
Genomic Sequence Resource of Talaromyces albobiverticillius, the Causative Pathogen of Pomegranate Pulp Rot Disease

Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Material and Culture Conditions
2.2. DNA Extraction and Genome Sequencing
2.3. Genome Assembly and Prediction
2.4. Phylogenetic Analysis
2.5. Dispersed Repeat Sequences and Noncoding tRNA Annotation
2.6. Gene Annotation and Functional Analyses
3. Results
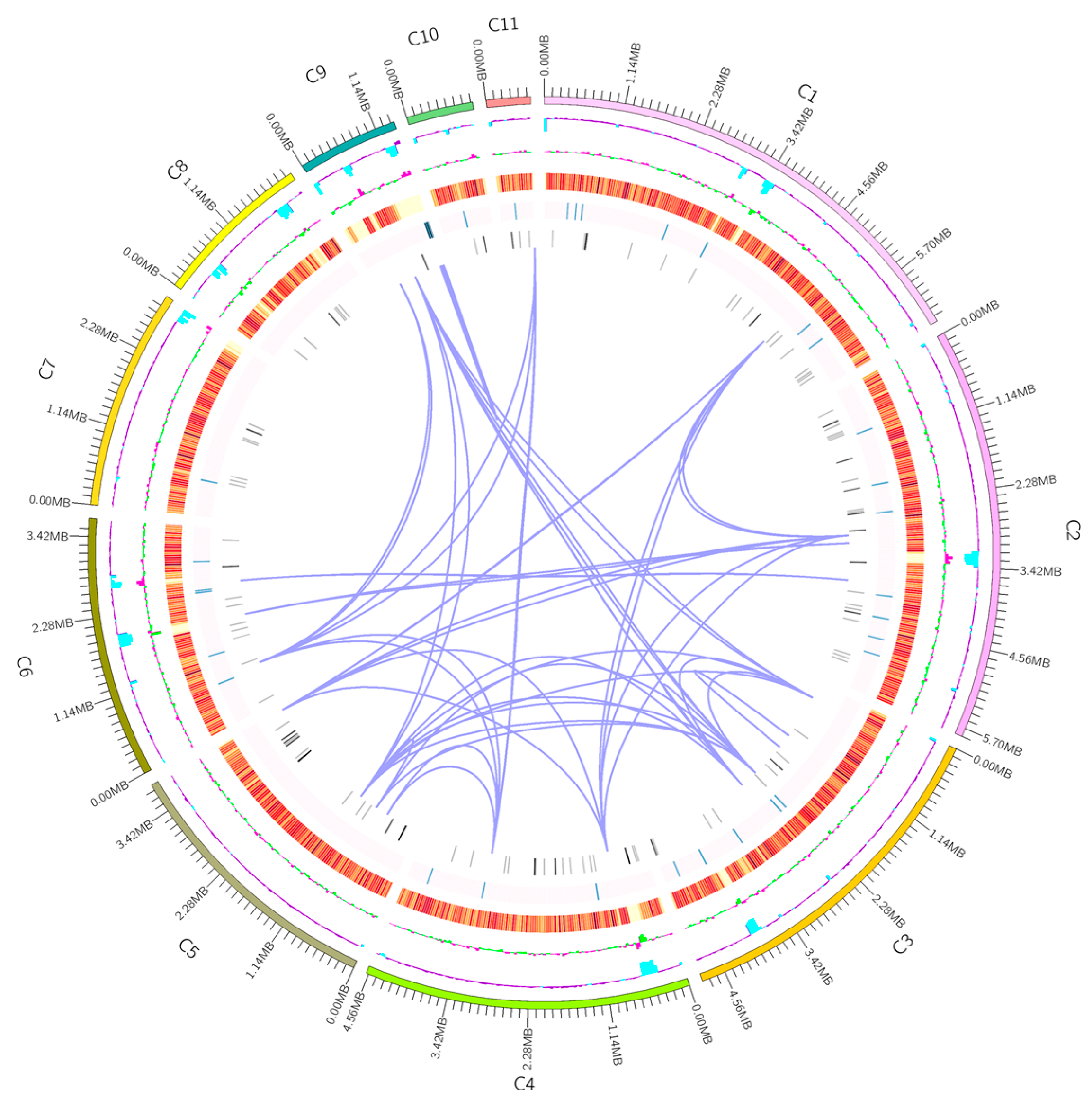
3.1. Genome Assembly and Genomic Characteristics
3.2. Gene Prediction and Functional Annotation
3.2.1. GO Annotation
3.2.2. Annotation of CAZymes
3.2.3. Annotation of Transport Proteins
3.2.4. Annotation of Secreted Proteins
3.2.5. Annotation of Putative Virulence-Associated Genes
3.2.6. Annotation of Secondary Metabolites Gene Clusters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yilmaz, N.; Visagie, C.M.; Houbraken, J.; Frisvad, J.C.; Samson, R.A. Polyphasic taxonomy of the genus Talaromyces. Stud. Mycol. 2014, 78, 175–341. [Google Scholar] [CrossRef] [PubMed]
- Houbraken, J.; Samson, R.A. Phylogeny of Penicillium and the segregation of Trichocomaceae into three families. Stud. Mycol. 2011, 70, 1–51. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.-D.; Chen, A.J.; Houbraken, J.; Frisvad, J.C.; Wu, W.-P.; Wei, H.-L.; Zhou, Y.-G.; Jiang, X.-Z.; Samson, R.A. New section and species in Talaromyces. MycoKeys 2020, 68, 75–113. [Google Scholar] [CrossRef] [PubMed]
- Limper, A.H.; Adenis, A.; Le, T.; Harrison, T.S. Fungal infections in HIV/AIDS. Lancet Infect. Dis. 2017, 17, e334–e343. [Google Scholar] [CrossRef] [PubMed]
- Kumari, M.; Taritla, S.; Sharma, A.; Jayabaskaran, C. Antiproliferative and Antioxidative Bioactive Compounds in Extracts of Marine-Derived Endophytic Fungus Talaromyces purpureogenus. Front. Microbiol. 2018, 9, 1777. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-J.; Shim, C.-K.; Kim, Y.-K.; Hong, S.-J.; Park, J.-H.; Han, E.-J.; Kim, S.-C. Enhancement of Seed Dehiscence by Seed Treatment with Talaromyces flavus GG01 and GG04 in Ginseng (Panax ginseng). Plant Pathol. J. 2017, 33, 1–8. [Google Scholar] [CrossRef]
- Li, H.-L.; Li, X.-M.; Li, X.; Wang, C.-Y.; Liu, H.; Kassack, M.U.; Meng, L.-H.; Wang, B.-G. Antioxidant Hydroanthraquinones from the Marine Algal-Derived Endophytic Fungus Talaromyces islandicus EN-501. J. Nat. Prod. 2017, 80, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Zerva, A.; Antonopoulou, I.; Enman, J.; Iancu, L.; Rova, U.; Christakopoulos, P. Cross-Linked Enzyme Aggregates of Feruloyl Esterase Preparations from Thermothelomyces thermophila and Talaromyces wortmannii. Catalysts 2018, 8, 208. [Google Scholar] [CrossRef]
- Varriale, S.; Houbraken, J.; Granchi, Z.; Pepe, O.; Cerullo, G.; Ventorino, V.; Chin-A-Woeng, T.; Meijer, M.; Riley, R.; Grigoriev, I.V.; et al. Talaromyces borbonicus, sp. nov., a novel fungus from biodegraded Arundo donax with potential abilities in lignocellulose conversion. Mycologia 2018, 110, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Frisvad, J.C.; Yilmaz, N.; Thrane, U.; Rasmussen, K.B.; Houbraken, J.; Samson, R.A. Talaromyces atroroseus, a new species efficiently producing industrially relevant red pigments. PLoS ONE 2013, 8, e84102. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, M.; Zelena, M.; Cacciola, F.; Ceslova, L.; Girard-Valenciennes, E.; Clerc, P.; Dugo, P.; Mondello, L.; Fouillaud, M.; Rotondo, A.; et al. Partial characterization of the pigments produced by the marine-derived fungus Talaromyces albobiverticillius 30548. Towards a new fungal red colorant for the food industry. J. Food Compost. Anal. 2018, 67, 38–47. [Google Scholar] [CrossRef]
- Venkatachalam, M.; Gérard, L.; Milhau, C.; Vinale, F.; Dufossé, L.; Fouillaud, M. Salinity and Temperature Influence Growth and Pigment Production in the Marine-Derived Fungal Strain Talaromyces albobiverticillius 30548. Microorganisms 2019, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, M.; Shum-Chéong-Sing, A.; Dufossé, L.; Fouillaud, M. Statistical Optimization of the Physico-Chemical Parameters for Pigment Production in Submerged Fermentation of Talaromyces albobiverticillius 30548. Microorganisms 2020, 8, 711. [Google Scholar] [CrossRef]
- Mincuzzi, A.; Sanzani, S.M.; Garganese, F.; Ligorio, A.; Ippolito, A. First Report of Talaromyces albobiverticillius Causing Postharvest Fruit Rot on Pomegranate in Italy. J. Plant Pathol. 2017, 99, 303. [Google Scholar]
- Huang, S.; Li, T.; Yang, T.; Zheng, X.; Yang, D.; Fu, G.; Hu, G.; Tao, A.; Wang, L.; Xu, T.; et al. First Report of Pulp Rot in Externally Asymptomatic Pomegranate Fruit Caused by Talaromyces albobiverticillius in Henan Province, China. Plant Dis. 2022, 106, 1989. [Google Scholar] [CrossRef] [PubMed]
- Mincuzzi, A.; Sanzani, S.M.; Caputo, M.; D’Ambrosio, P.; Palou, L.; Ragni, M.; Ippolito, A. Effectiveness of alternative means for controlling pomegranate postharvest pathogens. Acta Hortic. 2023, 1363, 181–186. [Google Scholar] [CrossRef]
- Labuda, R.; Hudec, K.; Piecková, E.; Mezey, J.; Bohovic, R.; Mátéová, S.; Lukác, S.S. Penicillium implicatum causes a destructive rot of pomegranate fruits. Mycopathologia 2004, 157, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Stošić, S.; Ristić, D.; Gašić, K.; Starović, M.; Ljaljević Grbić, M.; Vukojević, J.; Živković, S. Talaromyces minioluteus: New Postharvest Fungal Pathogen in Serbia. Plant Dis. 2020, 104, 656–667. [Google Scholar] [CrossRef] [PubMed]
- El-Ashram, S.; Al Nasr, I.; Suo, X. Nucleic acid protocols: Extraction and optimization. Biotechnol. Rep. 2016, 12, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Saha, S.; Bridges, S.; Magbanua, Z.V.; Peterson, D.G. Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res. 2008, 36, 2284–2294. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Gardner, P.P.; Daub, J.; Tate, J.G.; Nawrocki, E.P.; Kolbe, D.L.; Lindgreen, S.; Wilkinson, A.C.; Finn, R.D.; Griffiths-Jones, S.; Eddy, S.R.; et al. Rfam: Updates to the RNA families database. Nucleic Acids Res. 2009, 37, D136–D140. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015, 43, D261–D269. [Google Scholar] [CrossRef] [PubMed]
- Saier, M.H.; Reddy, V.S.; Tsu, B.V.; Ahmed, M.S.; Li, C.; Moreno-Hagelsieb, G. The Transporter Classification Database (TCDB): Recent advances. Nucleic Acids Res. 2016, 44, D372–D379. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Blin, K.; Cimermancic, P.; de Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. antiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39, W339–W346. [Google Scholar] [CrossRef]
- Urban, M.; Pant, R.; Raghunath, A.; Irvine, A.G.; Pedro, H.; Hammond-Kosack, K.E. The Pathogen-Host Interactions database (PHI-base): Additions and future developments. Nucleic Acids Res. 2015, 43, D645–D655. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Bai, W.; Jing, L.-L.; Guan, Q.-Y.; Tan, R.-X. Two new azaphilone pigments from Talaromyces albobiverticillius and their anti-inflammatory activity. J. Asian Nat. Prod. Res. 2021, 23, 325–332. [Google Scholar] [CrossRef]
- Ruiz-Sánchez, J.P.; Morales-Oyervides, L.; Giuffrida, D.; Dufossé, L.; Montañez, J.C. Production of Pigments under Submerged Culture through Repeated Batch Fermentation of Immobilized Talaromyces atroroseus GH2. Fermentation 2023, 9, 171. [Google Scholar] [CrossRef]
- Thrane, U.; Rasmussen, K.B.; Petersen, B.; Rasmussen, S.; Sicheritz-Pontén, T.; Mortensen, U.H. Genome Sequence of Talaromyces atroroseus, Which Produces Red Colorants for the Food Industry. Genome Announc. 2017, 5, e01736-16. [Google Scholar] [CrossRef]
- Chadni, Z.; Rahaman, M.H.; Jerin, I.; Hoque, K.; Reza, M.A. Extraction and optimisation of red pigment production as secondary metabolites from Talaromyces verruculosus and its potential use in textile industries. Mycology 2017, 8, 48–57. [Google Scholar] [CrossRef]
- He, L.; Mei, X.; Lu, S.; Ma, J.; Hu, Y.; Mo, D.; Chen, X.; Fan, R.; Xi, L.; Xie, T. Talaromyces marneffei infection in non-HIV-infected patients in mainland China. Mycoses 2021, 64, 1170–1176. [Google Scholar] [CrossRef]
- Ning, C.; Lai, J.; Wei, W.; Zhou, B.; Huang, J.; Jiang, J.; Liang, B.; Liao, Y.; Zang, N.; Cao, C.; et al. Accuracy of rapid diagnosis of Talaromyces marneffei: A systematic review and meta-analysis. PLoS ONE 2018, 13, e0195569. [Google Scholar] [CrossRef] [PubMed]
- Fukaya, M.; Ozaki, T.; Minami, A.; Oikawa, H. Biosynthetic machineries of anthraquinones and bisanthraquinones in Talaromyces islandicus. Biosci. Biotechnol. Biochem. 2022, 86, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Schafhauser, T.; Wibberg, D.; Rückert, C.; Winkler, A.; Flor, L.; van Pée, K.-H.; Fewer, D.P.; Sivonen, K.; Jahn, L.; Ludwig-Müller, J.; et al. Draft genome sequence of Talaromyces islandicus (“Penicillium islandicum”) WF-38-12, a neglected mold with significant biotechnological potential. J. Biotechnol. 2015, 211, 101–102. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Variables | Statistics |
|---|---|
| Genome assembly size (bp) | 38,354,882 |
| Number of contigs | 14 |
| Contigs N50 (bp) | 4,594,200 |
| Maximum contigs length (bp) | 6,575,826 |
| GC content (%) | 45.78 |
| BUSCO completeness (%) * | 98.3 |
| Fragmented BUSCOs (%) | 0.3 |
| Missing BUSCOs (%) | 1.4 |
| Total BUSCOs searched | 758 |
| SINEs (bp) | 2322 |
| LINEs (bp) | 32,971 |
| LTR (bp) | 175,050 |
| DNA transposons (bp) | 116,497 |
| Total length of Repeats (bp) | 321,868 |
| TR (bp) | 244,896 |
| Minisatellite DNA (bp) | 161,135 |
| Microsatellite DNA (bp) | 18,152 |
| Protein-coding genes | 10,380 |
| Mean gene length (bp) | 1432 |
| Genome Statistics | Talaromyces albobiverticillius | Talaromyces atroroseus | Talaromyces islandicus | Talaromyces marneffei | Talaromyces verruculosus |
|---|---|---|---|---|---|
| strain | Tp-2 | IBT 11181 | WF-38-12 | ATCC 18224 | TS63-9 |
| host | Punica granatum | Capsicum annuum | Triticum aestivum | Homo sapiens | Nicotiana tabacum |
| country | China | Denmark | Germany | USA | China |
| sequencing technology | Oxford Nanopore | Illumina HiSeq | Illumina MiSeq | SOLiD | Illumina HiSeq |
| assembly size (Mb) | 38.4 | 30.8 | 34.7 | 28.6 | 37.6 |
| protein-coding | 10,380 | 9523 | 9927 | 10,023 | 11,447 |
| GC content (%) | 46 | 46 | 46 | 46 | 46 |
| unassigned genes 1 | 381 | 325 | 515 | 429 | 398 |
| one-to-one orthologous 2 | — | 6396 | 5809 | 5871 | 6493 |
| Variables | Statistics |
|---|---|
| Protein-coding genes | 10,380 |
| Genes annotated by Pfam | 7412 |
| Genes annotated by KEGG | 8844 |
| Genes annotated by KOG | 2160 |
| Genes annotated by NR | 9782 |
| Genes annotated by Swiss-Prot | 3657 |
| Type I polyketide synthases genes | 214 |
| Cytochrome P450 genes | 227 |
| Virulence genes | 400 |
| Carbohydrate active enzymes | 750 |
| Transmembrane proteins | 2057 |
| Transport proteins | 638 |
| Secreted Protein | 671 |
| Categories | Families | Genes |
|---|---|---|
| Glycoside Hydrolases (GHs) | 105 | 427 |
| Glycosyltransferases (GTs) | 37 | 121 |
| Polysaccharide Lyases (PLs) | 8 | 11 |
| Carbohydrate Esterases (CEs) | 10 | 40 |
| Auxiliary Activities (AAs) | 14 | 60 |
| Carbohydrate-Binding Modules (CBMs) | 12 | 91 |
| Pathogen Species | PHI Database | Associated with Virulence 1 |
|---|---|---|
| Alternaria alternata | 10 | 8 |
| Aspergillus flavus | 15 | 15 |
| Aspergillus fumigatus | 147 | 83 |
| Beauveria bassiana | 45 | 30 |
| Bipolaris maydis | 11 | 11 |
| Botrytis cinerea | 41 | 18 |
| Candida albicans | 44 | 35 |
| Colletotrichum gloeosporioides | 10 | 8 |
| Colletotrichum graminicola | 7 | 6 |
| Cryptococcus neoformans | 40 | 29 |
| Fusarium graminearum | 366 | 118 |
| Magnaporthe oryzae | 163 | 117 |
| Sclerotinia sclerotiorum | 17 | 11 |
| Verticillium dahliae | 19 | 11 |
| Zymoseptoria tritici | 17 | 8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, T.; Chen, S.; Niu, Q.; Xu, G.; Lu, C.; Zhang, J. Genomic Sequence Resource of Talaromyces albobiverticillius, the Causative Pathogen of Pomegranate Pulp Rot Disease. J. Fungi 2023, 9, 909. https://doi.org/10.3390/jof9090909
Wang T, Chen S, Niu Q, Xu G, Lu C, Zhang J. Genomic Sequence Resource of Talaromyces albobiverticillius, the Causative Pathogen of Pomegranate Pulp Rot Disease. Journal of Fungi. 2023; 9(9):909. https://doi.org/10.3390/jof9090909
Chicago/Turabian StyleWang, Tan, Shuchang Chen, Qiuhong Niu, Guangling Xu, Chenxu Lu, and Jin Zhang. 2023. "Genomic Sequence Resource of Talaromyces albobiverticillius, the Causative Pathogen of Pomegranate Pulp Rot Disease" Journal of Fungi 9, no. 9: 909. https://doi.org/10.3390/jof9090909
APA StyleWang, T., Chen, S., Niu, Q., Xu, G., Lu, C., & Zhang, J. (2023). Genomic Sequence Resource of Talaromyces albobiverticillius, the Causative Pathogen of Pomegranate Pulp Rot Disease. Journal of Fungi, 9(9), 909. https://doi.org/10.3390/jof9090909