2.1. Synthesis and Gelation Ability of (R)-H6L
The gelator (
R)-H
6L was synthesized (
Scheme 1 and
Figures S1–S5). In our previous work, the synthesized (
R)-H
3L-Cu
2+ metal–organic gel was able to achieve the sensitive visual chemoselective and enantioselective recognition of histidine [
21]. After increasing the carboxyl groups and hydroxyl groups and combining chiral binaphthol as the skeleton, (
R)-H
6L was expected to form enhanced hydrogen-bonding interactions, which contributed to strengthening its gelation abilities and further improving the selectivity and sensitivity of recognition.
The gelation properties of compound (
R)-H
6L in various solvents were systematically evaluated using the inverted vial method. As shown in
Table 1, (
R)-H
6L exhibited good solubility in polar organic solvents, including acetone, THF, DMF, DMSO, 1,4-dioxane, i-PrOH, EtOH, and MeOH. The high solubility in these solvents comes from its multiple carboxyl groups, which engage in hydrogen bonding with polar solvents. The addition of water to organic solutions of (
R)-H
6L may induce the formation of gels; therefore, mixed-solvent systems of these organic solvents and water were screened. It was found that both the type of organic solvent and the fraction of water affected the gelation process of (
R)-H
6L. As shown in
Figure S6, the stable gel formation of (
R)-H
6L in DMSO was induced exclusively under conditions containing 50% water by volume at equivalent concentrations. Deviations from this optimal ratio resulted in distinct phase behaviors: systems with <50% water remained as homogeneous solutions, while those with >50% water led to sol formation or precipitation. Among them, only in mixed solvents of DMF/H
2O (1/1,
v/
v) and DMSO/H
2O (1/1,
v/
v) did it form a stable gel, with a critical gelation concentration (CGC) at 20.3 mM and 14.8 mM, respectively. The lower CGC in the DMSO/H₂O system may be attributed to DMSO’s stronger hydrogen-bond-accepting capacity compared to DMF, which enhances the thermodynamic stability of the gel network. A partial gel was formed in EtOH/H
2O and MeOH/H
2O. For the remaining four solvents, the addition of water caused no change to the (
R)-H
6L solution. All the studied gels were formed by heating to completely dissolve them followed by cooling to room temperature, and they were judged using the inverted vial method.
2.2. Specific Recognition of Cu2+ by Gel Formation
Motivated by the pronounced metal-coordinating affinity of carboxyl groups, we then investigated the metal ion responses of (
R)-H
6L through its gelation behavior. All metal ions were employed as their chloride salts to eliminate interference from anions (except AgNO
3). The effects of metal ions on the gelation ability of (
R)-H
6L were first studied in EtOH/H
2O, since only a partial gel was formed in this solvent. When 2 equiv. of 20 common metal salts were added to the partial gel of (
R)-H
6L (14.8 mmol in EtOH/H
2O 1/1,
v/
v), it was found that only Cu
2+ induced the formation of a stable light green gel, while the other metal salts caused either a partial gel or precipitation (
Figure 1). The CGC was determined to be 4.5 mM. The other seven solvent systems mentioned above (organic solvent/H
2O, 1/1,
v/
v) were also tested using 14.8 mM (
R)-H
6L with 2 equiv. of Cu
2+ (
Figure S7). The stable gel remained and was strengthened in DMF/H
2O and DMSO/H
2O, and the CGC in DMSO/H
2O was significantly reduced from 14.8 mM to 1.8 mM. For the remaining solvent systems, it appeared as a solution or precipitation occurred with no gel formation. These results demonstrate that (
R)-H
6L achieved the visual specific recognition of Cu
2+ in EtOH/H
2O (1/1,
v/
v) through gel formation, which is ascribed to the strong metal coordination of carboxylic acid groups with Cu
2+, enhancing the stability of the gel and the fixation ability of the solvent. The gelation response of (
R)-H
6L to cuprous ions (Cu⁺) was also examined. Copper(I) halides are poorly soluble in ethanol and water; therefore, we modified the testing protocol by adding the solvent to a fixed amount of solid. Under these conditions, a turbid solution formed without gelation.
The pH-stimuli responses of the supramolecular gel (
R)-H
6L and the MOG (
R)-H
6L-Cu
2+ were tested. As shown in
Figure 2a, the addition of 20 μL triethylamine (TEA) to the (
R)-H
6L gel (14.8 mM) in DMSO/H
2O (1/1,
v/
v) resulted in a gel–sol transition, while the gel reformed after an equal amount of trifluoroacetic acid (TFA) was subsequently added to the above solution. However, the MOG (
R)-H
6L-Cu
2+ (4.5 mM) in EtOH/H
2O (1/1,
v/
v) did not exhibit this reversible response (
Figure 2b). The addition of 20 μL TEA transformed the gel into an olive-colored solution, and then it precipitated and formed a white suspension after the addition of 20 μL TFA. The difference between them was based on their main interactions: hydrogen bonding in supramolecular gels and metal–ligand coordination bonds in metal–organic gels. The continuous addition of alkalis and acids did not change the pH and hardly affected the hydrogen bonding. For the MOG (
R)-H
6L-Cu
2+, TEA competed with (
R)-H
6L to coordinate with Cu
2+ and thus disrupted the gel. Following this, a systematic investigation was conducted on the gelation ability of (
R)-H
6L (4.5 mM) in EtOH/H
2O (1/1,
v/
v) and Cu
2+ in different pH environments. As shown in
Figure 2c, gel formation occurred only under neutral or weakly acidic conditions. Under either acidic or alkaline conditions, the formation of gels was notably hindered. When the pH was below 3, the solution exhibited a white turbid appearance, attributed to H⁺ interfering with the hydrogen-bonding interactions between (
R)-H
6L and the solvent molecules, thereby decreasing its solubility. It was unable to form a gel at a pH exceeding 8. Concurrently, the solution exhibited progressive color intensification with rising hydroxide ion (OH⁻) concentrations, which was attributed to the formation of copper(II)-based coordination complexes.
The quantitative gelation investigation of (
R)-H
6L with Cu
2+ was performed by gradually adding Cu
2+ (0–4 equiv.) to (
R)-H
6L (4.5 mmol in EtOH/H
2O 1/1,
v/
v) via the inverted vial method (
Figure S8). A suspension was formed with 0.05 equiv. of Cu
2+, and an unstable gel with weak fluidity was produced with 0.1 equiv. of Cu
2+. Starting from 0.2 equiv. of Cu
2+ addition, the gel became stable with no fluidity. The quantitative investigation of (
R)-H
6L with Cu
2+ in a gel was performed by gradually adding Cu
2+ to an EtOH/H
2O solution of (
R)-H
6L using CD spectroscopy (
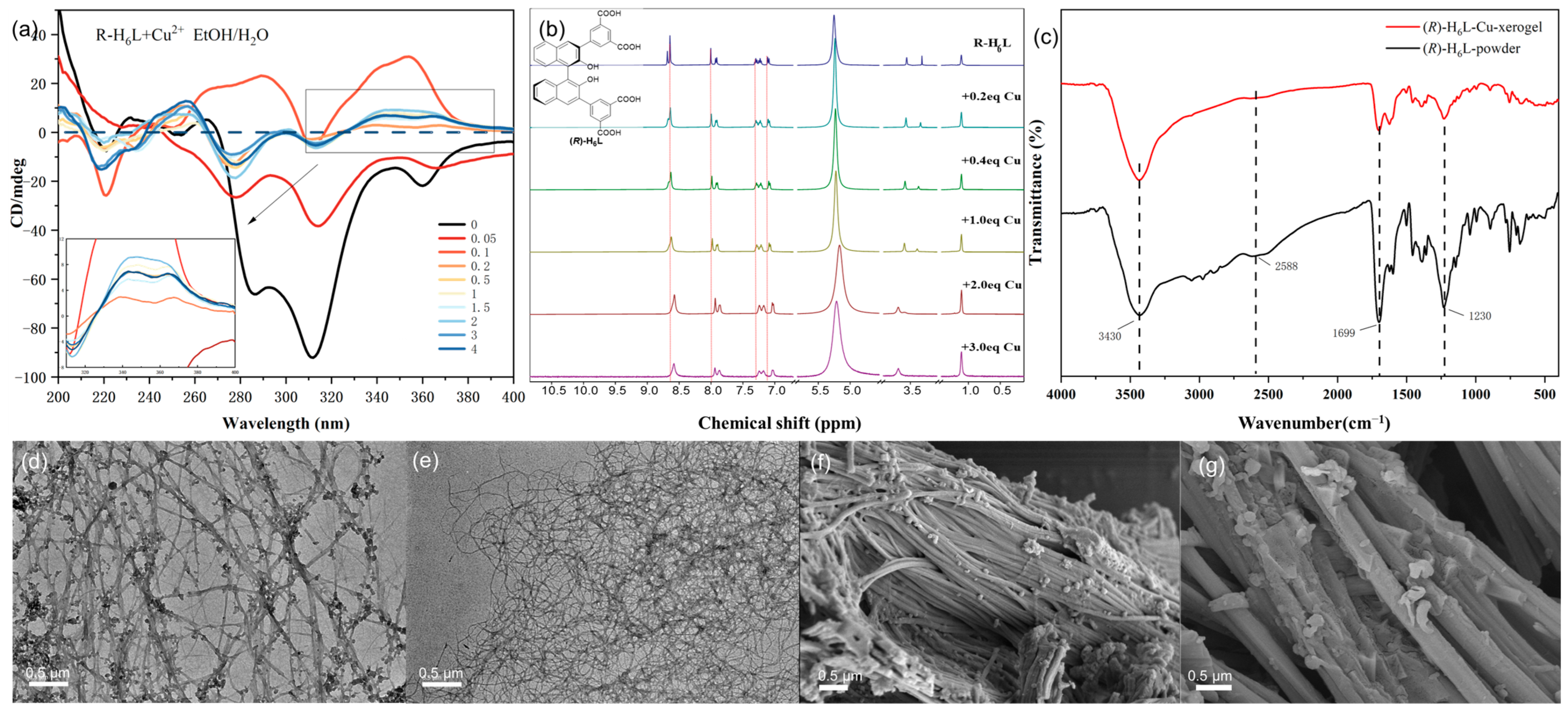
Figure 3a). (
R)-H
6L showed negative CD signals in the region of 270–380 nm, which were caused by the chiral binaphthalene group and benzene group. With the continuous addition of Cu
2+, this negative CD band was first significantly reduced with 0.05 equiv. of Cu
2+ and then reversed to positive CD signals at 290 nm and 354 nm with 0.1 equiv. of Cu
2+, which meant that Cu
2+ changed from its previous self-assembly structure with a negative CD band and formed a new chiral assembly structure (
R)-H
6L-Cu
2+ with a positive CD band. When the Cu
2+ addition was in the range of 0.2 equiv. to 4 equiv., the CD spectra exhibited similar patterns, and the CD intensity at 340 nm and 365 nm gradually increased from 0.2 equiv. to 2 equiv. and then decreased steadily, which indicated a stable gel and implies that the coordinated stoichiometry for Cu
2+ to (
R)-H
6L is 2:1. The gel with 2 equiv. Cu
2+ was named (
R)-H
6L-Cu
2 and that with 0.2 equiv. Cu
2+ was named (
R)-H
6L-Cu
0.2.
1H NMR studies of (
R)-H
6L with different equivalents of Cu
2+ (0–3 equiv.) in EtOD-
d6 were also conducted (
Figure 3b), which showed that the signals of all aromatic protons had an upfield shift with the addition of Cu
2+ from 0.2 to 2 equiv. and remained unchanged from 2 to 3 equiv., confirming that the coordination ratio for (
R)-H
6L: Cu
2+ is 1:2. The observed upfield shifts in the chemical shifts of all aromatic protons could be attributed to π–π stacking interactions between aromatic rings, which were likely induced by metal-coordination-driven supramolecular assembly. The coordination of Cu
2⁺ with (
R)-H
6L promoted structural organization, facilitating the closer and more ordered alignment of the aromatic moieties. This π–π stacking enhanced the ring current shielding effect, thereby shifting the aromatic proton resonances to higher fields in the
1H NMR spectrum.
The FT-IR spectrum of the xerogel of (
R)-H
6L-Cu
2 was compared with that of the powder of (
R)-H
6L (
Figure 3c). The FT-IR analysis revealed a significant decrease in intensity for the carboxylic acid C=O stretching vibration peak at 1699 cm⁻
1 and the C-O stretching vibration peak at 1230 cm⁻
1, while the O-H stretching vibration peak in phenolic hydroxyl groups at 3430 cm
−1 remained unchanged, indicating that carboxylic acid groups coordinate with Cu
2+ while phenolic hydroxyl groups do not. The absence of hydroxyl group coordination can likely be attributed to the steric hindrance exerted by substituents located at the ortho positions of the binaphthyl framework, which restricts access to the metal center.
Transmission electron microscopy (TEM) and scanning electron microscopy (SEM) were utilized to characterize the morphology of the gels. TEM showed that both the (
R)-H
6L-Cu
0.2 gel and (
R)-H
6L-Cu
2 gel assembled into a cross-linked fibrous structure; the former had small spherical aggregates attached to the nanofibers, while the latter had a more homogeneous and dense fiber network structure (
Figure 3d,e). The TEM data of dilute (
R)-H
6L (0.45 mM) and (
R)-H
6L (0.45 mM) with 2 equiv. Cu
2+ in EtOH/H
2O (1/1,
v/
v) were also recorded for comparison. In a dilute solution, (
R)-H
6L presented as nanovesicles or spheres, and the addition of 2 equiv. Cu
2+ strung these particles together (
Figure S9). The SEM of the (
R)-H
6L-Cu
0.2 xerogel revealed that there was a large bundle of fine fibers arranged together, with some small spheres adhered to them, while it formed relatively coarse and straight fibers in the (
R)-H
6L-Cu
2 xerogel. We can conclude that, under the gelation conditions, (
R)-H
6L coordinated with Cu
2+ to form a cross-linked fiber network structure, resulting in the formation of a gel. (
R)-H
6L with 0.2 equiv. of Cu
2+ partially assembled into fibers due to the incomplete coordination, and the remaining molecules aggregated into spheres to connect with the fibers, while 2 equiv. of Cu
2+ rendered the fibrosis more complete.
According to the CD, NMR, and FT-IR experiments, we confirm the coordination between Cu
2+ and the carboxyl groups in compound (
R)-H
6L, establishing a metal-to-ligand stoichiometric ratio of 2:1 (M:L = 2:1). In this case, it forms a three-dimensional network structure of (
R)-H
6L-Cu
2+, which is supported by SEM and TEM. In other words, Cu
2+ connects the (
R)-H
6L molecules in series to form long chains; meanwhile, the spare carboxyl groups of ligands on the different chains are connected through Cu
2+ [
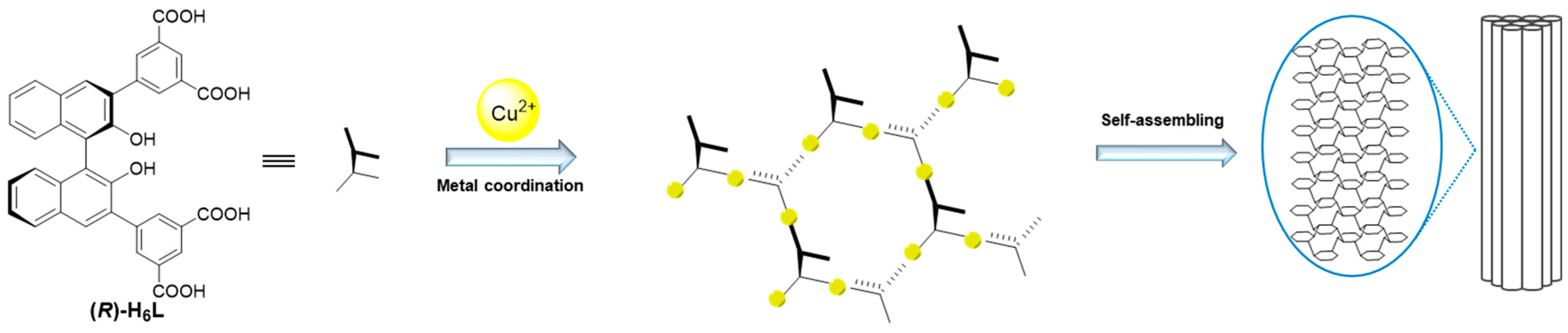
22]. Then, self-assembly dominated by hydrogen bond interactions and π–π stacking results in the formation of gels (
Figure 4). This model explains the opposite chirality and weak intensity in the CD spectrum. The antiparallel arrangement of (
R)-H
6L modified the inherent chirality of the self-assembly originally formed by hydrogen-bonding interactions and π–π stacking interactions, while the network structure led to the diminished chiral amplification of the assembly. This model further explains the gelation mechanism with low equiv. of metal ions. Not all carboxyl groups of (
R)-H
6L participate in metal coordination, and a minimal amount of Cu
2⁺ (0.2 equiv.) suffices to induce structural assembly, while higher concentrations (2 equiv.) promote more complete planar-directed assembly. At the molecular level, this enhanced coordination manifests as increased structural stability, as evidenced by the CD and NMR titration experiments. Macroscopically, it results in thicker fibrous networks, as directly visualized through SEM.
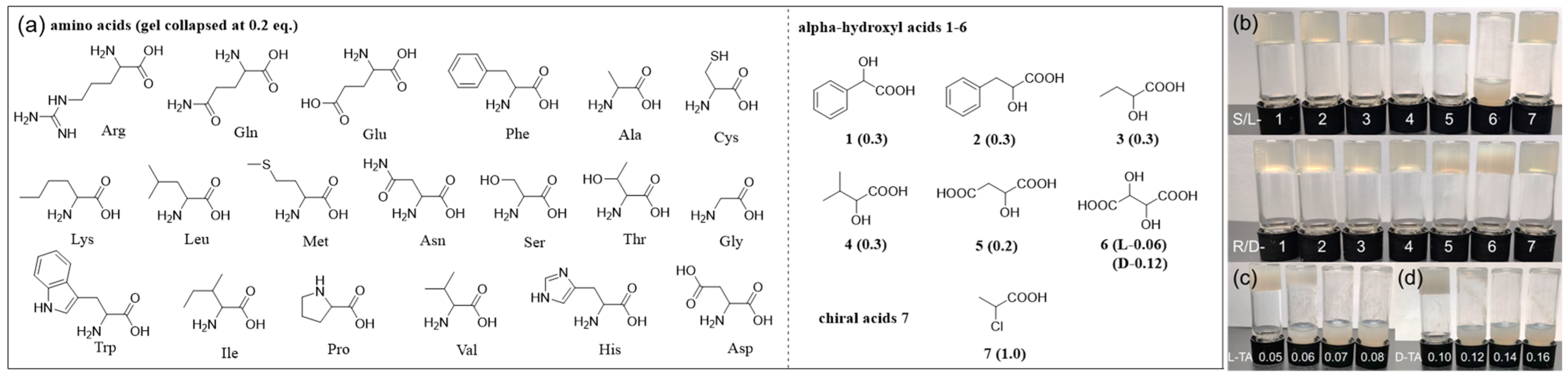
2.3. Chemoselective and Enantioselective Recognition of Tartaric Acid by Gel Collapse
(
R)-H
6L obtained stable chiral MOGs with Cu
2+ through the coordination of carboxyl groups. To explore its potential for enantiomer recognition, we systematically investigated its response to the enantiomers of various chiral acids, including 19 common amino acids, α-hydroxyl carboxylic acids
1–
6, and regular chiral acid 2-chloropropanoic acid
7 (
Figure 5a). In order to achieve more sensitive recognition, the equivalent of Cu
2+ was reduced to the minimum of 0.2, which still formed a stable gel with (
R)-H
6L (4.5 mM in EtOH/H
2O 1/1,
v/
v) (
Figure 5a and
Figure S8). Chiral acids of different equivalents (0.05–0.5 equiv.) were added to the (
R)-H
6L-Cu
0.2 gel, and it was found that all tested carboxylic acids induced gel collapse, except 2-chloropropanoic acid
7, proving that amino groups or hydroxyl groups at the α-position play an important role in gel collapse (
Table S2). The abilities of various chiral acids to induce gel collapse are also different. All 19 amino acids collapsed the gel at 0.2 equivalents, while mono hydroxyl acids
1–
4 caused gel collapse at 0.3 equivalents, ascribed to the stronger coordinating ability of amino groups with Cu
2+ than hydroxyl groups. Malic acid
5 features an extra carboxylic acid group; it disrupted the gel at lower equivalents (0.2 equiv.) than mono acids
1–
4. The gel responses caused by amino acids and hydroxyl acids
1–
5 are not enantioselective. Of all tested chiral acids, tartaric acid (TA)
6, featuring two hydroxyl groups and two carboxylic groups, exhibited the most unique and sensitive gel response, exhibiting enantioselective gel collapse at 0.1 equiv. with the L-enantiomer only (
Figure 5b).
Therefore, the response behavior of the (
R)-H
6L-Cu
0.2 gel (4.5 mM) in EtOH/H
2O (1/1,
v/
v) toward 0.1 equiv. of 26 chiral carboxylic acids was studied, and it was found that only L-TA caused gel collapse, while D-TA and both enantiomers of the remaining 25 chiral acids caused no change to the gel, indicating that (
R)-H
6L-Cu
0.2 has both chemoselective and enantioselective recognition abilities toward L-TA by simple visual gel collapse. The disrupting capacity of TA on the (
R)-H
6L-Cu
0.2 gel was further carefully examined through a series of concentration gradients. The gel underwent rapid collapse upon the addition of merely 0.06 equivalents of L-TA (
Figure 5c), in stark contrast to its D-enantiomer, which required twice the amount to induce similar gel collapse (
Figure 5d). The enantiomer (
S)-H
6L was synthesized and exhibited identical gelation properties. The resulting (
S)-H
6L-Cu
0.2 gel demonstrated enantioselective recognition toward D-TA, undergoing collapse at a remarkably low concentration of 0.06 equiv. In contrast, the L-enantiomer required twice the amount (0.12 equiv.) to induce gel collapse, mirroring the chiral recognition behavior observed in its (
R)-H
6L enantiomer.
In order to study the process of (
R)-H
6L-Cu
0.2’s enantioselective recognition of tartaric acid, the CD spectra were measured (
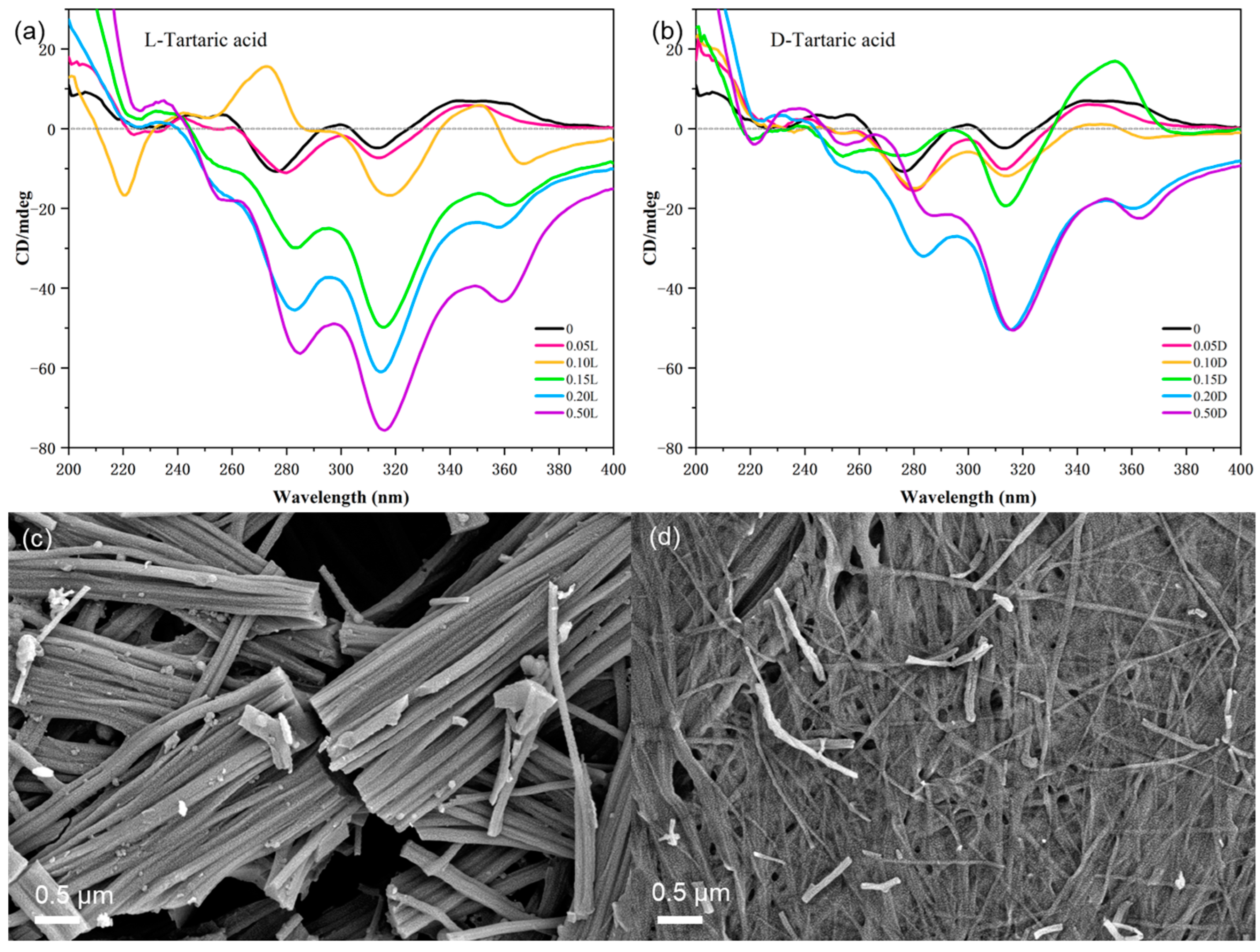
Figure 6). The addition of 0.05 equiv. of L-TA had no effect on the gel and did not change the CD signal. The gel collapsed after adding 0.1 equiv. of L-TA, with a new negative peak appearing at 368 nm and a positive peak appearing at 270 nm (
Figure 6a), indicating that L-TA entered the chiral sites of (
R)-H
6L-Cu
2+ and formed an intermediate structure. With the addition of more than 0.15 equiv. of L-TA, the CD spectra recovered to the CD signals of (
R)-H
6L only, showing that L-TA completely displaced Cu
2+ from (
R)-H
6L via metal coordination. For D-TA, the addition of 0.05 equiv. did not change the CD signal in the same way. The CD signal of (
R)-H
6L-Cu
0.2 with 0.15 equiv. D-TA was similar to that of (
R)-H
6L with 0.1 equiv. Cu
2+ (
Figure 3a). Excessive D-TA restored the negative CD signal and maintained it (
Figure 6b), which could indicate that D-TA competed with (
R)-H
6L for the coordination of Cu
2+.
Malic acid (MA)
5, with fewer hydroxyl groups, was also measured (
Figure S11). It was found that both L-MA and D-MA had the same effect on the (
R)-H
6L-Cu
0.2 gel, indicating that the additional hydroxyl group played an important role in chiral recognition. Meanwhile, succinic acid, with no hydroxyl groups, and 2,3-butanediol, with no carboxyl groups, were also tested (
Figure S12). Neither of them had an effect on the CD signal or caused the gel to collapse with 0.2 equiv. addition, proving that visual recognition via the collapse of the gel requires the interaction of hydroxyl and carboxyl groups.
Scanning electron microscopy (SEM) studies provided the direct visualization of the distinct morphological impacts exerted by D- and L-TA enantiomers on the (
R)-H
6L-Cu
0.2 gel. As shown in
Figure 6c, the addition of 0.1 equiv. of L-TA cracked the fibers and thereby collapsed the gel. In contrast, D-TA formed some broken fibers while retaining the cross-linked fibrous structure (
Figure 6d), which was similar to the effects seen in (
R)-H
6L-Cu
0.1 (
Figure S13). This similarity suggests that D-TA destabilizes the gel by competitively coordinating with Cu
2⁺, thereby disrupting the (
R)-H
6L-Cu
2+ gel architecture.
The formation constants were determined to compare the coordination abilities of tartaric acid and (
R)-H
6L with Cu
2+. By monitoring the chemical shift changes of specific protons in (
R)-H
6L as a function of the Cu
2⁺ concentration, the fitting yielded a high correlation coefficient (R
2 = 0.99), and the calculated formation constant (K) was determined to be 2.45 × 10
3 M
−2 with lgK = 3.39 (
Figures S14 and S15). According to reference [
23], under neutral conditions, tartaric acid coordinates with Cu
2+ in a 1:2 ratio, and the corresponding lgK as a critical stability constant is 5.11. These data demonstrate that tartaric acid coordinates more readily with Cu
2+ compared to (
R)-H
6L.
Therefore, a possible mechanism for the chemoselective and enantioselective gel collapse was proposed. Carboxylic acid with alpha-hydroxyl or amino groups is more likely to coordinate with Cu
2+ by chelation than carboxyl groups alone. It can compete with (
R)-H
6L for Cu
2+ coordination and then cause the gel to collapse. It is easier for chirality-matched L-TA to enter the chiral pores or chiral sites of the (
R)-H
6L-Cu
2+ gel and capture Cu
2+ [
21], while D-TA with chirality mismatch requires a greater amount to compete with (
R)-H
6L for Cu
2+ coordination, thus realizing enantioselective gel collapse.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}