

Microgel with a Core—Shell Particulate Structure Formed via Spinodal Decomposition of a Diblock Ionomer Containing a Doped Hydrophobic Moiety


Abstract

1. Introduction
2. Results and Discussion
2.1. Monitoring the Chain Growth of (AxGy)Sz in One-Pot ATRP System
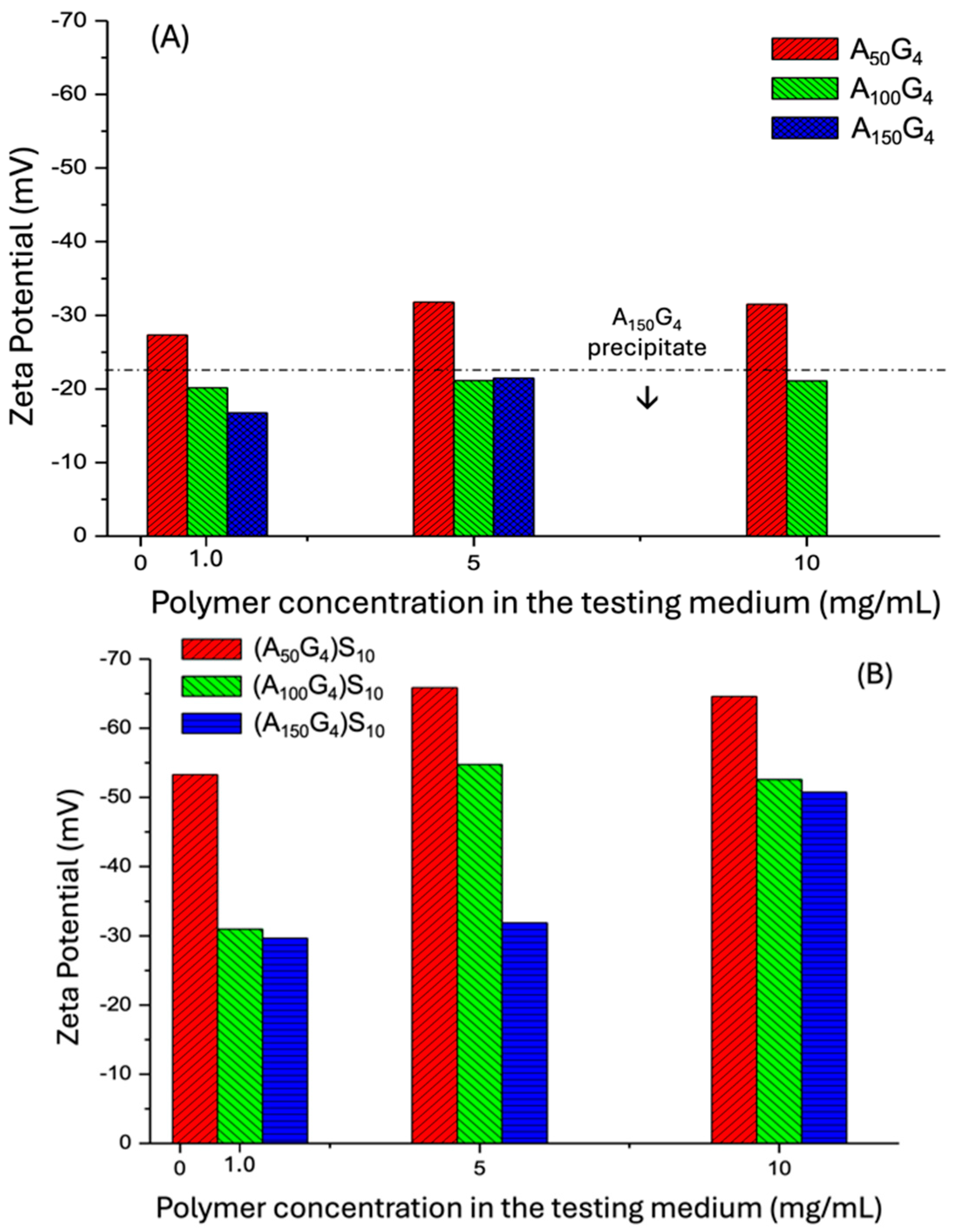
2.2. The DI Chain Structures and Their Colloidal Dispersion States in Water Deduced from Zeta Potential Measurement
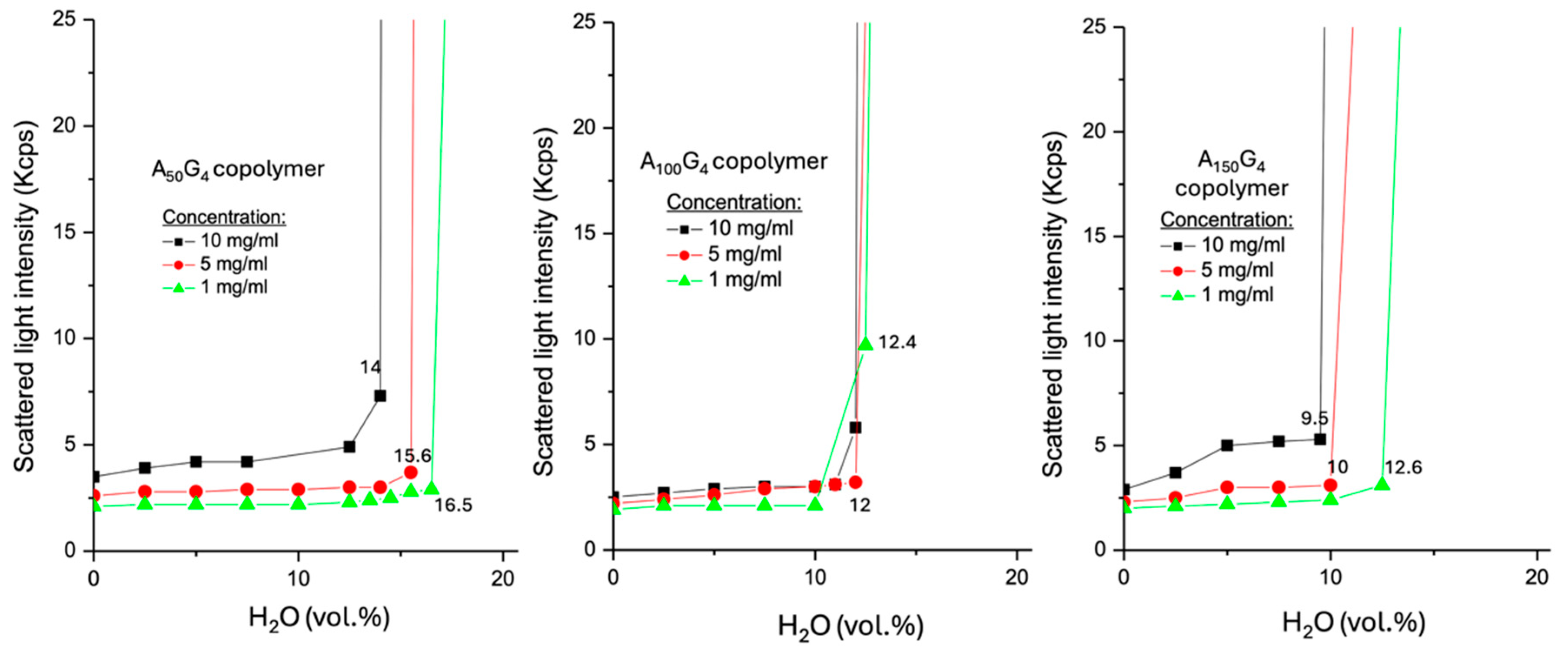
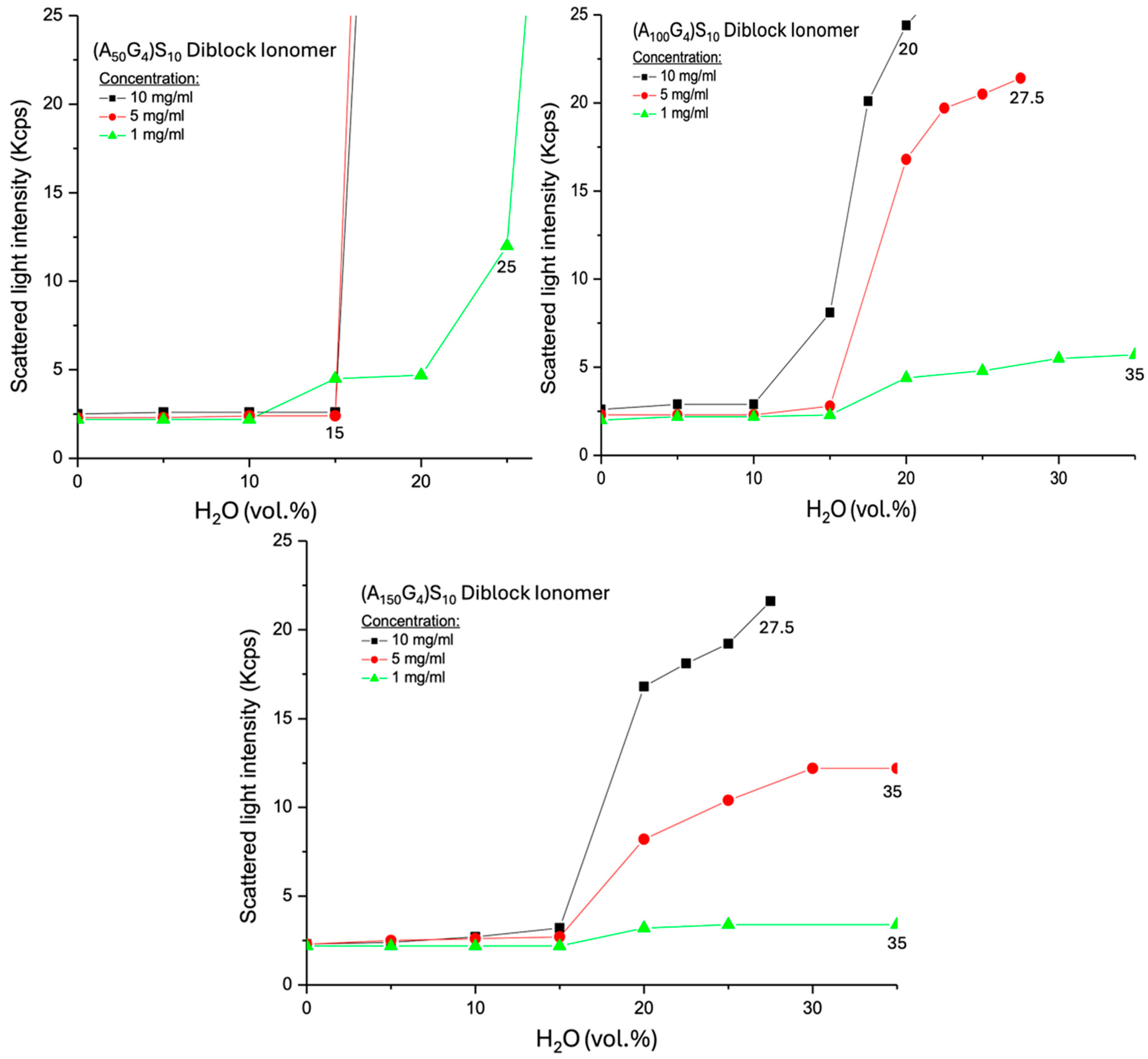
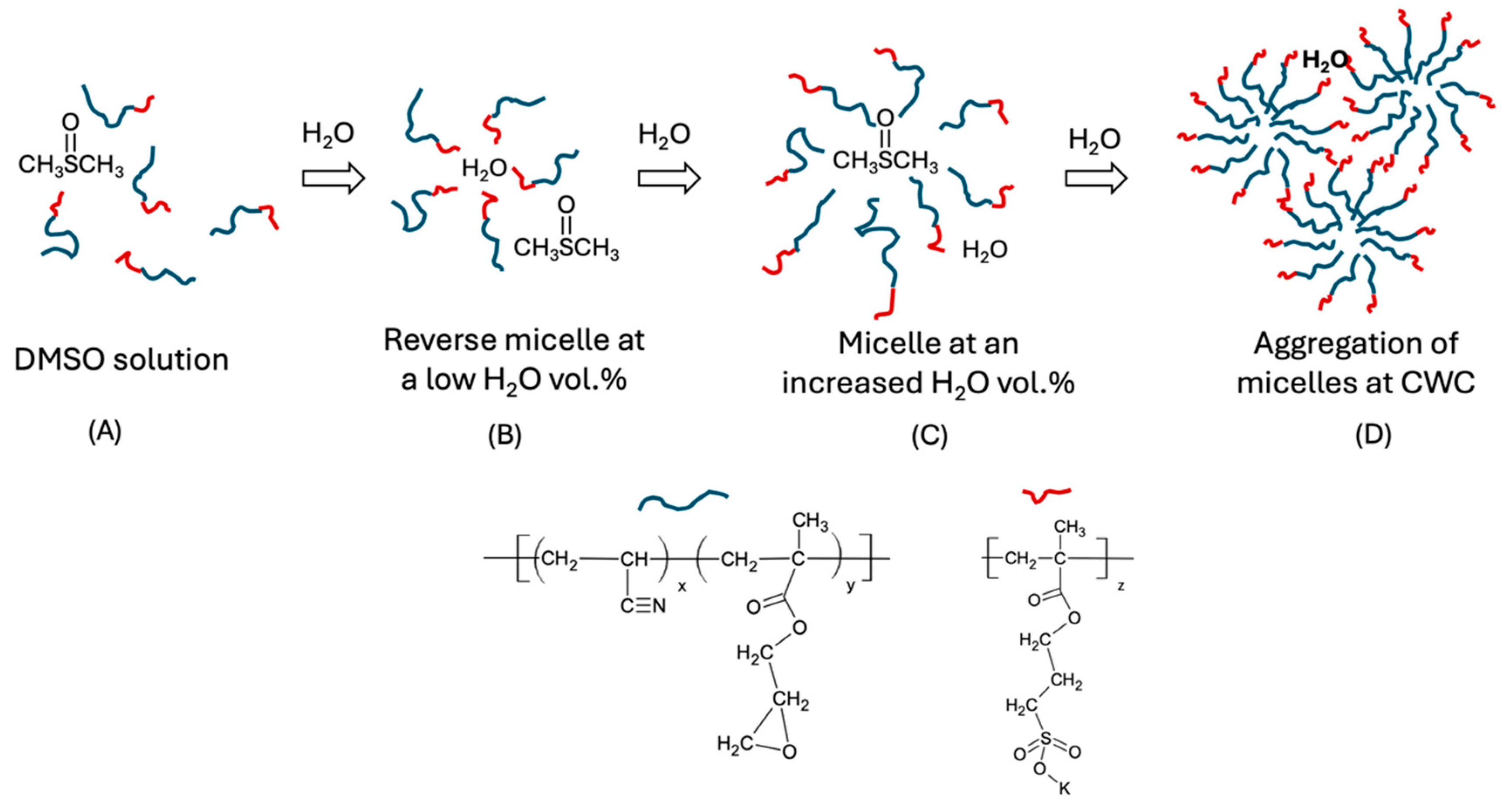
2.3. Analysis of the Spinodal Decomposition Behaviors of DIs in a DMSO-Water Medium
- (i)
- For a copolymer AxGy, an increase in concentration results in a lower CWC. This reflects the number-driven aggregation behavior.
- (ii)
- At the low concentration, A100G4 shows the highest SLI (ca. 9.9 Kcps) at its CWC compared to the other two copolymers, whose SLI values are comparable. This is attributed to the balance between the ease of chain tangling and the realized interfacial area of the colloidal particles, which both contribute to SLI to different extents. This result aligns with their moderately negative ζ potential, as previously observed. At the middle concentration, all three copolymers exhibit comparable SLI at their CWCs due to a tradeoff between the surface of the colloidal particles and their surface characteristic. For example, A150G4 colloidal particles exhibit the lowest CWC, indicating the strongest occurrence of hydrophobic coagulation of many small colloidal particle sizes, and such a light aggregation assisted by the DMSO component should maintain a relatively higher portion of the initial interfacial area, which favors SLI [46]. Similarly, the higher interfacial exposure allows more nitrile groups to contribute to the surface charge as well.
- (iii)
- At the highest concentration, A50G4 exhibits the highest SLI at its CWC among the three copolymers under identical conditions. This can be attributed to the impact of the highest colloidal particle surface associated with the maximum CWC (14%), as well as a more efficient surface chain packing extent facilitated by the reduced steric hindrance.
- (iv)
- A100G4 exhibits concentration-independent CWCs, which can be attributed to the balance between competing factors: raising concentration promotes chain aggregation, while steric hindrance counteracts this effect. In contrast, both A50G4 and A150G4 show more pronounced variations in CWCs across different concentrations due to shifts in this balance as well as the leverage of their obviously diverse hydrophobic traits.
- The higher concentration leads to greater SLI following the formation of normal micelles.
- (A150G4)S10 micelles exhibit weaker SLI than (A100G4)S10 micelles at equivalent water content.
- No observable precipitation occurs in the dispersions of these two DIs.
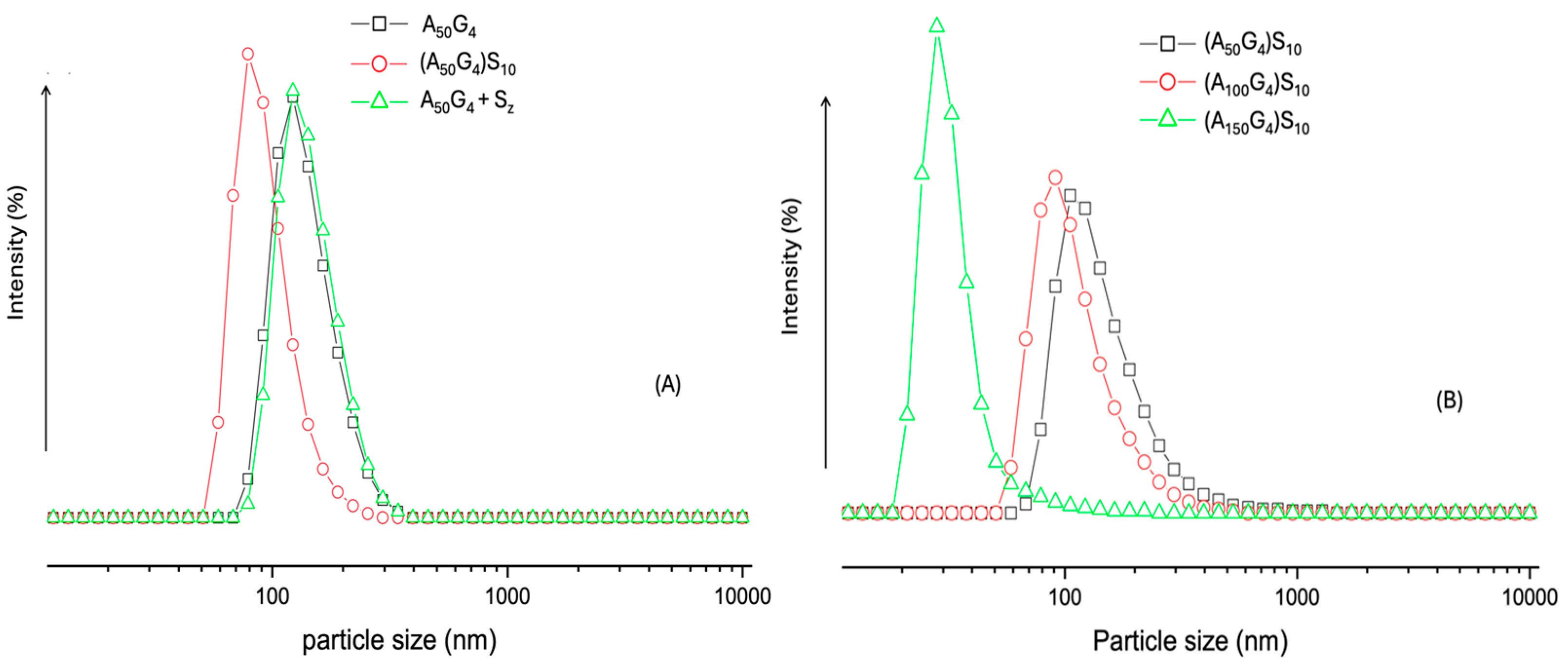
2.4. Determination of Copolymer Colloidal Particle Size Distribution in Water Using DLS
3. Conclusions
- Both types of copolymers dissolve in DMSO as solvated coils and transition to colloidal particles upon water incorporation. In the case of AxGy, the colloidal particles form as random aggregates of copolymer chains, whereas for (AxGy)Sz, they assemble into micelles, which subsequently coagulate to form colloidal particles. The resulting colloidal dispersions remain in a metastable state when DMSO is dialyzed from the medium.
- The colloidal particles of AxGy dispersed in water exhibit a weak negative ζ potential, which increases with a higher mole fraction of the amphiphilic unit G (viz. a higher y/x), favoring the generation of surface charge. Reciprocally, a higher x/y ratio enhances the ζ potential by promoting looser chain packing and, hence, surface area due to increased conformational flexibility. A similar trend is observed in aqueous dispersions of (AxGy)Sz, though these exhibit significantly more negative ζ potentials.
- Scattered light intensity (SLI) analysis reveals the coagulation process of AxGy chains as water content increases in the DMSO-H2O dispersion medium. Clear spinodal decomposition (phase separation) occurs at critical water contents, generally following the trend that a higher x/y ratio corresponds to a lower critical water content, though this is slightly influenced by chain conformation. In contrast, (AxGy)Sz exhibits no distinct phase separation thresholds due to the hydrated exterior, except for the dispersion with the smallest x/y ratio, where compact micelles form, which triggers sedimentation.
- Dynamic light scattering analysis reveals the colloidal particle size distribution of the three (AxGy)Sz in water, showing that the largest x/y ratio produces the smallest particles, while the smallest x/y ratio results in the largest particles. The hydrophobic chain conformational flexibility plays a key role in leveraging the colloidal particle sizes. Notably, the colloidal particles with the largest sizes are highly stable, leaving behind nano-spherical structures (ca. 20–200 nm) after drying. The nanospheres function effectively as microgels, featuring a hydrophilic shell and a hydrophobic core.
4. Materials and Methods
4.1. Chemicals
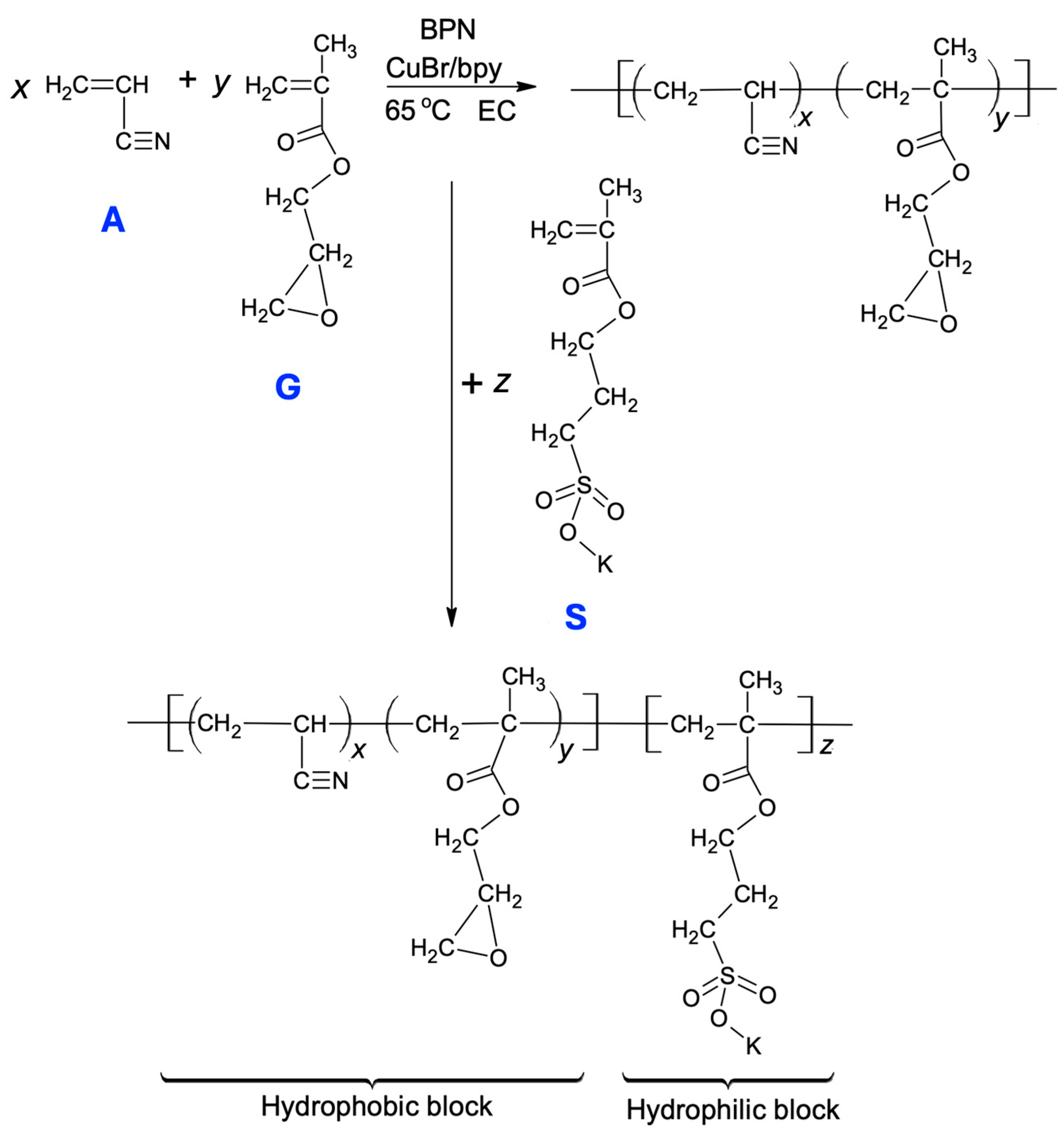
4.2. One-Pot Synthesis of the Block Copolymer (AxGy)Sz by Atom Transfer Radical Polymerization (ATRP)
4.3. Characterization Methods
4.3.1. Chromatography Analysis
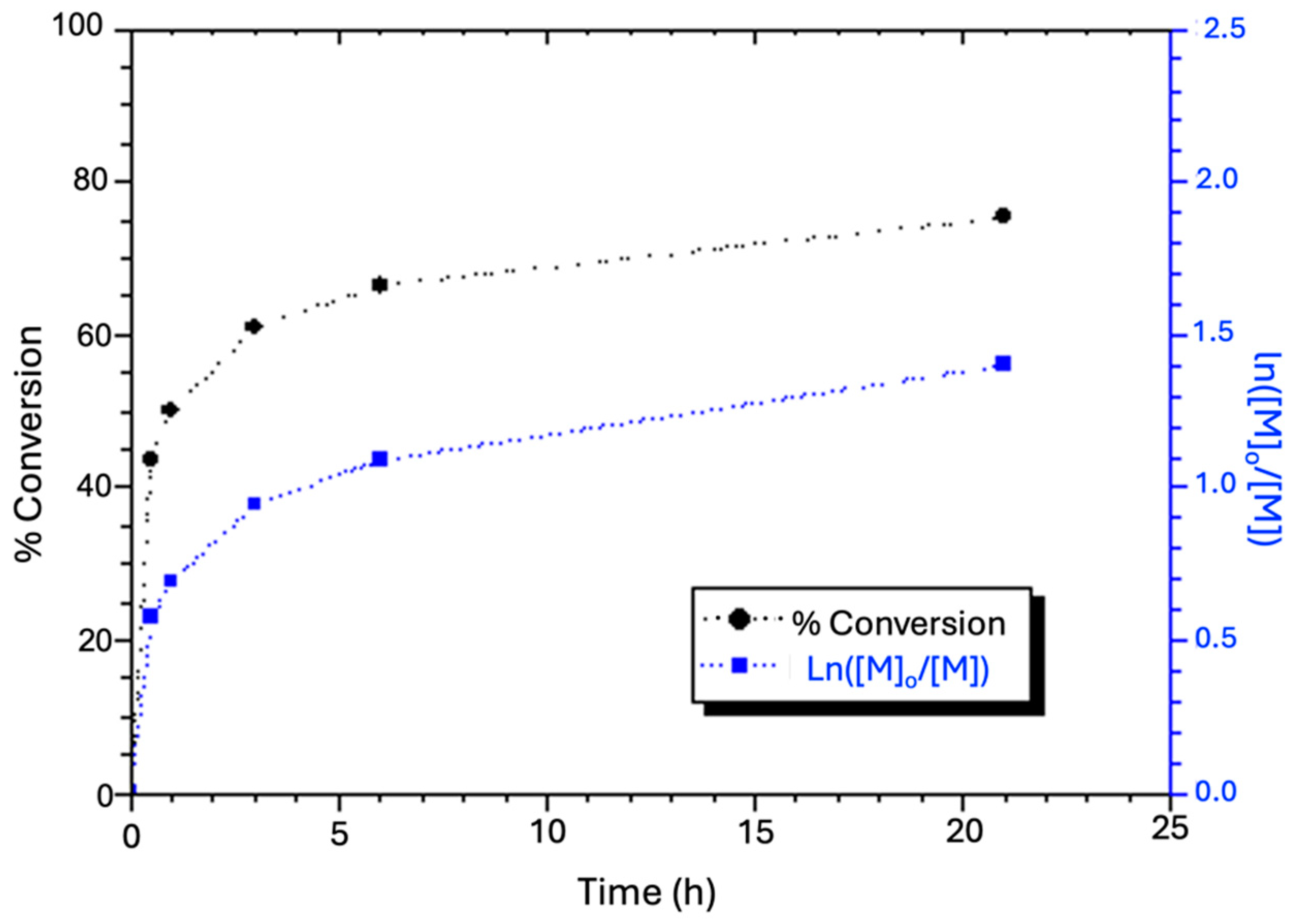
- The time-dependent conversion of the two monomers, AN and GMA, during ATRP to synthesize the hydrophobic block (AxGy) was monitored using gas chromatography (GC) analysis. This polymerization followed the method established by Matyjaszewski et al. [53]. Samples (1 mL) were withdrawn from the Schlenk flask at predetermined intervals (0, 0.5, 1, 3, 6, and 23 h) using a glass syringe. Each sample was immediately introduced in 2 mL THF to precipitate the polymer component and keep the monomers in THF. The resulting suspension was filtered through 0.45-μm Millipore Millex® HN filtration paper, and the filtrate was analyzed using a Shimadzu GC-2010 equipment (Kyoto, Japan) equipped with a flame ionization detector, for which THF acted as the internal standard. The calibration standards were prepared by mixing a series of AN (0.0152–6.076 mmol), respectively, with 4 mole % GMA in 2 mL THF in sample vials. After the same filtration purification as described above, the clear solution in each vial was injected into the GC to establish the calibration curve. Quantitatively, the monomer conversion was the mean of the conversions of the two monomers.
- b.
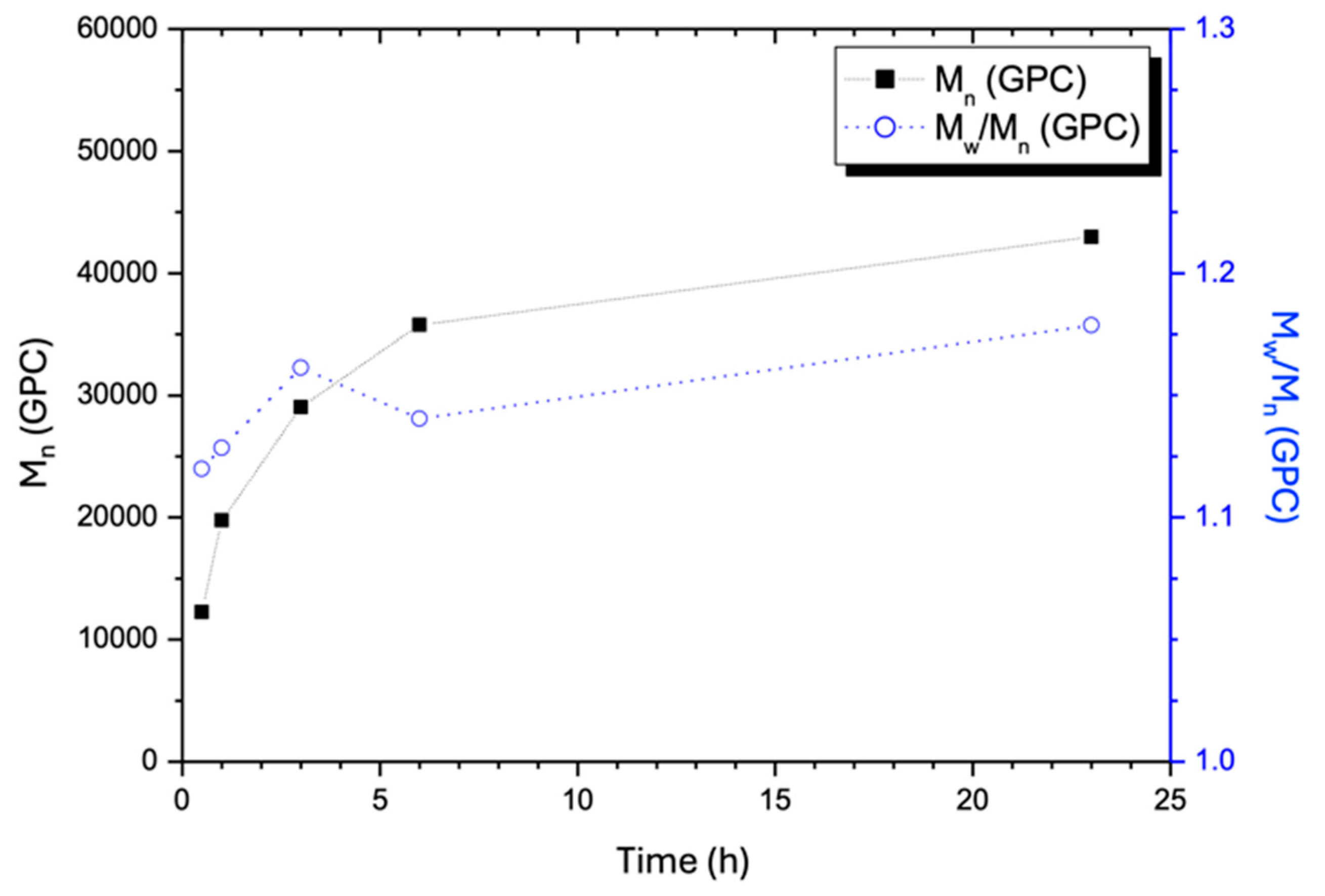
- Determination of the molecular weights, and , of copolymers AxGy by the gel permeation chromatography (GPC). The molecular weights of the copolymers AxGy were determined using the gel permeation chromatography (GPC) method. The measurements were carried out on a Waters GPC system (Milford, MA, USA) equipped with Waters Styragel columns, a Waters—2487 dual wavelength UV detector, and a Waters—2414 refractive index detector. DMF was used as the eluent at a flow rate of 1.0 mL/min.
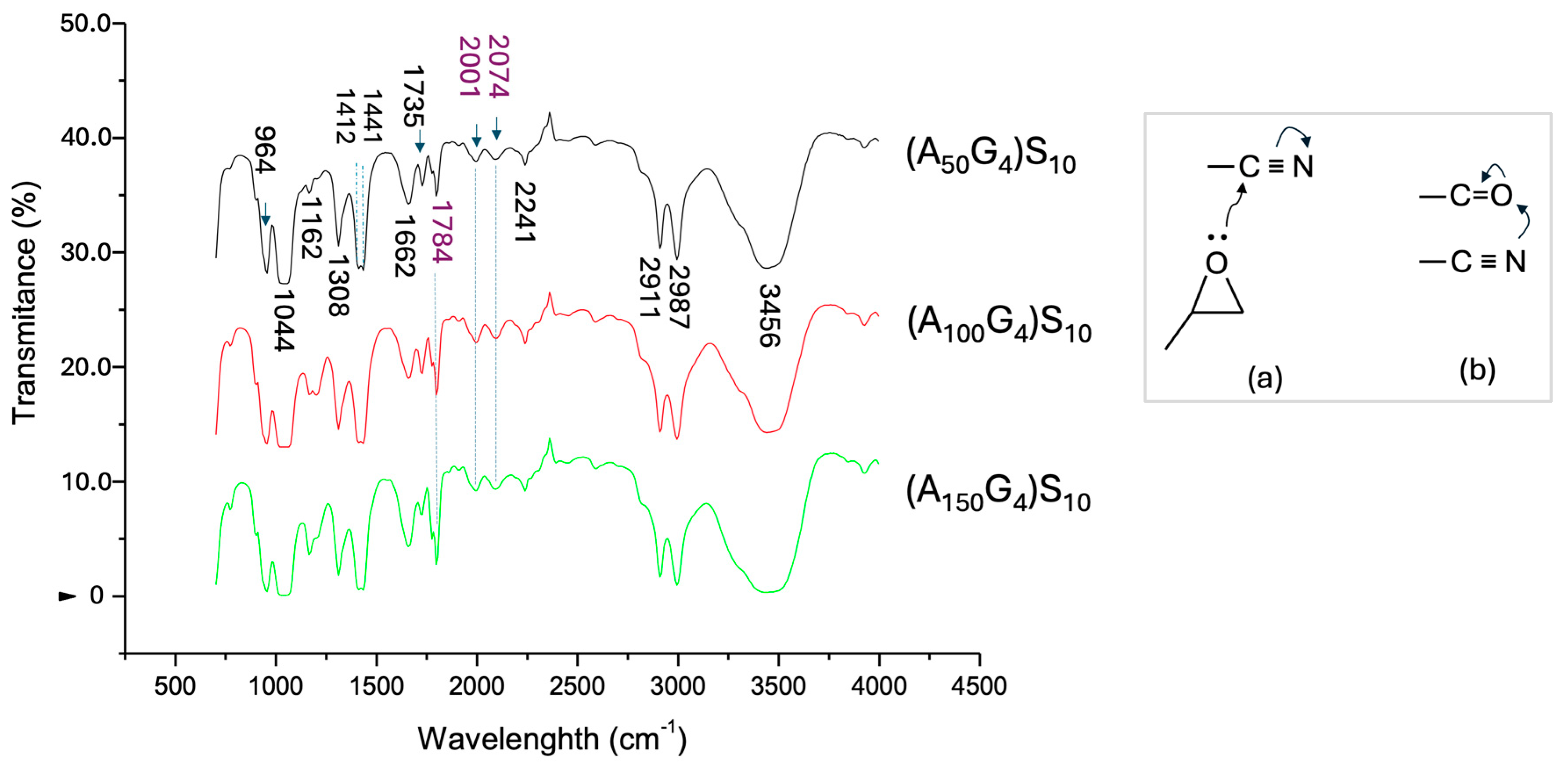
4.3.2. Infrared Spectroscopy Analysis of the Copolymers of AxGy and (AxGy)Sz
4.3.3. Zeta Potential Measurement of the Copolymers of AxGy and (AxGy)Sz
4.3.4. Static Light Scattering (SLS) and Dynamic Light Scattering (DLS) Measurements
4.3.5. Transmission Electron Microscopy (TEM)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Use of Artificial Intelligence
References
- Dodda, J.M.; Deshmukh, K.; Bezuidenhout, D. (Eds.) Multicomponent Hydrogels Smart Materials for Biomedical Applications; RSC: London, UK, 2023; Chapter 1; pp. 1–25. [Google Scholar] [CrossRef]
- Ahmed, E.M. Hydrogel: Preparation, characterization, and applications: A review. J. Adv. Res. 2015, 6, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.-C.; Chang, C.-C.; Chan, H.-P.; Chung, T.-W.; Shu, C.-W.; Chuang, K.-P.; Duh, T.-H.; Yang, M.-H.; Tyan, Y.-C. Hydrogels: Properties and applications in biomedicine. Molecules 2022, 27, 2902. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Cid, P.; Jiménez-Rosado, M.; Romero, A.; Pérez-Puyana, V. Novel trends in hydrogel development for biomedical applications: A review. Polymers 2022, 14, 3023. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yu, C.; Kumar, H.; He, X.; Lu, Q.; Bai, H.; Kim, K.; Hu, J. The effect of crosslinking degree of hydrogels on hydrogel adhesion. Gels 2022, 8, 682. [Google Scholar] [CrossRef]
- Picchioni, F.; Muljana, H. Hydrogels based on dynamic covalent and non-covalent bonds: A chemistry perspective. Gels 2018, 4, 21. [Google Scholar] [CrossRef]
- Hu, W.; Wang, Z.; Zhang, S.; Wang, J. Advances in crosslinking strategies of biomedical hydrogels. Biomater. Sci. 2019, 7, 843. [Google Scholar] [CrossRef]
- Agrawal, G.; Agrawal, R. Stimuli-responsive microgels and microgel-based systems: Advances in the exploitation of microgel colloidal properties and their interfacial activity. Polymers 2018, 10, 418. [Google Scholar] [CrossRef]
- Bertsch, P.; Andrée, L.; Besheli, N.H.; Leeuwenburgh, S.C.G. Colloidal hydrogels made of gelatin nanoparticles exhibit fast stress relaxation at strains relevant for cell activity. Acta Biomater. 2022, 138, 124–132. [Google Scholar] [CrossRef]
- Daly, A.C.; Riley, L.; Segura, T.; Burdick, J.A. Hydrogel microparticles for biomedical applications. Nat. Rev. Mater. 2020, 5, 20–43. [Google Scholar] [CrossRef]
- Xu, J.; Liu, X.; Ren, X.; Gao, G. The role of chemical and physical crosslinking in different deformation stages of hybrid hydrogels. Eur. Polym. J. 2018, 100, 86–95. [Google Scholar] [CrossRef]
- North, S.M.; Armes, S.P. Aqueous solution behaviour of stimulus-responsive poly(methacrylic acid)-poly (2-hydroxypropyl methacrylate) diblock copolymer nanoparticles. Polym. Chem. 2020, 11, 2147–2156. [Google Scholar] [CrossRef]
- Larrañeta, E.; Stewart, S.; Ervine, M.; Al-Kasasbeh, R.; Donnelly, R.F. Hydrogels for hydrophobic drug delivery. classification, synthesis and applications. J. Funct. Biomater. 2018, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, H. Micro hydrogels: Preparation, properties, and applications. J. Oleo Sci. 2013, 62, 865–871. Available online: https://pubmed.ncbi.nlm.nih.gov/24200933/ (accessed on 1 November 2013). [PubMed]
- Liu, Y.; Chen, Z.; Xu, J. Recent advances in the microfluidic generation of shape-controllable hydrogel microparticles and their applications. Green Chem. Eng. 2024, 5, 16–30. [Google Scholar] [CrossRef]
- Gibson, M.I.; O’Reilly, R.K. To aggregate, or not to aggregate? Considerations in the design and application of polymeric thermally responsive nanoparticles. Chem. Soc. Rev. 2013, 42, 7204. [Google Scholar] [CrossRef]
- Karg, M.; Pich, A.; Hellweg, T.; Hoare, T.; Lyon, L.A.; Crassous, J.J.; Suzuki, D.; Gumerov, R.A.; Schneider, S.; Potemkin, I.I.; et al. Nanogels and microgels: From model colloids to applications, recent developments, and future trends. Langmuir 2019, 35, 6231–6255. [Google Scholar] [CrossRef]
- Song, W.; Li, L.; Liu, X.; Zhu, Y.; Yu, S.; Wang, H.; Wang, L. Hydrogel microrobots for biomedical applications. Front. Chem. 2024, 12, 1416314. [Google Scholar] [CrossRef]
- Cao, Q.; Chen, W.; Zhong, Y.; Ma, X.; Wang, B. Biomedical applications of deformable hydrogel microrobots. Micromachines 2023, 14, 1824. [Google Scholar] [CrossRef]
- Thorne, J.B.; Vine, G.J.; Snowden, M.J. Microgel applications and commercial considerations. Collod. Polym. Sci. 2011, 289, 625–646. [Google Scholar] [CrossRef]
- Hussain, I.; Shahid, M.; Faisal Ali, F.; Irfan, A.; Farooqi, Z.H.; Begum, R. Methacrylic acid based microgels and hybrid microgels. Rev. Chem. Eng. 2023, 39, 1061–1083. [Google Scholar] [CrossRef]
- Begum, R.; Farooqi, Z.H.; Khan, S.R. Poly(N-isopropylacrylamide-acrylic acid) copolymer microgels for various applications: A review. Int. J. Polym. Mater. Polym. Biomater. 2016, 65, 841–852. [Google Scholar] [CrossRef]
- Scheffold, F. Pathways and challenges towards a complete characterization of microgels. Nat. Comm. 2020, 11, 4315. [Google Scholar] [CrossRef]
- Truong, N.P.; Jones, G.R.; Bradford, K.G.E.; Konkolewicz, D.; Anastasaki, A. A comparison of RAFT and ATRP methods for controlled radical polymerization. Nat. Rev. Chem. 2021, 5, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Grubbs, R.B. Nitroxide-mediated radical polymerization: Limitations and versatility. Polym. Rev. 2011, 51, 104–137. [Google Scholar] [CrossRef]
- Masci, G.; Bontempo, D.; Tiso, N.; Diociaiuti, M.; Mannina, L.; Capitani, D.; Crescenzi, V. Atom transfer radical polymerization of potassium 3-sulfopropyl methacrylate: direct synthesis of amphiphilic block copolymers with methyl methacrylate. Macromolecules 2004, 37, 4464–4473. [Google Scholar] [CrossRef]
- Tsarevsky, N.V.; Matyjaszewski, K. “Green” atom transfer radical polymerization: from process design to preparation of well-defined environmentally friendly polymeric materials. Chem. Rev. 2007, 107, 2270–2299. [Google Scholar] [CrossRef] [PubMed]
- Abu-Thabit, N.Y.; Azad, A.K.; Mezghani, K.; Hakeem, A.S.; Drmosh, Q.A.; Akhtar, S.; Adesina, A.Y. Facile and green fabrication of superhydrophobic polyacrylonitrile nonwoven fabric with iron hydroxide. ACS Appl. Polym. Mater. 2022, 4, 8450–8460. [Google Scholar] [CrossRef]
- Lv, C.-J.; Hao, B.; Yasin, A.; Yue, X.; Ma, P.-C. H2O2-assisted preparation of superhydrophilic polyacrylonitrile fabric and its application for the separation of oil/water mixture. Colloids Surf. A 2022, 646, 129004. [Google Scholar] [CrossRef]
- Zhou, Q.; Liu, Q.; Song, N.; Yang, J.; Ni, L. Amphiphilic reactive poly(glycidyl methacrylate)-block-poly(dimethyl siloxane)-block-poly(glycidyl methacrylate) triblock copolymer for the controlling nanodomain morphology of epoxy thermosets. Eur. Polym. J. 2019, 120, 109236. [Google Scholar] [CrossRef]
- Tigner, T.J.; Scull, G.; Brown, A.C.; Alge, D.L. Microparticle hydrogel material properties emerge from mixing-induced homogenization in a poly(ethylene glycol) and dextran aqueous two-phase system. Macromolecules 2023, 56, 8518–8528. [Google Scholar] [CrossRef]
- Lamch, Ł.; Gancarz, R.; Tsirigotis-Maniecka, M.; Moszyńska, I.M.; Ciejka, J.; Wilk, K.A. Studying the “rigid-flexible” properties of polymeric micelle core-forming segments with a hydrophobic phthalocyanine probe using NMR and UV spectroscopy. Langmuir 2021, 37, 4316–4330. [Google Scholar] [CrossRef] [PubMed]
- Matyjaszewski, K.; Jo, S.M.; Paik, H.-J.; Shipp, D.A. An investigation into the CuX/2,2′-Bipyridine (X = Br or Cl) mediated atom transfer radical polymerization of acrylonitrile. Macromolecules 1999, 32, 6431–6438. [Google Scholar] [CrossRef]
- Liu, R.C.W.; Pallier, A.; Brestaz, M.; Pantoustier, N.; Tribet, C. Impact of polymer microstructure on the self-assembly of amphiphilic polymers in aqueous solutions. Macromolecules 2007, 40, 4276–4286. [Google Scholar] [CrossRef]
- Julius, D.; Lee, J.Y.; Hong, L. Hydrophilic cross-linked aliphatic hydrocarbon diblock copolymer as proton exchange membrane for fuel cells. Materials 2021, 14, 1617. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Raghupathi, K.; Song, C.; Prasad, P.; Thayumanavan, S. Self-assembly of random copolymers. Chem. Commun. 2014, 50, 13417–13432. [Google Scholar] [CrossRef]
- Eom, Y.; Kim, B.C. Solubility parameter-based analysis of polyacrylonitrile solutions in N,N-dimethyl formamide and dimethyl sulfoxide. Polymer 2014, 55, 2570–2577. [Google Scholar] [CrossRef]
- Kaya, İ.; İlter, Z.; Şenol, D. Thermodynamic interactions and characterisation of poly[(glycidyl methacrylate-co-methyl, ethyl, butyl) methacrylate] by inverse gas chromatography. Polymer 2002, 43, 6455–6463. [Google Scholar] [CrossRef]
- Krys, P.; Matyjaszewski, K. Kinetics of atom transfer radical polymerization. Eur. Polym. J. 2017, 89, 482–523. [Google Scholar] [CrossRef]
- Chennam, P.K.; Kachlík, M.; Říhová, M.; Čičmancová, V.; Maca, K.; Macak, J.M. Synthesis of centrifugally spun polyacrylonitrile-carbon fibers. J. Mater. Res. Technol. 2024, 28, 2199–2205. [Google Scholar] [CrossRef]
- Shishlov, M.M.; Khursan, S.L. Effect of ion interactions on the IR spectrum of benzenesulfonate ion. Restoration of sulfonate ion symmetry in sodium benzenesulfonate dimer. J. Mol. Struct. 2016, 1123, 360–366. [Google Scholar] [CrossRef]
- Kishore Chand, A.A.; Bajer, B.; Schneider, E.S.; Mantel, T.; Ernst, M.; Filiz, V.; Glass, S. Modification of polyacrylonitrile ultrafiltration membranes to enhance the adsorption of cations and anions. Membranes 2022, 12, 580. [Google Scholar] [CrossRef] [PubMed]
- Nakatuka, Y.; Yoshida, H.; Fukui, K.; Matuzawa, M. The effect of particle size distribution on effective zeta-potential by use of the sedimentation method. Adv. Powder Technol. 2015, 26, 650–656. [Google Scholar] [CrossRef]
- Zhang, L.F.; Shen, H.; Eisenberg, A. Phase separation behavior and crew-cut micelle formation of polystyrene-b-poly(acrylic acid) copolymers in solutions. Macromolecules 1997, 30, 1001–1011. [Google Scholar] [CrossRef]
- Yu, Y.; Eisenberg, A. Control of morphology through polymer-solvent interactions in crew-cut aggregates of amphiphilic block copolymers. J. Am. Chem. Soc. 1997, 119, 8383–8384. [Google Scholar] [CrossRef]
- Vacha, M.; Habuchi, S. Conformation and physics of polymer chains: A single-molecule perspective. NPG Asia Mater. 2010, 2, 134–142. [Google Scholar] [CrossRef]
- Alexandridis, P.; Andersson, K. Reverse micelle formation and water solubilization by polyoxyalkylene block copolymers in organic solvent. Phys. Chem. B 1997, 101, 8103–8111. [Google Scholar] [CrossRef]
- Glatter, O.; Salentinig, S. Inverting structures: From micelles via emulsions to internally self-assembled water and oil continuous nanocarriers. Curr. Opin. Colloid Interface Sci. 2020, 49, 82–93. [Google Scholar] [CrossRef]
- Hashidzume, A.; Kawaguchi, A.; Tagawa, A.; Hyoda, K.; Sato, T. Synthesis and structural analysis of self-associating amphiphilic statistical copolymers in aqueous media. Macromolecules 2006, 39, 1135–1143. [Google Scholar] [CrossRef]
- Yukioka, S.; Kitadume, T.; Chatterjee, S.; Ning, G.; Ooya, T.; Yusa, S.-I. Amphiphilic block copolymers bearing hydrophobic γ-tocopherol groups with labile acetal bond. Polymers 2020, 12, 36. [Google Scholar] [CrossRef]
- Topel, Ö.; Çakır, B.A.; Budama, L.; Hoda, N. Determination of critical micelle concentration of polybutadiene-block-poly(ethyleneoxide) diblock copolymer by fluorescence spectroscopy and dynamic light scattering. J. Mol. Liq. 2017, 177, 40–43. [Google Scholar] [CrossRef]
- Pires, J.M.; Osmair, V.; de Oliveira, O.V.; Freitas, L.C.; da S Filho, E.A.; Prado, A.R. Molecular dynamic simulations of polyacrylonitrile in ethanol and water solvents. Compt. Theor. Chem. 2012, 995, 75–78. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Jo, S.M.; Paik, H.; Gaynor, S.G. Synthesis of well-defined polyacrylonitrile by atom transfer radical polymerization. Macromolecules 1997, 30, 6398–6400. [Google Scholar] [CrossRef]
- Yu, K.; Eisenberg, A. Multiple morphologies in aqueous solutions of aggregates of polystyrene-block-poly(ethylene oxide) diblock copolymers. Macromolecules 1996, 29, 6359–6361. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Copolymer | Concentration (mg/mL) | Dispersion Medium | SLI (Kcps) |
|---|---|---|---|
| (A50G4)S10 | 8.7 a | water | 2.7 |
| (A100G4)S10 | 8.7 | - | 5.8 |
| (A150G4)S10 | 8.7 | - | 3.3 |
| PEG54-P(AA/VE6/γTCP29)140 | 0.1 | water | 300 |
| PEO4000-b-PB1800 | 1.18 | water | 180 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Julius, D.; Lee, J.Y.; Hong, L. Microgel with a Core—Shell Particulate Structure Formed via Spinodal Decomposition of a Diblock Ionomer Containing a Doped Hydrophobic Moiety. Gels 2025, 11, 231. https://doi.org/10.3390/gels11040231
Julius D, Lee JY, Hong L. Microgel with a Core—Shell Particulate Structure Formed via Spinodal Decomposition of a Diblock Ionomer Containing a Doped Hydrophobic Moiety. Gels. 2025; 11(4):231. https://doi.org/10.3390/gels11040231
Chicago/Turabian StyleJulius, David, Jim Yang Lee, and Liang Hong. 2025. "Microgel with a Core—Shell Particulate Structure Formed via Spinodal Decomposition of a Diblock Ionomer Containing a Doped Hydrophobic Moiety" Gels 11, no. 4: 231. https://doi.org/10.3390/gels11040231
APA StyleJulius, D., Lee, J. Y., & Hong, L. (2025). Microgel with a Core—Shell Particulate Structure Formed via Spinodal Decomposition of a Diblock Ionomer Containing a Doped Hydrophobic Moiety. Gels, 11(4), 231. https://doi.org/10.3390/gels11040231