolecules 2005, 6, 994–999. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Madsen, J.; Armes, S.P.; Lewis, A.L. A New Class of Biochemically Degradable, Stimulus-Responsive Triblock Copolymer Gelators. Angew. Chem. Int. Ed. 2006, 45, 3510–3513. [Google Scholar] [CrossRef] [PubMed]
- Vogt, A.P.; Sumerlin, B.S. Temperature and redox responsive hydrogels from ABA triblock copolymers prepared by RAFT polymerization. Soft Matter 2009, 5, 2347–2351. [Google Scholar] [CrossRef]
- Yu, L.; Chang, G.T.; Zhang, H.; Ding, J.D. Injectable block copolymer hydrogels for sustained release of a PEGylated drug. Int. J. Pharm. 2008, 348, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Determan, M.D.; Guo, L.; Thiyagarajan, P.; Mallapragada, S.K. Supramolecular self-assembly of multiblock copolymers in aqueous solution. Langmuir 2006, 22, 1469–1473. [Google Scholar] [CrossRef] [PubMed]
- Bossard, F.; Aubry, T.; Gotzamanis, G.; Tsitsilianis, C. pH-Tunable rheological properties of a telechelic cationic polyelectrolyte reversible hydrogel. Soft Matter 2006, 2, 510–516. [Google Scholar] [CrossRef]
- O’Lenick, T.G.; Jiang, X.; Zhao, B. Thermosensitive Aqueous Gels with Tunable Sol–Gel Transition Temperatures from Thermo-and pH-Responsive Hydrophilic ABA Triblock Copolymer. Langmuir 2010, 26, 8787–8796. [Google Scholar] [CrossRef] [PubMed]
- Hamley, I.W.; Cheng, G.; Castelletto, V. A Thermoresponsive Hydrogel Based on Telechelic PEG End-Capped with Hydrophobic Dipeptides. Macromol. Biosci. 2011, 11, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Appel, E.A.; del Barrio, J.; Loh, X.J.; Scherman, O.A. Supramolecular polymeric hydrogels. Chem. Soc. Rev. 2012, 41, 6195–6214. [Google Scholar] [CrossRef] [PubMed]
- Voorhaar, L.; Hoogenboom, R. Supramolecular polymer networks: Hydrogels and bulk materials. Chem. Soc. Rev. 2016, 45, 4013–4031. [Google Scholar] [CrossRef] [PubMed]
- Chiappisi, L.; Hoffmann, I.; Gradzielski, M. Complexes of oppositely charged polyelectrolytes and surfactants—Recent developments in the field of biologically derived polyelectrolytes. Soft Matter 2013, 9, 3896–3909. [Google Scholar] [CrossRef]
- Lindman, B.; Antunes, F.; Aidarova, S.; Miguel, M.; Nylander, T. Polyelectrolyte-surfactant association from fundamentals to applications. Colloid J. 2014, 76, 585–594. [Google Scholar] [CrossRef]
- Kästner, U.; Hoffmann, H.; Dönges, R.; Ehrler, R. Interactions between modified hydroxyethyl cellulose (HEC) and surfactants. Colloids Surf. A Physicochem. Eng. Asp. 1996, 112, 209–225. [Google Scholar] [CrossRef]
- Hoffmann, H.; Kästner, U.; Dönges, R.; Ehrler, R. Gels from modified hydroxyethyl cellulose and ionic surfactants. Polym. Gels Netw. 1996, 4, 509–526. [Google Scholar] [CrossRef]
- Annable, T.; Buscall, R.; Ettelaie, R. Influence of surfactants on the rheology of associating polymers in solution. Langmuir 1994, 10, 1060–1070. [Google Scholar] [CrossRef]
- Appell, J.; Rawiso, M. Interactions between Nonionic Surfactant Micelles Introduced by a Telechelic Polymer A Small Angle Neutron Scattering Study. Langmuir 1998, 14, 4409–4414. [Google Scholar] [CrossRef]
- Piculell, L.; Egermayer, M.; Sjöström, J. Rheology of mixed solutions of an associating polymer with a surfactant. Why are different surfactants different? Langmuir 2003, 19, 3643–3649. [Google Scholar]
- Jiménez-Regalado, E.; Selb, J.; Candau, F. Effect of Surfactant on the Viscoelastic Behavior of Semidilute Solutions of Multisticker Associating Polyacrylamides. Langmuir 2000, 16, 8611–8621. [Google Scholar] [CrossRef]
- Tabuteau, H.; Ramos, L.; Nakaya-Yaegashi, K.; Imai, M.; Ligoure, C. Nonlinear rheology of surfactant wormlike micelles bridged by telechelic polymers. Langmuir 2009, 25, 2467–2472. [Google Scholar] [CrossRef] [PubMed]
- Nakaya-Yaegashi, K.; Ramos, L.; Tabuteau, H.; Ligoure, C. Linear viscoelasticity of entangled wormlike micelles bridged by telechelic polymers: An experimental model for a double transient network. J. Rheol. 2008, 52, 359–377. [Google Scholar] [CrossRef]
- Ramos, L.; Ligoure, C. Structure of a new type of transient network: Entangled wormlike micelles bridged by telechelic polymers. Macromolecules 2007, 40, 1248–1251. [Google Scholar] [CrossRef]
- Gradzielski, M. Recent developments in the characterisation of microemulsions. Curr. Opin. Colloid Interface Sci. 2008, 13, 263–269. [Google Scholar] [CrossRef]
- Langevin, D. Microemulsions. Acc. Chem. Res. 1988, 21, 255–260. [Google Scholar] [CrossRef]
- Gradzielski, M.; Langevin, D.; Sottmann, T.; Strey, R. Small angle neutron scattering near the wetting transition: Discrimination of microemulsions from weakly structured mixtures. J. Chem. Phys. 1996, 104, 3782–3787. [Google Scholar] [CrossRef]
- Gradzielski, M.; Raucher, A.; Hoffmann, H. Hydrophobically cross-linked micellar solutions: Microstructure and properties of the solutions. J. Phys. IV 1993, 3, C1-65–C1-79. [Google Scholar] [CrossRef]
- Bagger-Jörgensen, H.; Coppola, L.; Thuresson, K.; Olsson, U.; Mortensen, K. Phase Behavior, Microstructure, and Dynamics in a Nonionic Microemulsion on Addition of Hydrophobically End-Capped Poly(ethylene oxide). Langmuir 1997, 13, 4204–4218. [Google Scholar] [CrossRef]
- Maccarrone, S.; Frielinghaus, H.; Allgaier, J.; Richtery, D.; Lindner, P. SANS study of polymer-linked droplets. Langmuir 2007, 23, 9559–9562. [Google Scholar] [CrossRef] [PubMed]
- Malo de Molina, P.; Appavou, M.-S.; Gradzielski, M. Oil-in-water microemulsion droplets of TDMAO/decane interconnected by the telechelic C18-EO150-C18: Clustering and network formation. Soft Matter 2014, 10, 5072–5084. [Google Scholar] [CrossRef] [PubMed]
- Hed, G.; Safran, S.A. The immunity of polymer-microemulsion networks. Eur. Phys. J. 2006, 19, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Porte, G.; Ligoure, C.; Appell, J.; Aznar, R. Bridging interactions due to telechelic linkers balanced by screened Coulombic repulsions. J. Stat. Mech. Theory Exp. 2006, 2006, P05005. [Google Scholar] [CrossRef]
- Zilman, A.; Kieffer, J.; Molino, F.; Porte, G.; Safran, S.A. Entropic phase separation in polymer-microemulsion networks. Phys. Rev. Lett. 2003, 91, 15901. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.R.; Russel, W.B. End-Capped Associative Polymer Chains between Nanospheres: Attractions in Ideal Solutions. Macromolecules 2000, 33, 5713–5720. [Google Scholar] [CrossRef]
- Malo de Molina, P.; Ihlefeldt, F.S.; Prévost, S.; Herfurth, C.; Appavou, M.-S.; Laschewsky, A.; Gradzielski, M. Phase Behavior of Nonionic Microemulsions with Multi-end-capped Polymers and Its Relation to the Mesoscopic Structure. Langmuir 2015, 31, 5198–5209. [Google Scholar] [CrossRef] [PubMed]
- Michel, E.; Filali, M.; Aznar, R.; Porte, G.; Appell, J. Percolation in a Model Transient Network: Rheology and Dynamic Light Scattering. Langmuir 2000, 16, 8702–8711. [Google Scholar] [CrossRef]
- Malo de Molina, P.; Herfurth, C.; Laschewsky, A.; Gradzielski, M. Structure and dynamics of networks in mixtures of hydrophobically modified telechelic multiarm polymers and oil in water microemulsions. Langmuir 2012, 28, 15994–16006. [Google Scholar] [CrossRef] [PubMed]
- Meier, W.; Hotz, J.; GuntherAusborn, S. Vesicle and cell networks: Interconnecting cells by synthetic polymers. Langmuir 1996, 12, 5028–5032. [Google Scholar] [CrossRef]
- Antunes, F.E.; Marques, E.F.; Gomes, R.; Thuresson, K.; Lindman, B.; Miguel, M.G. Network formation of catanionic vesicles and oppositely charged polyelectrolytes. Effect of polymer charge density and hydrophobic modification. Langmuir 2004, 20, 4647–4656. [Google Scholar] [PubMed]
- dos Santos, T.; Medronho, B.; Antunes, F.E.; Lindman, B.; Miguel, M. How does a non-ionic hydrophobically modified telechelic polymer interact with a non-ionic vesicle? Rheological aspects. Colloids Surf. A Physicochem. Eng. Asp. 2008, 319, 173–179. [Google Scholar] [CrossRef]
- Lee, J.H.; Gustin, J.P.; Chen, T.; Payne, G.F.; Raghavan, S.R. Vesicle-biopolymer gels: Networks of surfactant vesicles connected by associating biopolymers. Langmuir 2005, 21, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Ruocco, N.; Frielinghaus, H.; Vitiello, G.; D’Errico, G.; Leal, L.G.; Richter, D.; Ortona, O.; Paduano, L. How hydrophobically modified chitosans are stabilized by biocompatible lipid aggregates. J. Colloid Interface Sci. 2015, 452, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Ashbaugh, H.S.; Boon, K.; Prud’homme, R.K. Gelation of “catanionic” vesicles by hydrophobically modified polyelectrolytes. Colloid Polym. Sci. 2002, 280, 783–788. [Google Scholar] [CrossRef]
- Marques, E.F.; Regev, O.; Khan, A.; Miguel, M.G.; Lindman, B. Interactions between catanionic vesicles and oppositely charged polyelectrolytes phase behavior and phase structure. Macromolecules 1999, 32, 6626–6637. [Google Scholar] [CrossRef]
- Lee, J.-H.; Oh, H.; Baxa, U.; Raghavan, S.R.; Blumenthal, R. Biopolymer-Connected Liposome Networks as Injectable Biomaterials Capable of Sustained Local Drug Delivery. Biomacromolecules 2012, 13, 3388–3394. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Javvaji, V.; Yaraghi, N.A.; Abezgauz, L.; Danino, D.; Raghavan, S.R. Light-induced transformation of vesicles to micelles and vesicle-gels to sols. Soft Matter 2013, 9, 11576–11584. [Google Scholar] [CrossRef]










© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malo de Molina, P.; Gradzielski, M. Gels Obtained by Colloidal Self-Assembly of Amphiphilic Molecules. Gels 2017, 3, 30. https://doi.org/10.3390/gels3030030
Malo de Molina P, Gradzielski M. Gels Obtained by Colloidal Self-Assembly of Amphiphilic Molecules. Gels. 2017; 3(3):30. https://doi.org/10.3390/gels3030030
Chicago/Turabian StyleMalo de Molina, Paula, and Michael Gradzielski. 2017. "Gels Obtained by Colloidal Self-Assembly of Amphiphilic Molecules" Gels 3, no. 3: 30. https://doi.org/10.3390/gels3030030
APA StyleMalo de Molina, P., & Gradzielski, M. (2017). Gels Obtained by Colloidal Self-Assembly of Amphiphilic Molecules. Gels, 3(3), 30. https://doi.org/10.3390/gels3030030