1. Introduction
Cancer therapy remains an enduring challenge due to the limitations of conventional treatment modalities, including surgery, radiotherapy, and chemotherapy [
1,
2,
3,
4,
5]. While these approaches have achieved notable clinical success, they are often associated with significant drawbacks, such as non-specific cytotoxicity, incomplete tumor ablation, and severe side effects that compromise the therapeutic outcomes. Therefore, there is a pressing need for the development of novel therapeutic strategies that combine precision, efficiency, and minimal invasiveness to enhance cancer treatment efficacy. Photothermal therapy (PTT) has emerged as a transformative approach to cancer treatment, offering distinct advantages over conventional methods [
6,
7,
8]. By using lasers of specific wavelengths to activate photothermal agents, PTT induces localized hyperthermia, effectively ablating tumor cells with minimal invasiveness [
9,
10]. This approach exhibits high specificity, controllability, and reduced collateral damage to surrounding healthy tissues. However, PTT faces challenges, such as limited penetration depth of laser light and uneven heat distribution, which constrain its efficacy, particularly in treating heterogeneous or deep-seated tumors [
11,
12].
To address these limitations, combined therapeutic strategies that combine PTT with chemotherapy have gained research interest [
13,
14]. This dual-modality approach leverages the localized thermal effects of PTT to enhance the sensitivity of tumor cells to chemotherapeutic agents while enabling temperature-controlled drug release. By integrating the precision of PTT with the systemic therapeutic effects of chemotherapy, these strategies can achieve enhanced tumor ablation at reduced drug dosages, minimizing systemic toxicity [
15,
16]. Therefore, the combined effects of PTT and chemotherapy hold promise for overcoming the intrinsic limitations of each modality, offering a robust and versatile framework for cancer therapy.
Hydrogels, as a class of soft materials with three-dimensional network structures, are gaining attention for their applications in drug delivery and tumor therapy [
17,
18]. With high water content and excellent biocompatibility, hydrogels can provide a favorable microenvironment for therapeutic interventions [
19,
20,
21]. Their tunable porous structures allow precise control over drug loading and release kinetics, while their responsiveness to external stimuli, such as temperature, pH, and light, further enhances their utility as multifunctional therapeutic platforms [
22,
23,
24]. Moreover, their capacity to encapsulate bioactive compounds and photothermal agents makes them ideal candidates for combined therapeutic strategies [
25,
26,
27]. Recent advancements have explored the incorporation of photothermal agents, such as PDA nanoparticles, into hydrogels to achieve combined therapeutic outcomes [
28]. These integrations not only enhance the photothermal conversion efficiency but also enable controlled and localized drug delivery, addressing key limitations in conventional treatment methods. Compared to conventional hydrogel matrices such as alginate or chitosan, γ-PGA offers superior water solubility, controlled biodegradability, and pH-responsive behavior [
29], making it highly suitable for tumor-targeted drug delivery. Its anionic nature also allows for efficient interaction with cationic therapeutic agents, enhancing drug retention and release profiles [
30,
31].
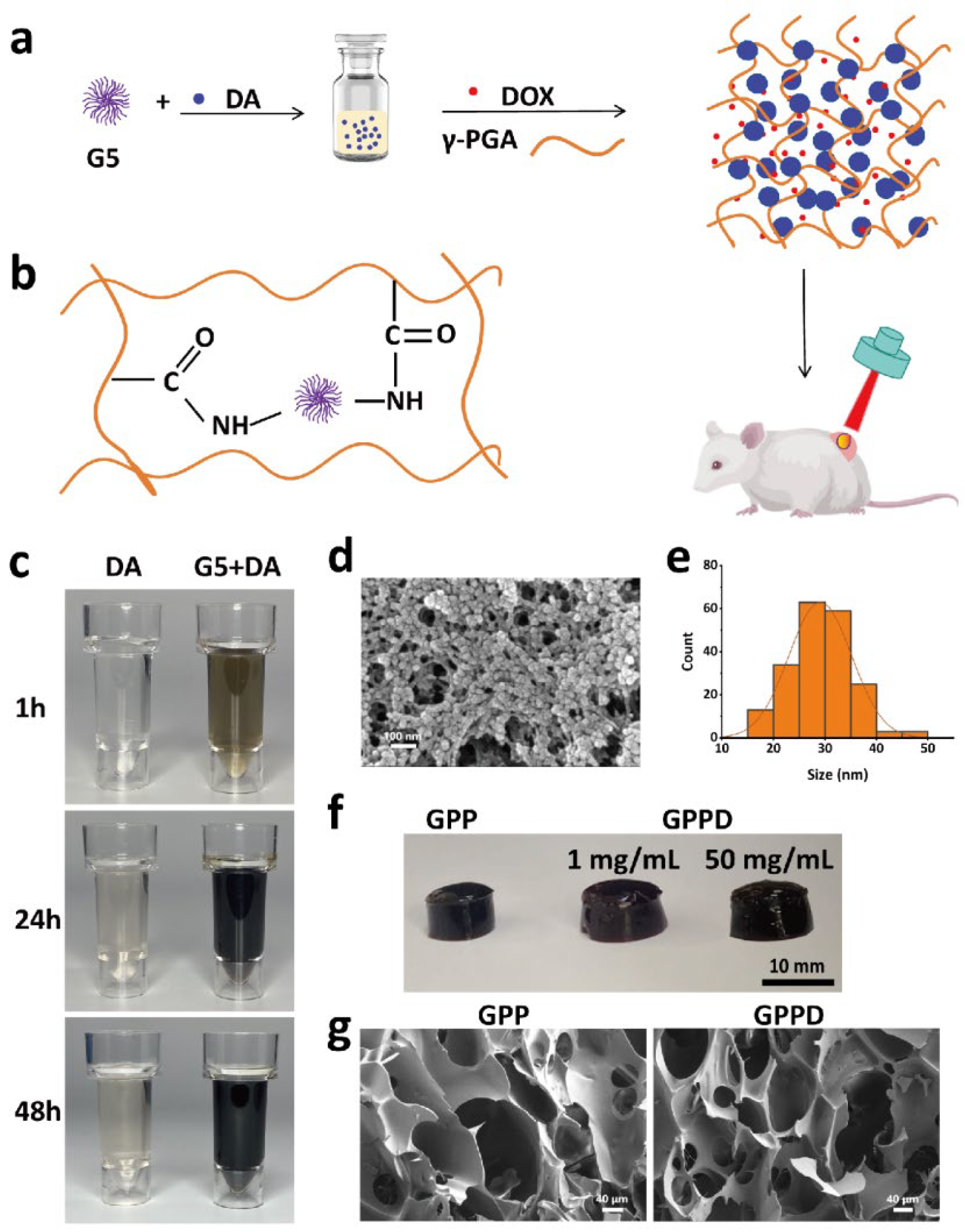
In this study, we developed a novel multifunctional hydrogel incorporating γ-polyglutamic acid (γ-PGA), fifth-generation dendrimers (G5), and PDA nanoparticles. This hydrogel system integrates photothermal conversion and controlled drug release, enabling combined PTT and chemotherapy. The hydrogel leverages the amino groups of G5 to facilitate the polymerization of dopamine into PDA nanoparticles, which are subsequently embedded in the hydrogel matrix through the chemical cross-linking between amino groups of G5 and carboxyl of γ-PGA. Before the hydrogel formation, DOX was first dissolved in the γ-PGA solution, enabling direct drug encapsulation. This unique structure allows precise control over the release kinetics of encapsulated drugs, while the PDA nanoparticles provide robust photothermal conversion under near-infrared (NIR) laser. The resulting system exhibits excellent photothermal responsiveness, controlled degradation, and sustained drug release capabilities, making it a versatile platform for cancer treatment. Systematic evaluations of the hydrogel demonstrate its biocompatibility, efficient photothermal conversion, and significant tumor cell ablation both in vitro and in vivo. In vitro studies revealed rapid temperature increases under laser irradiation, facilitating targeted drug release and tumor cell apoptosis. In vivo experiments further validated the hydrogel’s therapeutic efficacy, showcasing pronounced tumor suppression with minimal systemic toxicity. Additionally, the hydrogel’s biodegradability eliminates the need for secondary removal post-treatment, simplifying clinical applications and reducing patient risk. Our findings highlight the potential of this multifunctional hydrogel as a next-generation therapeutic platform, combining the precision of PTT with the systemic benefits of chemotherapy. Therefore, this study aims to develop a γ-PGA-based hydrogel system that integrates photothermal therapy (PTT) and chemotherapy for improved cancer treatment. The novelty of this system lies in its dual-responsive nature, where NIR irradiation not only induces localized hyperthermia but also enhances drug release in a controlled manner.
4. Materials and Methods
4.1. Materials
All chemicals were used without further purification. G5 was purchased from Weihai Chenyuan Molecular New Materials Co., Ltd. (Weihai, China). γ-PGA was purchased from Shanghai Yika Biotechnology Co., Ltd. (Shanghai, China). Dopamine hydrochloride, doxorubicin hydrochloride, EDC, and NHS were purchased from Shanghai Aladdin Reagent Co., Ltd. (Shanghai, China). Sodium chloride was purchased from Greagent. Phosphate buffer was purchased from Sigma-Aldrich (Shanghai, China). CBS was purchased from Shanghai Titan Technology Co., Ltd. (Shanghai, China). Mouse fibroblasts and CT26 were purchased from Wuhan Pricella Biotechnology Co., Ltd. (Wuhan, China). Modified Eagle medium (DMEM) was purchased from Corning Incorporated (Shanghai, China). CCK-8 and live/dead cell staining kit were purchased from Shanghai Weiao Biotechnology Co., Ltd. (Shanghai, China). KM mice and BALB/c-Nude nude mice were purchased from GemPharmatech Co., Ltd. (Nanjing, China). The Institutional Review Board of Changhai Hospital Affiliated with Naval Medical University (SYXK (Shanghai) 2020–0033) approved the animal study protocol.
4.2. Synthesis of PDA Nanoparticles
To synthesize PDA nanoparticles, 1 g of G5 dendrimer was dissolved in 5 mL of deionized water to prepare a 200 mg/mL solution. Separately, 0.1 g of dopamine hydrochloride was dissolved in 10 mL of deionized water. To initiate the polymerization, 100 μL of the G5 solution was added to the dopamine solution under continuous stirring at room temperature. The reaction mixture was allowed to stir for 24 h, leading to the formation of PDA nanoparticles. The resulting suspension was dialyzed against deionized water for 3 days to remove unreacted dopamine and byproducts. The purified PDA nanoparticles were freeze-dried to obtain a powder. The morphology of the nanoparticles was characterized using scanning electron microscopy (SEM, Zeiss Sigma 300, Carl Zeiss AG, Oberkochen, Germany).
4.3. Preparation of Hydrogel
Hydrogels were synthesized by cross-linking γ-PGA with G5 dendrimer in the presence of EDC and NHS. First, 0.1 g of dopamine hydrochloride was dissolved in 8.9 mL of deionized water, followed by the addition of 100 μL of G5 solution. After stirring for 24 h, the PDA nanoparticle suspension was dialyzed against deionized water for 3 days to remove unreacted dopamine and byproducts. Then, 1 g of γ-PGA was added and thoroughly mixed to prepare the precursor solution. Separately, 0.1 g each of EDC and NHS were dissolved in 1 mL of deionized water to create an activator solution. The activator solution was quickly added to the precursor solution under vigorous stirring for 30 s. The mixture was then left undisturbed for 5 min to form the γ-PGA/G5/PDA composite hydrogel (GPP) hydrogel. For drug-loaded hydrogels, DOX was added to the above γ-PGA solution with a final concentration of 1 mg/mL. The other steps were identical to those described above. The final drug-loaded hydrogel was designated as γ-PGA/G5/PDA composite (GPPD) hydrogel. The same method was used to prepare GPPD with a DOX loading concentration of 50 mg/mL. The hydrogel samples were subjected to freeze-drying using a laboratory-grade freeze dryer to remove residual water while preserving the structural integrity. The dried hydrogels were then analyzed using SEM to examine their microstructural morphology.
4.4. Rheological Study
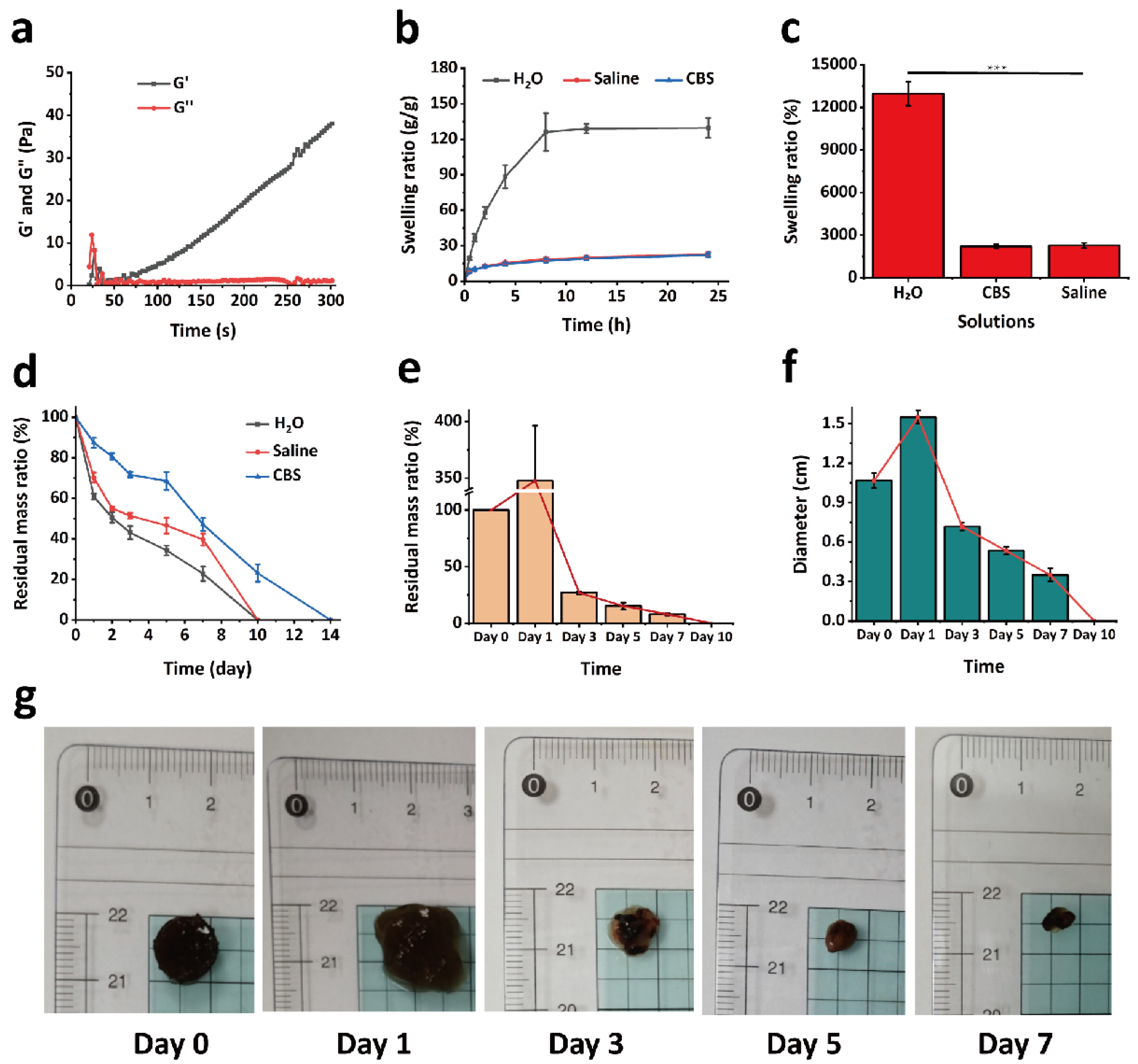
To investigate the dynamic changes of the hydrogel during the gelation process, a rotational rheometer (MARS III HAAKE, Thermo Fisher Scientific, Karlsruhe, Germany) was employed to evaluate its rheological properties. The experimental parameters were set as follows: temperature at 37 °C, a fixed oscillation frequency of 1 Hz, and a strain amplitude of 1%. A 200 μL aliquot of GPPD hydrogel precursor solution was placed onto a P20 TiL parallel plate rotor with a diameter of 20 mm and a gap of 1 mm, followed by the addition of 20 μL of activator solution. The storage modulus (G′) and loss modulus (G″) were recorded and analyzed using a time sweep test to monitor their evolution over the gelation period.
4.5. Swelling Behavior of Hydrogel
To evaluate the swelling performance of the hydrogel under different environmental conditions, experiments were conducted using saline to simulate the physiological environment and CBS buffer (pH = 5.4) to mimic the tumor microenvironment. Freeze-dried GPPD hydrogel samples of similar dimensions were initially weighed, and their dry weight was recorded as W0. Subsequently, the hydrogels were immersed in 20 mL of deionized water, saline, and CBS buffer (n = 3 for each condition) and incubated at 37 °C. At predetermined time intervals (0.5 h, 1 h, 2 h, 4 h, 8 h, 12 h, and 24 h), the hydrogels were removed, and the excess surface liquid was gently blotted using filter paper before weighing. The weight after water absorption was recorded as Wt. The swelling behavior was analyzed by calculating the swelling rate (%) of the hydrogel, using Equation (1). A swelling kinetic curve over 24 h was plotted to illustrate and compare the hydrogel’s swelling behavior and kinetics under different conditions.
4.6. In Vitro Photothermal Experiment
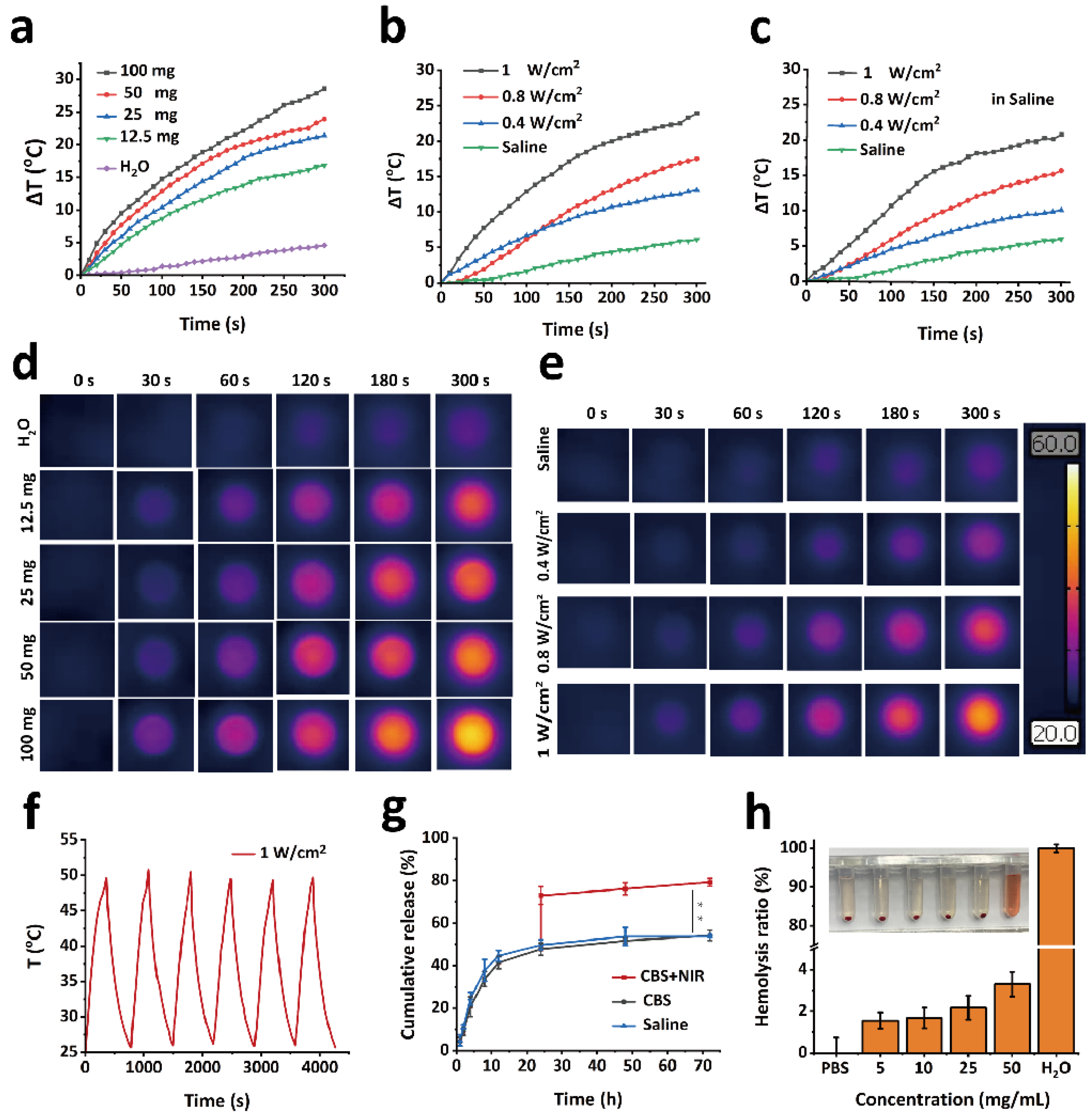
GPPD hydrogels of varying masses (12.5, 25, 50, and 100 mg) were placed in individual wells of a 96-well plate. Each hydrogel was irradiated with an 808 nm laser for 5 min at a power density of 1 W/cm2. Temperature changes during irradiation were recorded using an infrared thermal imaging camera. Deionized water (100 μL) was used as a blank control. To evaluate the impact of laser power, 50 mg of GPPD hydrogel was placed in a 96-well plate and irradiated with an 808 nm laser at power densities of 0.4, 0.8, and 1 W/cm2 for 5 min. The temperature changes were recorded using an infrared thermal imaging camera. For comparison, 50 μL of deionized water was used as a blank control. The photothermal behavior of the hydrogel in a saline environment was assessed by weighing 50 mg of GPPD hydrogel, placing it in a 96-well plate, and adding 100 μL of saline. The hydrogels were irradiated with an 808 nm laser at power densities of 0.4, 0.8, and 1 W/cm2 for 5 min. Temperature changes were monitored using an infrared thermal imaging camera. To evaluate the photothermal stability of the GPPD hydrogel, 50 mg of hydrogel was irradiated with an 808 nm laser at a power density of 1 W/cm2 for 5 min. The hydrogel was then allowed to cool naturally to room temperature. This heating–cooling cycle was repeated for six consecutive cycles, and temperature changes during each irradiation process were recorded using an infrared thermal imaging camera.
4.7. Drug Release
A specified amount of GPDD hydrogel was placed in a dialysis bag with a molecular weight cutoff of 3500 Da and immersed in 20 mL of saline or CBS buffer (pH 5.4) (n = 3 for each condition). The dialysis bags were incubated in a shaker at 37 °C with an agitation speed of 120 rpm. At predefined time intervals (0.5 h, 1 h, 2 h, 4 h, 8 h, 12 h, 24 h, 48 h, and 72 h), 1 mL of the supernatant was collected, and an equal volume of fresh corresponding buffer was added to maintain the total volume. The absorbance of the collected supernatant was measured at 480 nm using a UV-visible spectrophotometer (Shimadzu UV-3600, Shimadzu Corporation, Kyoto, Japan), and the cumulative release efficiency of DOX was calculated based on a pre-established standard calibration curve. To assess the influence of photothermal conditions on DOX release, additional samples from the CBS buffer group (n = 3) were subjected to photothermal stimulation. After 24 h of incubation under the same conditions, the samples were exposed to 50 °C water. The release efficiency of DOX was subsequently determined.
4.8. In Vitro Degradation of Hydrogel
The in vitro degradation performance of the GPPD hydrogel was evaluated by measuring the residual mass ratio over time under different conditions. Freeze-dried hydrogel samples were prepared, and their initial weight was recorded as W0. The samples were fully immersed in 10 mL of deionized water, saline, or CBS buffer (pH 5.4) and incubated at 37 °C in a constant temperature environment (n = 3 for each condition). At predetermined time points (1 day, 2 days, 3 days, 5 days, 7 days, 10 days, and 14 days), the hydrogels were retrieved from the solution and gently rinsed three times with deionized water to remove surface residues. The samples were then freeze-dried and weighed as Wt. The degradation behavior was analyzed by calculating the residual mass ratio (%) of the hydrogel using Equation (2):
4.9. In Vitro Blood Compatibility
The hemolytic performance of the GPPD hydrogel was evaluated using Kunming mouse whole blood. A total of 2 mL of whole blood was collected and centrifuged at 5000 rpm for 5 min to remove the plasma. The RBCs were washed three times with PBS until the residual plasma was completely removed. The washed RBCs were resuspended in 50 mL of PBS for further use. The experimental groups consisted of 7.5 mg, 15 mg, 37.5 mg, and 75 mg of GPPD hydrogel (n = 3 for each concentration) mixed with 1.5 mL of PBS, followed by the addition of 0.3 mL of the RBCs suspension. The negative control group consisted of 1.5 mL PBS and 0.3 mL of the RBCs suspension, while the positive control group contained 1.5 mL of deionized water and 0.3 mL of the RBCs suspension. All samples were incubated in a 37 °C incubator for 2 h. After incubation, all samples were centrifuged at 5000 rpm for 5 min, and the supernatant was carefully collected. The absorbance of the supernatant at 541 nm was measured using a UV-visible spectrophotometer. The hemolysis ratio (%) was calculated using Equation (3):
Ds is the absorbance of the supernatant of the experimental group, Dn is the absorbance of the negative control group, and Dp is the absorbance of the positive control group.
4.10. In Vitro Cytocompatibility
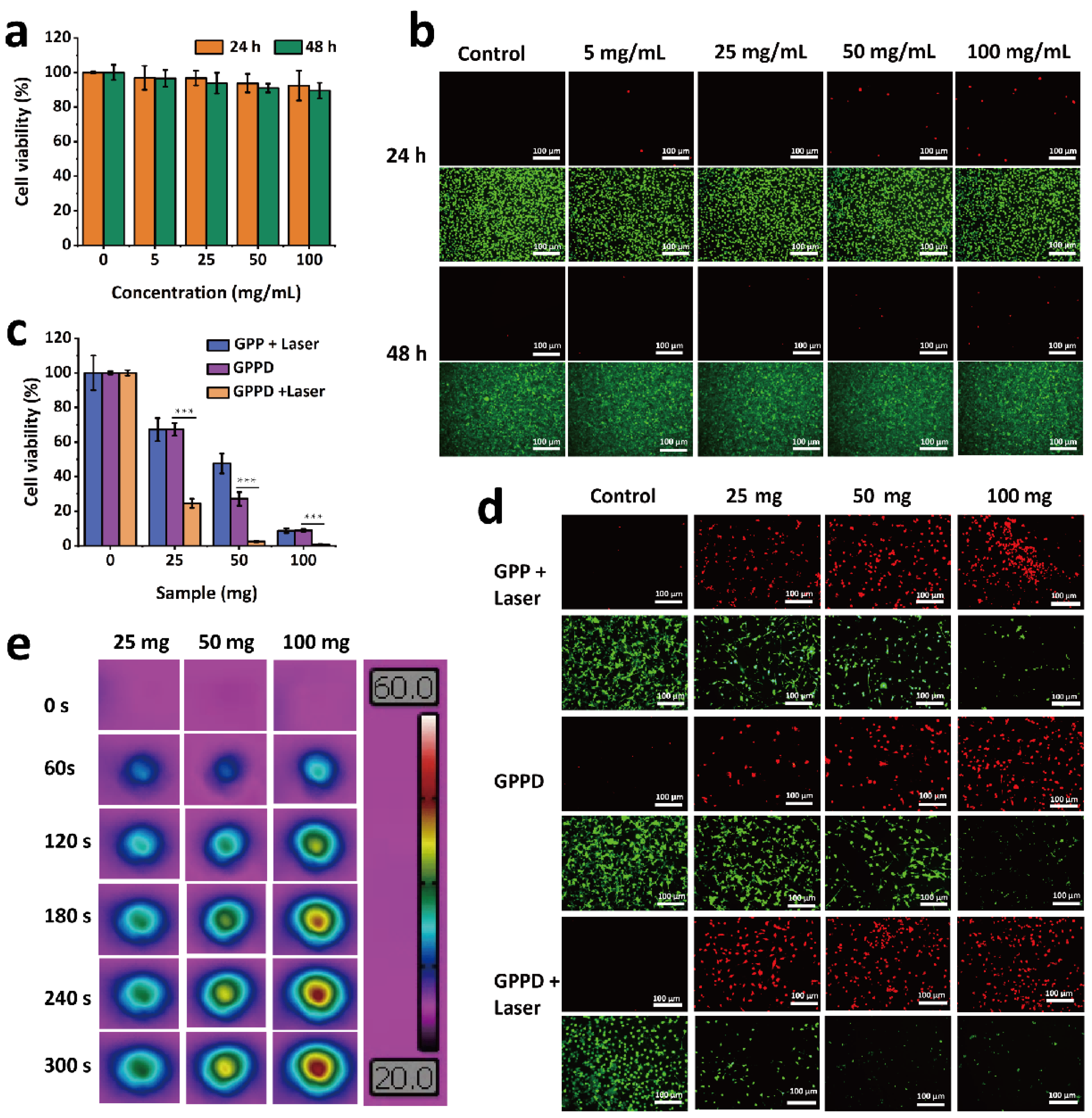
The cytocompatibility of the GPP hydrogel was assessed using the CCK-8 assay and live/dead cell staining. Mouse fibroblast cells were seeded into 96-well plates at a density of 1 × 104 cells per well and incubated at 37 °C in a humidified atmosphere containing 5% CO2 for 12 h to allow for cell adhesion. The GPP hydrogel was sterilized, and hydrogel extracts were prepared using DMEM at the GPP hydrogel concentrations of 0, 5, 25, 50, and 100 mg/mL. After removing the supernatant from each well, 100 μL of hydrogel extract at the corresponding concentration was added (n = 3 per group). DMEM alone was used as the blank control. The cells were cultured for 24 and 48 h. Following the incubation, the cells were washed three times with PBS to remove residual hydrogel extract. Cell viability was assessed using the CCK-8 assay kit (Dojindo Laboratories, Kumamoto, Japan), with absorbance measured at 450 nm. The relative cell viability under different hydrogel concentrations and incubation times was calculated to evaluate the cytotoxicity of the GPP hydrogel. Subsequently, the cells were stained using Calcein-AM (green fluorescence for live cells) and propidium iodide (PI, red fluorescence for dead cells) following the manufacturer’s instructions. The stained cells were observed and imaged using an inverted phase contrast fluorescence microscope (MARS III HAAKE) to evaluate the hydrogel’s cytotoxicity.
4.11. In Vitro Tumor Treatment Experiments
To evaluate the therapeutic effect of GPP and GPPD hydrogels on tumor cells, mouse CT26 cells were inoculated into 96-well plates with a cell density of 1 × 104 cells per well, incubated at 37 °C, 5% CO2 environment for 12 h, and discarded the supernatant. After sterilization, GPP and GPPD hydrogels were directly added into 96-well plates with hydrogels of 0, 25, 50, and 100 mg per well (n = 3). Then, an 808 nm laser was used to illuminate the perforating plates for 5 min with a laser power of 1 W/cm2. After irradiation, the culture continued for 24 h. A set of GPPD hydrogels (0, 25, 50, and 100 mg (n = 3)) were obtained and incubated directly with cells in 96-well plates for 24 h. The hydrogel extract was discarded, the cells were washed with PBS three times, the cell survival rate was calculated using CCK-8, and the cells were stained by live/dead staining. The appearance of live and dead cells was observed and photographed using an inverted phase contrast microscope, and the effect of GPP and GPPD hydrogels on tumor cells in vitro was evaluated.
4.12. In Vivo Degradation
To investigate the in vivo degradation of GPP hydrogel, a degradation study was conducted in KM mice. A total of 200 mg of GPP hydrogel was subcutaneously implanted into the dorsal region of the mice (n = 3). At predetermined time points (1, 3, 5, 7, and 10 days), the hydrogel samples were carefully retrieved, cleaned, and weighed. The percentage of mass loss over time was calculated to evaluate the in vivo degradation profile of the hydrogel.
4.13. In Vivo Biosafety
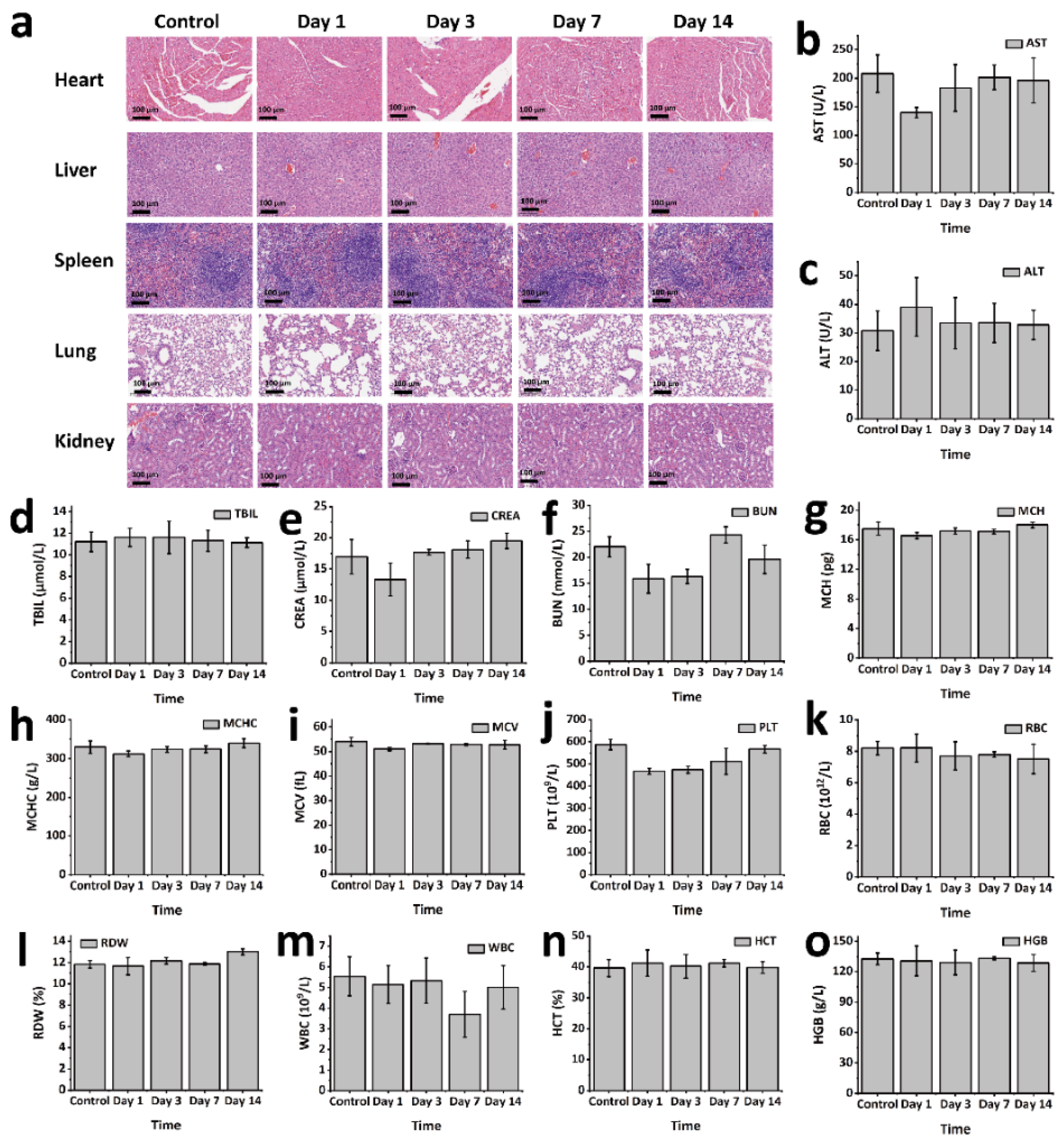
To evaluate the in vivo biosafety of GPP hydrogel, an animal model using KM mice was employed. GPP hydrogel blocks (0.2 g) were subcutaneously implanted into the dorsal region of the mice (n = 3). Mice were sacrificed at predetermined time points (1, 3, 7, and 14 days post-implantation). The main organs were harvested for histological analysis through hematoxylin and eosin (H&E) staining. Tissue sections were observed and imaged using an inverted phase contrast microscope to assess any histopathological changes induced by the hydrogel. Additionally, blood samples were collected for both serum biochemistry and routine blood analysis. Serum biochemical parameters were measured using the Beckman Coulter Unicel DxC 800 fully automated biochemical analyzer (Beckman Coulter, Inc., Brea, CA, USA). Routine blood tests were conducted using the Sysmex XS-800i fully automated blood analyzer (Sysmex Corporation, Kobe, Japan).
4.14. In Vivo Tumor Treatment Experiments
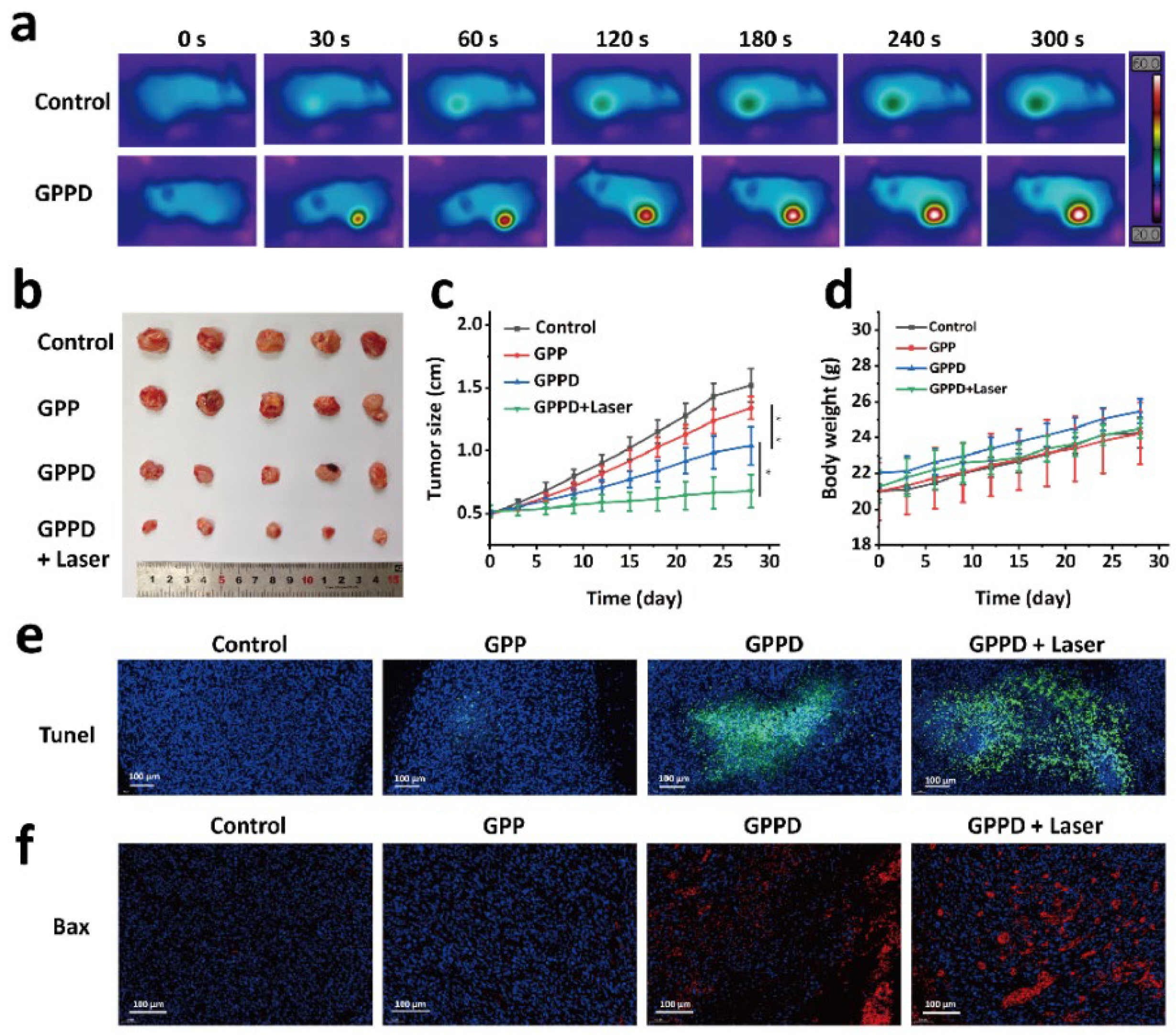
CT26 tumor cells were subcutaneously inoculated into the flanks of nude mice. When the tumor volume reached approximately 0.5 cm in diameter, the mice were randomly assigned to four treatment groups, with six mice per group. The mice were anesthetized before receiving the following treatments: Group 1 was injected with 200 mg of GPP hydrogel; Group 2 was injected with 200 mg of GPPD hydrogel; Group 3 received 200 mg of GPPD hydrogel and underwent laser irradiation (808 nm NIR laser, 1 W/cm2, 5 min); and Group 4 was injected with 200 μL of saline (control group). Throughout the experiment, the initial body weight and tumor volume of each mouse were recorded, and images were taken for visual tracking. Tumor growth was monitored at time points 0, 3, 6, 9, 12, 15, 18, 21, 24, and 28 days. Key evaluation parameters included relative tumor volume (V/V0, where V0 represents the tumor volume at day 0), changes in body weight, and the visual appearance of the tumors. After the study, tumors were excised and subjected to TUNEL staining and Bax protein immunohistochemistry to assess the impact of the various treatments on tumor cell apoptosis.
4.15. Statistical Analysis
All data were represented as mean ± standard deviation (SD). One-way ANOVA was used to evaluate comparisons between several groups. A p-value below 0.05 was deemed statistically significant (* p < 0.05, ** p < 0.01, *** p < 0.001).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}