
New Supramolecular Hydrogels Based on Diastereomeric Dehydrotripeptide Mixtures for Potential Drug Delivery Applications

 ,
,  ,
,  ,
,  , , , and
, , , and
Abstract

1. Introduction
2. Results and Discussion
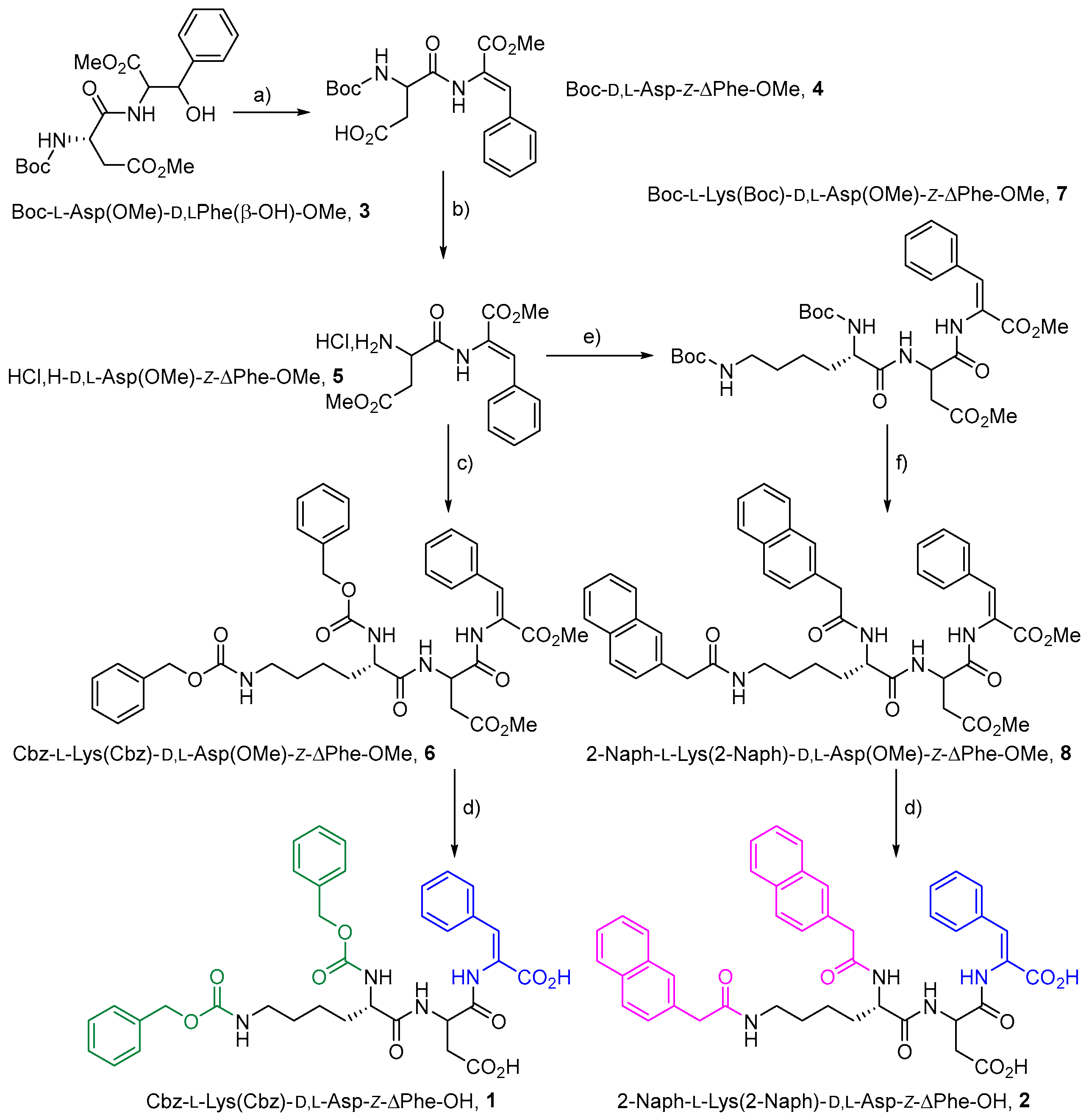
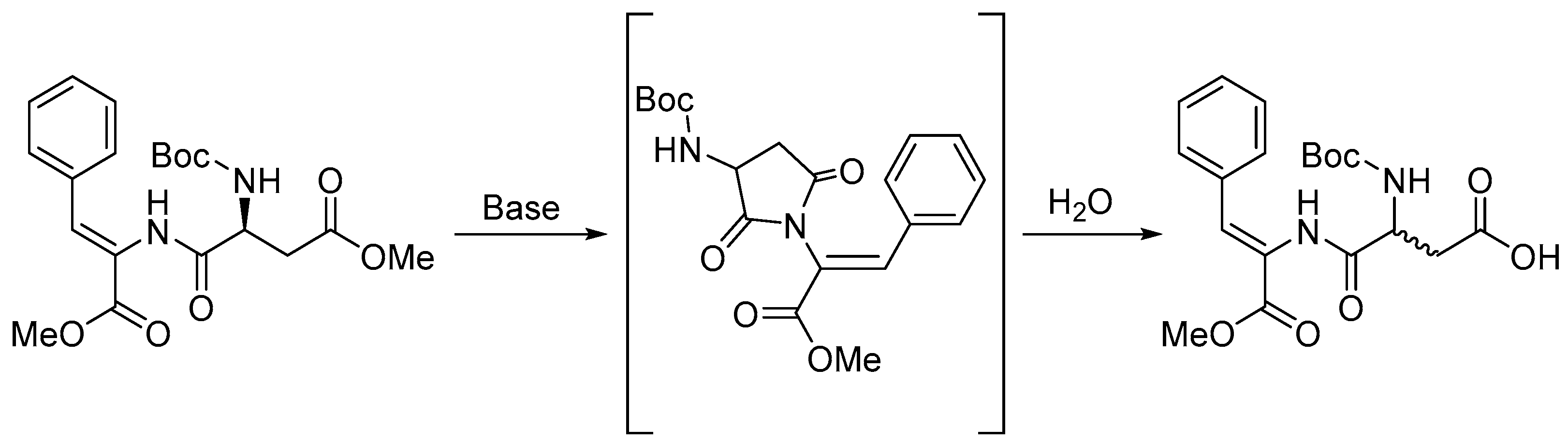
2.1. Synthesis and Gelation Studies
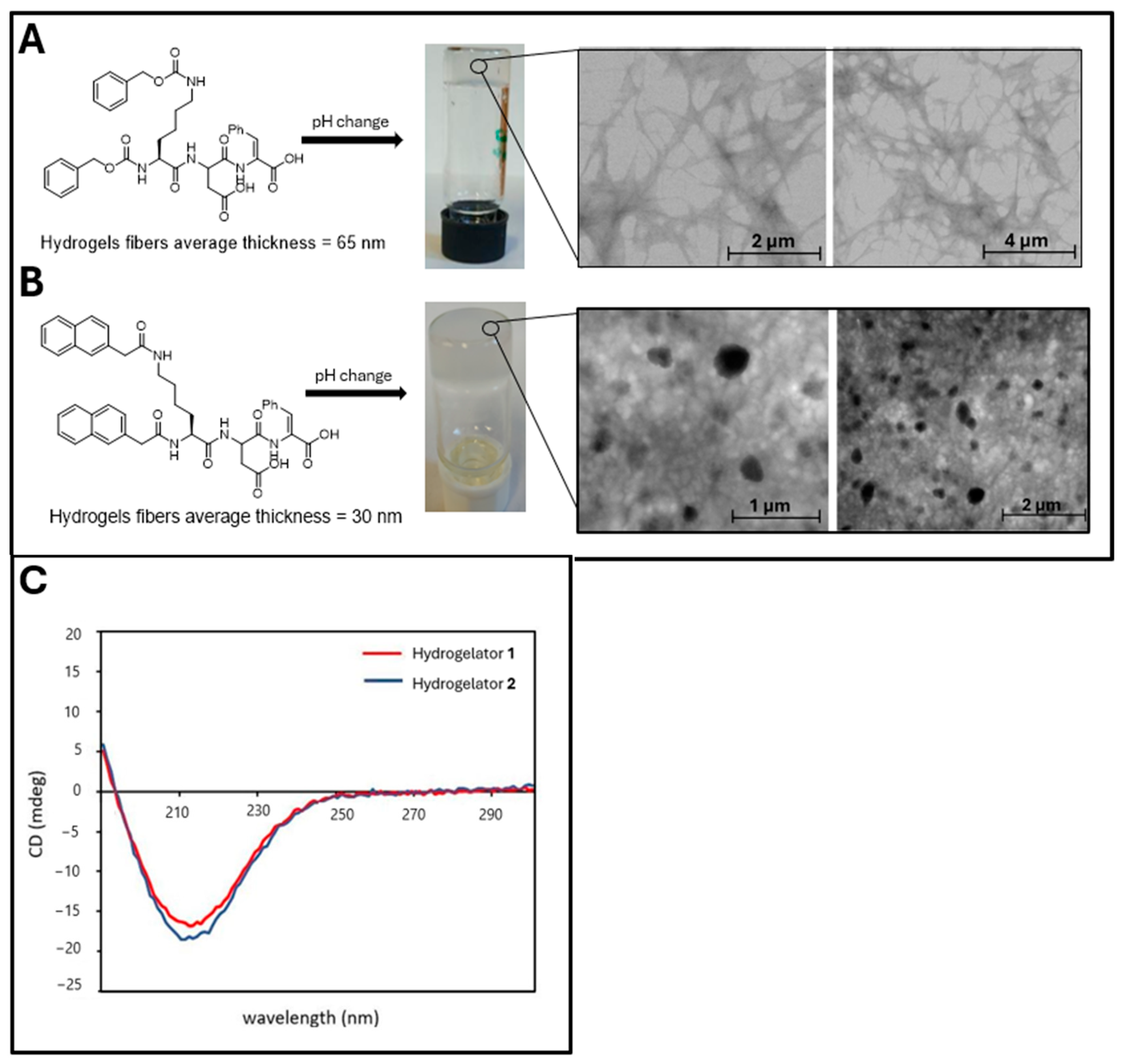
2.2. Scanning Transmission Electron Microscopy (STEM)
2.3. Circular Dichroism (CD) Studies
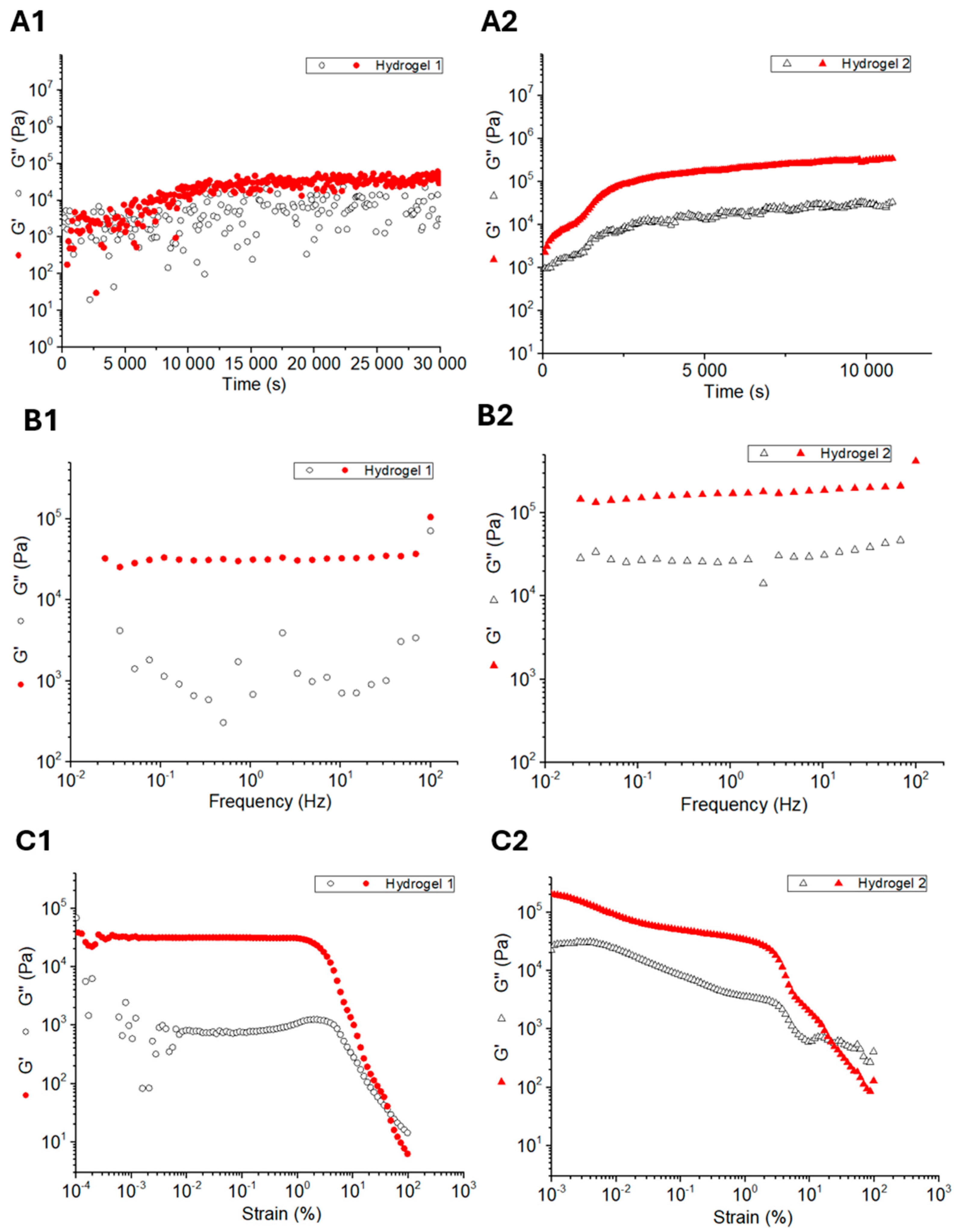
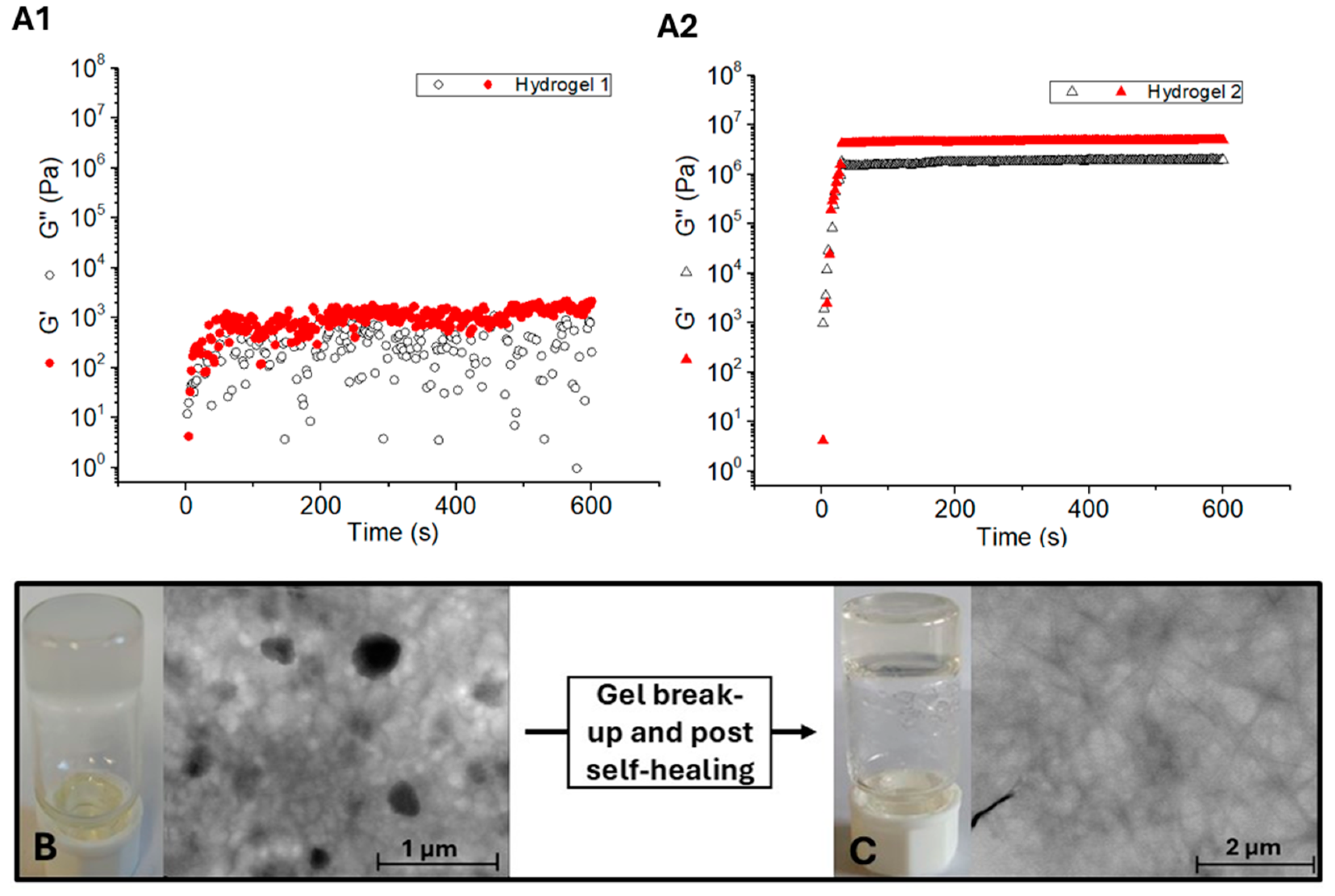
2.4. Rheological Studies
2.5. Biocompatibility and Cytotoxicity Studies
2.6. Drug Delivery Studies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hydrogelator | k | n | R2 |
|---|---|---|---|
| 1 | 6.1203 | 0.4459 | 0.9892 |
| 2 | 9.2804 | 0.4147 | 0.9713 |
3. Conclusions
4. Materials and Methods
4.1. Synthesis
4.1.1. Synthesis of Cbz-L-Lys(Cbz)-D,L-Asp(OH)-Z-ΔPhe-OH, 1
4.1.2. Synthesis of Naph-L-Lys(Naph)-D,L-Asp(OH)-Z-ΔPhe-OH, 2
4.2. Preparation of Hydrogels and Determination of the Critical Gelation Concentration (cgc)
4.3. Scanning Transmission Electron Microscopy (STEM)
4.4. Circular Dichroism
4.5. Rheological Studies
4.6. Sustained Release Assays
4.7. Cell Culture
4.8. MTT Assay
4.9. LDH Leakage
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yadav, N.; Chauhan, M.K.; Chauhan, V.S. Short to ultrashort peptide-based hydrogels as a platform for biomedical applications. Biomater. Sci. 2020, 8, 84–100. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, N.N.; Ferreira, L.M.B.; Cardoso, V.M.O.; Boni, F.I.; Souza, A.L.R.; Gremião, M.P.D. Recent advances in smart hydrogels for biomedical applications: From self-assembly to functional approaches. Eur. Polym. J. 2018, 99, 117–133. [Google Scholar] [CrossRef]
- Revete, A.; Aparicio, A.; Cisterna, B.A.; Revete, J.; Luis, L.; Ibarra, E.; Gonzalez, E.A.S.; Molino, J.; Reginensi, D. Advancements in the Use of Hydrogels for Regenerative Medicine: Properties and Biomedical Applications. Int. J. Biomater. 2022, 2022, 3606765. [Google Scholar] [CrossRef] [PubMed]
- Fleming, D.; Ulijn, R.V. Design of nanostructures based on aromatic peptide amphiphiles. Chem. Soc. Rev. 2014, 43, 8150–8177. [Google Scholar] [CrossRef] [PubMed]
- Florit, M.G.; Pardo, A.; Domingues, R.M.A.; Graça, A.L.; Babo, P.S.; Reis, R.L.; Gomes, M.E. Natural-Based Hydrogels for Tissue Engineering Applications. Molecules 2020, 25, 5858. [Google Scholar] [CrossRef]
- Rana, M.M.; Siegler, H.D.L.H. Evolution of Hybrid Hydrogels: Next-Generation Biomaterials for Drug Delivery and Tissue Engineering. Gels 2024, 10, 216. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Hu, Y.; Wang, S.; Chen, X.; Jiang, Y.; Su, J. Fabrication of physical and chemical crosslinked hydrogels for bone tissue engineering. Bioact. Mater. 2022, 12, 327–339. [Google Scholar] [CrossRef]
- Thang, N.H.; Chien, T.B.; Cuong, D.X. Polymer-Based Hydrogels Applied in Drug Delivery: An Overview. Gels 2023, 9, 523. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Yang, D.; Xu, B. Emerging Applications of Supramolecular Peptide Assemblies. Trends. Chem. 2020, 2, 71–83. [Google Scholar] [CrossRef]
- Wisniewska, J.S.; Flor, S.; Kozlowska, J. From Supramolecular Hydrogels to Multifunctional Carriers for Biologically Active Substances. Int. J. Mol. Sci. 2021, 22, 7402. [Google Scholar] [CrossRef]
- Dou, X.; Mehwish, N.; Zhao, C.; Liu, J.; Xing, C.; Feng, C. Supramolecular Hydrogels with Tunable Chirality for Promising Biomedical Applications. Acc. Chem. Res. 2020, 53, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhu, X.; Zhao, J.; Ling, G.; Zhang, P. Biomedical applications of supramolecular hydrogels with enhanced mechanical properties. Adv. Colloid Interface Sci. 2023, 321, 103000. [Google Scholar] [CrossRef] [PubMed]
- Raeburn, J.; Cardoso, A.Z.; Adams, D.J. The importance of the self-assembly process to control mechanical properties of low molecular weight hydrogels. Chem. Soc. Rev. 2013, 42, 5143–5156. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.J. Dipeptide and Tripeptide Conjugates as Low-Molecular-Weight Hydrogelators. Macromol. Biosci. 2010, 11, 160–173. [Google Scholar] [CrossRef]
- Adams, D.J.; Butler, M.F.; Frith, W.J.; Kirkland, M.; Mullen, L.; Sanderson, P. A new method for maintaining homogeneity during liquid-hydrogel transitions using low molecular weight hydrogelators. J. Soft Matter 2009, 5, 1856–1862. [Google Scholar] [CrossRef]
- Zhou, J.; Du, X.; Wang, J.; Yamagata, N.; Xu, B. Enzyme-instructed self-assembly of peptides containing phosphoserine to form supramolecular hydrogels as potential soft materials. Front. Chem. Sci. Eng. 2017, 11, 509–515. [Google Scholar] [CrossRef]
- Raeburn, J.; Pont, G.; Chen, L.; Cesbron, Y.; Lévy, R.; Adams, D.J. Fmoc-diphenylalanine hydrogels: Understanding the variability in reported mechanical properties. J. Soft Matter 2012, 8, 1168–1174. [Google Scholar] [CrossRef] [PubMed]
- Haines, L.A.; Rajagopal, K.; Ozbas, B.; Salick, D.A.; Pochan, D.J.; Schneider, J.P. Light-Activated Hydrogel Formation via the Triggered Folding and Self-Assembly of a Designated Peptide. J. Am. Chem. Soc. 2005, 127, 17025–17029. [Google Scholar] [CrossRef] [PubMed]
- Okesola, B.O.; Wu, Y.; Derkus, B.; Gani, S.; Wu, D.; Knani, D.; Smith, D.K.; Adams, D.J.; Mata, A. Supramolecular Self-Assembly To Control Structural and Biological Properties of Multicomponent Hydrogels. Chem. Mater. 2019, 31, 7883–7897. [Google Scholar] [CrossRef]
- Yuan, Y.; Shi, Y.; Banerjee, J.; Sadeghpour, A.; Azevedo, H.S. Structuring supramolecular hyaluronan hydrogels via peptide self-assembly for modulating the cell microenvironment. Mater. Today Commun. 2023, 19, 100598. [Google Scholar] [CrossRef]
- Du, X.; Zhou, J.; Shi, J.; Xu, B. Supramolecular Hydrogelators and Hydrogels: From Soft Matter to Molecular Biomaterials. Chem. Rev. 2015, 115, 13165–13307. [Google Scholar] [CrossRef]
- Fichman, G.; Gazit, E. Self-assembly of short peptides to form hydrogels: Design of building blocks, physical properties and technological applications. Acta Biomater. 2014, 10, 1671–1682. [Google Scholar] [CrossRef] [PubMed]
- Jervis, P.J.; Amorim, C.; Pereira, T.; Martins, J.A.; Ferreira, P.M.T. Dehydropeptide Supramolecular Hydrogels and Nanostructures as Potential Peptidomimetic Biomedical Materials. Int. J. Mol. Sci. 2021, 22, 2528. [Google Scholar] [CrossRef]
- Amorim, C.; Veloso, S.R.S.; Castanheira, E.M.S.; Hilliou, L.; Pereira, R.B.; Pereira, D.M.; Martins, J.A.; Jervis, P.J.; Ferreira, P.M.T. Bolaamphiphilic Bis-Dehydropeptide Hydrogels as Potential Drug Release Systems. Gels 2021, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Keasling, J.D.; Muller, S.J. Effects of the sequence and size of non-polar residues on self-assembly of amphiphilic peptides. Int. J. Biol. Macromol. 2005, 36, 232–240. [Google Scholar] [CrossRef]
- Tian, Y.; Wang, H.; Liu, Y.; Mao, L.; Chen, W.; Zhu, Z.; Liu, W.; Zheng, W.; Zhao, Y.; Kong, D.; et al. A Peptide-Based Nanofibrous Hydrogel as a Promising DNA Nanovector for Optimizing the Efficacy of HIV Vaccine. Nano Lett. 2014, 14, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Tang, Y.; Yamamoto, T.; Yao, Y.; Guterman, T.; Zilberzwige-Tal, S.; Adadi, N.; Ji, W.; Dvir, T.; Ramamoorthy, A.; et al. Unusual Two-Step Assembly of a Minimalistic Dipeptide-Based Functional Hypergelator. Adv. Mater. 2020, 32, 9. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.B.P.; Pereira, R.B.; Pereira, D.M.; Hilliou, L.; Castro, T.G.; Martins, J.A.; Jervis, P.J.; Ferreira, P.M.T. Aryl-capped Lysine-Dehydroamino Acid Dipeptide Supergelators as Potential Drug Release Systems. Int. J. Mol. Sci. 2022, 23, 11811. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.B.P. Development of New Supramolecular Hypergelators Based on Non-Natural Amino Acid Residues. Master’s Thesis, University of Minho, Braga, Portugal, 2022. Available online: https://hdl.handle.net/1822/82034 (accessed on 30 August 2024).
- Carny, O.; Gazit, E. Creating Prebiotic Sanctuary: Self-Assembling Supramolecular Peptide Structures Bind and Stabilize RNA. Orig. Life Evol. B. 2010, 41, 121–132. [Google Scholar] [CrossRef]
- Thornalley, K.A.; Laurini, E.; Pricl, S.; Smith, D.K. Enantiomeric and Diastereomeric Self-Assembled Multivalent Nanostructures: Understanding the Effects of Chirality on Binding to Polyanionic Heparin and DNA. Angew. Chem. Int. Ed. 2018, 57, 8530–8534. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q.; McAulay, K.; Xu, T.; Rogers, S.E.; Gayle, C.E.; Schweins, R.; Cui, H.; Seddon, A.M.; Adams, D.J. Self-Sorting in Diastereomeric Mixtures of Functionalized Dipeptides. Biomacromolecules 2023, 24, 2847–2855. [Google Scholar] [CrossRef]
- Ferreira, P.M.T.; Monteiro, L.S.; Pereira, G.; Ribeiro, L.; Sacramento, J.; Silva, L. Reactivity of Dehydroamino Acids and Dehydrodipeptides Towards N-Bromosuccinimide: Synthesis of β-Bromo- and β,β-Dibromodehydroamino Acid Derivatives and of Subtsituted 4-Imidazolidinones. Eur. J. Org. Chem. 2007, 35, 5934–5949. [Google Scholar] [CrossRef]
- Gisin, B.F.; Merrifield, R.B. Carboxyl-catalyzed intramolecular aminolysis. Side Reaction in solid-phase peptide synthesis. J. Am. Chem. Soc. 1972, 94, 3102–3106. [Google Scholar] [CrossRef]
- Neumann, K.; Farnung, J.; Baldauf, S.; Bode, J.W. Prevention of aspartimide formation during peptide synthesis using cyanosulfurylides as carbocilic acid protecting groups. Nat. Commun. 2020, 11, 982. [Google Scholar] [CrossRef] [PubMed]
- Abraham, B.L.; Toriki, E.S.; Tucker, N.J.; Nilsson, B.L. Eletrostatic interactions regulate the release of small molecules from supramolecular hydrogels. J. Mater. Chem. B 2020, 8, 6366–6377. [Google Scholar] [CrossRef]
- Wu, I.Y.; Bala, S.; Basnet, N.S.; Cagno, M.P. Interpreting non-linear drug diffusion data: Utilizing Korsmeyer-Peppas model to study drug release from liposomes. Eur. J. Pharm. Sci. 2019, 138, 105026. [Google Scholar] [CrossRef]
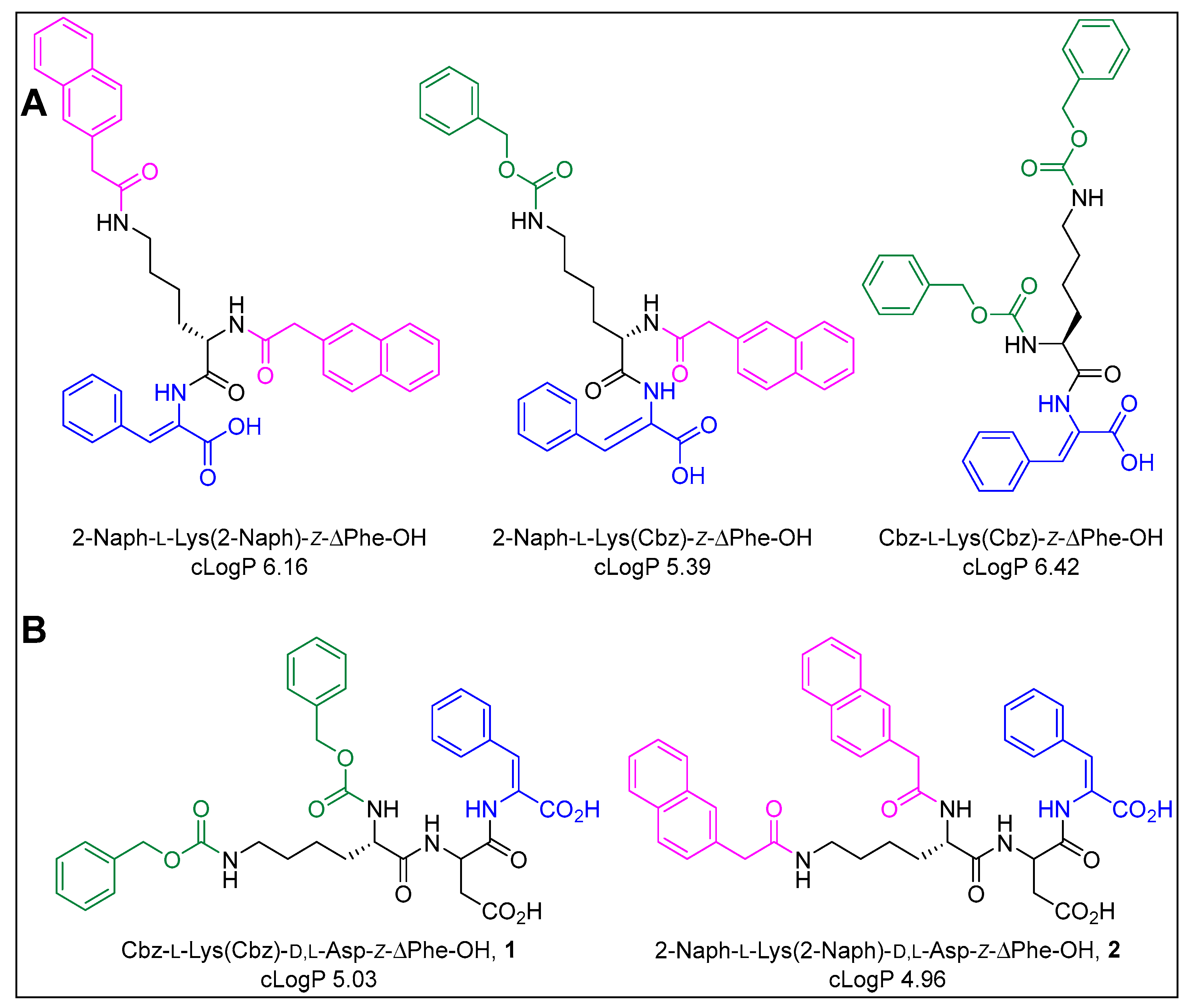


| Hydrogelator | Critical Gelation Concentration (CGC) a | pH | |
|---|---|---|---|
| wt% | mM | ||
| Cbz-L-Lys(Cbz)-Z-ΔPhe-OH b | 0.2 | 3.57 | 5.0 |
| 2-Naph-L-Lys(2-Naph)-Z-ΔPhe-OH b | 0.05 | 0.80 | 4.8 |
| Cbz-L-Lys(Cbz)-D,L-Asp-Z-ΔPhe-OH, 1 c | 0.1 | 0.002 | 5.2 |
| 2-Naph-L-Lys(2-Naph)-D,L-Asp-Z-ΔPhe-OH, 2 c | 0.04 | 0.0006 | 4.8 |
| Hydrogel | G’ (Pa) | G’’ (Pa) |
|---|---|---|
| 1 | 5.44 × 104 | 7.94 × 103 |
| 2 | 3.43 × 106 | 2.49 × 104 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliveira, C.B.P.; Carvalho, A.; Pereira, R.B.; Pereira, D.M.; Hilliou, L.; Jervis, P.J.; Martins, J.A.; Ferreira, P.M.T. New Supramolecular Hydrogels Based on Diastereomeric Dehydrotripeptide Mixtures for Potential Drug Delivery Applications. Gels 2024, 10, 629. https://doi.org/10.3390/gels10100629
Oliveira CBP, Carvalho A, Pereira RB, Pereira DM, Hilliou L, Jervis PJ, Martins JA, Ferreira PMT. New Supramolecular Hydrogels Based on Diastereomeric Dehydrotripeptide Mixtures for Potential Drug Delivery Applications. Gels. 2024; 10(10):629. https://doi.org/10.3390/gels10100629
Chicago/Turabian StyleOliveira, Carlos B. P., André Carvalho, Renato B. Pereira, David M. Pereira, Loic Hilliou, Peter J. Jervis, José A. Martins, and Paula M. T. Ferreira. 2024. "New Supramolecular Hydrogels Based on Diastereomeric Dehydrotripeptide Mixtures for Potential Drug Delivery Applications" Gels 10, no. 10: 629. https://doi.org/10.3390/gels10100629
APA StyleOliveira, C. B. P., Carvalho, A., Pereira, R. B., Pereira, D. M., Hilliou, L., Jervis, P. J., Martins, J. A., & Ferreira, P. M. T. (2024). New Supramolecular Hydrogels Based on Diastereomeric Dehydrotripeptide Mixtures for Potential Drug Delivery Applications. Gels, 10(10), 629. https://doi.org/10.3390/gels10100629