Abstract
Oudemansiella raphanipes, considered as a well-known culinary edible mushroom with a high content of natural bioactive substances, is widely cultivated in China with the commercial name Changgengu. However, due to the lack of genomic data, molecular and genetic study on O. raphanipes is rare. To obtain a comprehensive overview of genetic characteristics and enhance the value of O. raphanipes, two mating-compatible monokaryons isolated from the dikaryon were applied for de novo genome sequencing and assembly using Nanopore and /or Illumina sequencing platforms. One of the monokaryons, O. raphanipes CGG-A-s1, was annotated with 21,308 protein-coding genes, of which 56 were predicted to be involved in the biosynthesis of secondary metabolites such as terpene, type I PKS, NRPS, and siderophore. Phylogenetic and comparative analysis of multiple fungi genomes revealed a close evolutionary relationship between O. raphanipes and Mucidula mucid based on single-copy orthologous protein genes. Significant collinearity was detected between O. raphanipes and Flammulina velutipes on the synteny of inter-species genomes. 664 CAZyme genes in CGG-A-s1 were identified with GHs and AAs families significantly elevated when compared with the other 25 sequenced fungi, indicating a strong wood degradation ability. Furthermore, the mating type locus analysis revealed that CGG-A-s1 and CGG-A-s2 were conserved in the gene organization of the mating A locus but various in that of the mating B locus. The genome resource of O. raphanipes will provide new insights into its development of genetic studies and commercial production of high-quality varieties.
1. Introduction
Oudemansiella raphanipes is a well-known culinary edible mushroom with an excellent unique flavor, as well as a medicinal mushroom with high economic value [1,2]. In China, it was originally produced in Yunnan province and was commercially known as Changgengu or Heipijizong [2]. As for the taxonomic name, it was first described by M. J. Berkeley in India and effectively published in 1850 named as Agaricus raphanipes [2]. However, it was confusingly classified as Hymenopellis raphanipes, Hymenopellis furfuracea, Oudemansiella furfuracea, or Oudemansiella radicata in China during the past decades [1,2,3], but until 2016 its classification was determined to be O. raphanipes based on morphological and molecular data [2]. Furthermore, no O. radicata or O. furfuracea species were detected within over 300 samples collected from 13 provinces in China since 2013 [2], indicating that these two previously reported species should also classified as O. raphanipes. Since then, O. raphanipes has been used to refer to Heipijizong in domestic academic research to avoid naming confusion.
O. raphanipes has been recently cultivated throughout China since its artificial cultivation in large quantities was realized [3]. Numerous bioactive compounds produced by O. raphanipes have been discovered, including polysaccharides, enzymes, orcinol, ergosterol, triterpenes, and other nutrients, indicating that it plays a positive role in antioxidant, antitumor, immunomodulatory, and hepatoprotection [4,5,6,7]. Owing to its outstanding health-promoting properties, multiple studies have been focused on domestication and cultivation conditions, the liquid fermentation process, bioactive compound extraction, and medical function identification in O. raphanipes [6,7,8,9,10,11]. The huge demand makes O. raphanipes commercial cultivation profitable. However, molecular biological and genetic study on O. raphanipes is rare due to the lack of genomic information, which limits its development of molecular genetic features.
As sequencing technology advances, genome sequencing has been extensively applied to investigate the genetic traits of mushrooms [12,13,14]. In particular, numerous genomes of mushrooms, including Pleurotus giganteus [15], Clitopilus passeckerianus [16], Stropharia rugosoannulata [17], Gomphus purpuraceus [18], Inonotus obliquus [19], Inonotus hispidus [20], and Laetiporus sulphureus [21], among others, have been most recently reported, which will provide valuable genetic resources and molecular markers for biological and genetic studies of edible mushrooms and also disease. Therefore, the availability of high-quality whole genomic sequencing for O. raphanipes is imperative to carry out, as it would facilitate the identification of functional candidate genes and genetic breeding of the cultivars.
In the present study, the complete genomes of O. raphanipes CGG-A-s1 and O. raphanipes CGG-A-s2 were sequenced using Illumina and Nanopore sequencing platforms. Comparative analysis was conducted between the genome of O. raphanipes CGG-A-s1 and the other 25 published fungi genomes. Secondary metabolite-related genes, carbohydrate-active enzymes (CAZymes), and mating genes were also analyzed. The availability of a whole-genome sequence will help to classify the taxonomic status of O. raphanipes and aid future breeding efforts to improve its commercial cultivation.
2. Materials and Methods
2.1. Strains and Culture Conditions
The O. raphanipes strain CGG-A was provided by the Laboratory of Mushroom Precision Breeding (http://mushroomlab.cn/, accessed on 1 December 2022). The O. raphanipes strain was cultivated and maintained on potato dextrose agar (PDA) plates. For fruiting body production, the strain was inoculated into solid media (60% (w/w) sawdust, 20% (w/w) corncob, 18% (w/w) wheat bran, 1% (w/w) sucrose and 1% (w/w) gypsum powder) in a polypropylene bag. Vegetative growth of O. raphanipes mycelia was carried out at 25 °C with a humidity of 70%. After 35 to 40 days of cultivation, the mycelia occupied the full culture bag. The mycelia then continued cultivation under the same conditions for 30 to 35 days for maturation. The polypropylene bag was removed and the solid culture was buried into the ground and covered by 3–4 cm soil. At 20 to 30 days after earthing, the fruiting body formed under the stimulation of temperature, water, and light. The basidiospores were collected and the monokaryotic strains CGG-A-s1 and CGG-A-s2 were isolated from the spores as previously described [15].
2.2. Genome and Transcriptome Sequencing
The O. raphanipes CGG-A-s1 and CGG-A-s2 strains were inoculated on PDA plates covered with cellophane. The mycelia were scraped from the cellophane after they covered the entire plate. The genome was extracted using a NucleoBond HMW DNA kit (Macherey-Nagel, Düren, Germany). The O. raphanipes genome was sequenced using Illumina NovaSeq 6000 (paired-end, 2 × 150 bp) and Nanopore PromethION 48 sequencing platforms (Benagen Technology Co., Ltd., China, Wuhan), with a sequencing depth of ~60× for the Illumina platform and ~200× for the Nanopore platform. For RNA-Seq, total RNA was extracted from O. raphanipes mycelia and fruiting body using Trizol (Takara, Dalian, Liaoning Province, China) methods according to the manufacturer’s instructions. Paired-end sequencing libraries and analysis were performed on an Illumina NovaSeq 6000 platform (Illumina, CA, USA) by Benagen Co. (Wuhan, China).
2.3. Genome Assembling and Gene Prediction
The Nanopore reads were filtered using filtlong software and assembled using NECAT software [22]. The initial polishing was performed with racon v1.4.21 software (https://github.com/isovic/racon) for two rounds using Nanopore long reads. Then, pilon v1.24 was utilized to further correct the racon-corrected contigs with Illumina short reads for two rounds [23]. The assembly completeness was evaluated with QUAST v5.1.0rc1 software with the Illumina reads and Nanopore reads [24]. Gene prediction was performed by augustus software using Laccaria bicolor model [25] and by GeneMark-ES v 4.69 for self-training gene prediction. Meanwhile, the transcriptome data was filtered and assembled by trimmomatic, and Stringtie software was then re-trained by augustus for gene prediction. Finally, the software EVidenceModeler was used to integrate the gene prediction results mentioned above. Then the completeness of the assembled genome was also evaluated using BUSCO v5.1.2 software [26] with comparison to lineage dataset fungi_odb10 (creation date: 2020-09-10, number of BUSCO markers: 758). Protein functional annotation was performed using eggNGOmapper software [27], Pfam database, and SwissProt databases. In addition, gene clusters related to secondary metabolites were predicted by antiSMASH v6.0 [28]. Circular layouts were generated using Circos software (http://circos.ca/, accessed on 20 December 2022) [29].
2.4. Comparative Genomic Analysis
The pairwise average nucleotide identity (ANI) values between genomes were analyzed using FastANI software [30]. Collinearity analysis was performed using MCScanX software [31], based on location information from the GFF3 files of O. raphanipes CGG-A-s1, Flammulina velutipes (Genbank: PRJEB54953), and Lentinula edodes L808, as well as O. raphanipes CGG-A-s1, Agaricus bisporus var. burnettii H119, and Pleurotus ostreatus P15. Gene families and single-copy orthologous genes were analyzed using OrthoFinder v2.5.4 software as previously described [32]. Timetree was evaluated using single-copy orthologous protein sequences and performed using mcmctree software prom paml packages. The species tree was visualized using FastTree software.
2.5. Repeat Sequence Identification
Repeat sequence was analyzed using RepeatModeler and RepeatMasker software [33,34]. RepBase database was used to predict sequences similar to known repeat sequences. RepeatModeler was used for de novo constructing the candidate database of repetitive elements, by which the repeated sequences were annotated using RepeatMasker (http://www.repeatmasker.org/RepeatModeler/, accessed on 22 December 2022).
2.6. Identification of CAZymes
Annotation of carbohydrate-active enzymes (CAZymes) for the genome of O. raphanipes was performed using dbcan version v3.0.2 software [35]. The database was downloaded from the dbCAN meta server (http://bcb.unl.edu/dbCAN2/, accessed on 28 December 2022) (version of the database is V10). Hmmer software was used for the annotation of proteins with default parameters (HMMER E-Value < 1e-15, HMMER Coverage > 0.35).
2.7. Analysis of Mating Genes
The mating genes from Flammulina velutips [36,37,38] and Pleurotus giganteus [15] were used as query sequences for the Blastp alignment against the proteome of O. raphanipes strain CGG-A-s1 and strain CGG-A-s2.
3. Results and Discussion
3.1. Genome Assembly of Monokaryotic O. raphanipes
O. raphanipes is a well-known culinary edible mushroom that has been widely cultivated in China over the past few decades with the commercial name “Changgengu” or “Heipijizong”. We have succeeded in industrially cultivating the strain O. raphanipes CGG-1 in Shandong province (Figure 1). In this study, monokaryotic mycelia were germinated from the basidiospores of O. raphanipes CGG-1 by gradient dilution cultivation on the plate. Clamp connection was observed within the two monokaryons CGG-A-s1 and CGG-A-s2 after mating, which were used as sexually compatible monokaryotic strains and then for genome sequencing.

Figure 1.
The fruiting bodies of O. raphanipes CGG-1. (a) The fruiting body morphology of O. raphanipes. (b) Industrial cultivation of O. raphanipes. (c) The greenhouse for industrial cultivation of O. raphanipes.
The genome of CGG-A-s1 was sequenced using Nanopore and Illumina sequencing platforms. A total 1,313,194 reads were obtained by Nanopore, with an average length of 9980 bp and a total length of 13.1 Gb, whereas 23,017,968 reads up to 3.44 Gb of data were obtained by Illumina using the PE150 method. Finally, 37 contigs with an N50 of 2.55 Mb and an N90 of 879 Kb were obtained after removing the repeats. The overall length was 60.2 Mb and the length of the largest contig was 5.27 Mb (Table 1). Repeat sequences accounted for 15.84% of the whole genome of CGG-A-s1 and the majority were LTR elements (5.60%), the LINE (0.04%), and simple repeats (0.30%) (Table 2). The integrity of the genome was evaluated using QUAST v5.1.0 software and determined to be 99.25%. In order to conduct comprehensive genomic prediction, the transcriptome sequencing was performed by Illumina using the PE150 method and a total of 95.5M reads up to 14.32 Gb of data were obtained. The whole genome of monokaryon CGG-A-s2 generated from the Illumina platform was assembled by SPAdes software and 3,154 contigs (>500 bp) were obtained. The total length was 54.9 Mb with an N50 of 77.4 Kb and an N90 of 9.94 Kb (Table 1).

Table 1.
Genome assembly features of monokaryotic O. raphanipes CGG-A-s1 and CGG-A-s2.
Table 2.
Repeat element analysis in the P. giganteus genome.
3.2. Gene Prediction of O. raphanipes
A total of 21,308 CDSs were predicted for the CGG-A-s1 genome with an average length of 1,406 bp. The cumulative length of encoded genes was 30.0 Mb, which accounted for 49.8% of the whole genome. The average exon and intron numbers per gene were 5.78 and 4.78, respectively (Table 3). The completeness of the O. raphanipes CGG-A-s1 genome assembly and gene prediction was evaluated by the BUSCO software, which was analyzed using the fungi_odb10 (758 genes) database with a completeness of 94.4%, the basidiomycota_odb10 (1764 genes) database with a completeness of 93.9%, and the agaricales_odb10 (3870 genes) database with a completeness of 91.3% (Figure S1). The results suggested that we presented a high-quality genome sequence of O. raphanipes with preferable integrity and continuity.
Table 3.
Characteristics of the gene prediction of O. raphanipes CGG-A-s1.
3.3. Genome Annotation of O. raphanipes
The predicted gene sequence was functionally analyzed by EggNOGmapper, SwissProt, Pfam, and CAZymes databases as shown in Table S1. Among the annotated genes, 15,004 and 7753 genes were classified by the EggNOGmapper database and SwissProt database, respectively. A total of 11,289 genes were annotated by the Pfam database via Hmmer based on the similarity of protein domains, and 1152 secreted proteins, containing a signal peptide without transmembrane domains, were identified. In addition, a total of 56 gene clusters related to secondary metabolites were predicted by antiSMASH software. Among them, there are 24 gene regions that are associated with terpenes, 16 gene regions that are involved in non-ribosomal peptide synthase (NRPS) or NRPS-like, six gene regions that are related to type I polyketide synthase (T1PKS), two gene regions that are associated with indole, two regions that are associated with siderophore, and five gene region that are identified as fungal-RiPP-like. One region was predicted to be both fungal-RiPP-like and terpene. Table S2 and Figure 2 show the summary of predicted metabolite gene clusters. Finally, all genomic information of O. raphanipes CGG-A-s1 was circularly mapped by Circos software according to the results of genome assembly and functional annotation (Figure 2).
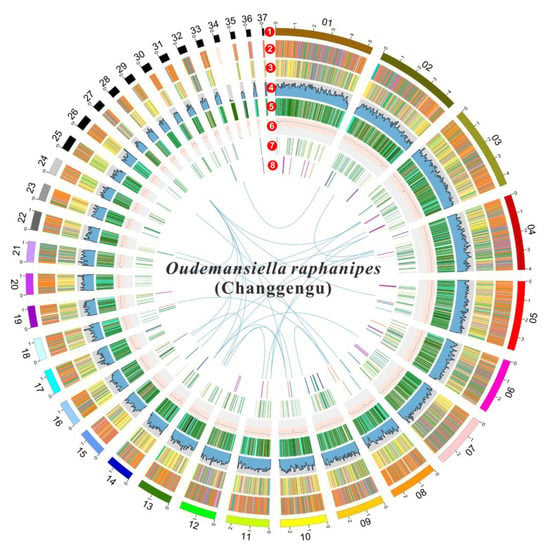
Figure 2.
Circle map of the genome assembly and gene prediction of O. raphanipes CGG-A-s1. A total of seven layers were plotted from the outside to the inside. The outmost layer one is a circular representation of the 37 contigs with size intervals of 1 Mb. Layers two and three represent the predicted genes in the forward and reverse strands of the genome. Layer four indicates the gene density. Layer five indicates the repeat sequences. Layer six represents the GC content. Layer seven represents the genes of CAZymes. Links within and between chromosomes indicate collinear blocks generated from MCScanX: terpene (purple), NRPS or NRPS-like (green), fungal-Ripp-like (blue), T1PKS (orange), NRPS-independent-siderophore (red), indole (light orange), both of fungal-RiPP-like and terpene (light blue).
3.4. Genome Evolutionary and Comparative Analysis
Phylogenetic analyses were carried out in order to investigate evolutionary relations between O. raphanipes and 25 other fungi species (23 Basidiomycetes and 2 Ascomycetes). 309 single-copy orthologous genes were found and used for phylogenetic reconstruction and species divergence time estimation (Figure 3). O. raphanipes was clustered in one clade with M. mucid, which indicated that they were closely related (Figure 3a). It was also shown that O. raphanipes had a close evolutionary relationship with cultivated edible mushrooms, such as Flammulina velutipes, Armillaria solidipes and Lentinula edodes. It was estimated that the divergence between species from O. raphanipes and M. mucid occurred ~33.1 million years ago (MYA). O. raphanipes split off from F. velutipes around 93.4 MYA, and it split off from A. solidipes and L. edodes around 122.5 and 152.3 MYA, respectively (Figure 3a). Considering that edible mushrooms with close relatives may be conserved in terms of cultivation methods and substrate utilization, the cultivation mode of O. raphanipes can be referred to as that of F. velutipes and A. solidipes.
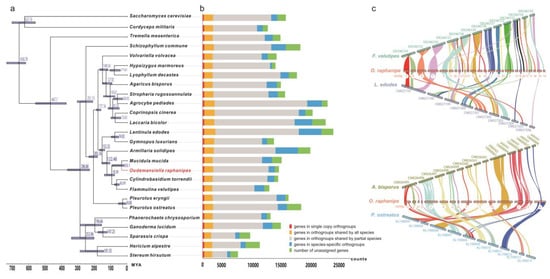
Figure 3.
Evolutionary and comparative genomic analysis of O. raphanipes. (a) Phylogenetic tree was constructed based on 309 single-copy orthologs from O. raphanipes and 25 other fungal species (23 Basidiomycetes and 2 Ascomycetes) using OrthoFinder. Divergence timings were indicated using transparent blue bars at the internodes with 95% highest posterior density using MYA (million years ago) as a unit at the x-axis. Single-copy orthologs are defined as orthologs that were present as a single-copy gene in all 26 species. (b) Number of different orthologous gene types was calculated in each fungal species and indicated with different colors. (c) The genome collinearity among O. raphanipes, F. velutipes and L. edodes (upper), as well as O. raphanipes, A. bisporus and P. ostreatus (lower). Each line connects a pair of collinearity blocks between two genomes.
For inter-species comparative genomic studies, the synteny of the O. raphanipes genome with two chromosome-scale assembled genomes in each of the two groups was analyzed. One group includes F. velutipes and L. edodes, the other includes Agaricus bisporus and Pleurotus ostreatus. Significant collinearity was detected between these species (Figure 3c). O. raphanipes and F. velutipe in the first group had the highest level of collinearity, covering 26 contigs, while O. raphanipes and A. bisporus in the second group had a smaller level, covering only 15 contigs. Based on the collinearity results from the two groups, the following contigs of O. raphanipes should belong to the same chromosome: contig 7 and contig 9; contig 11, 21, 27, and 32; contig 6, 8, and 14; as well as contig 15, 16, 22, and 31, in which 26 contigs of O. raphanipes CGG-A-s1 show collinearity with 12 chromosomes of F. velutipes. Rupture and fusion events were identified in contig 1, 2, 3, 5, 6, 7, and 9 of O. raphanipes compared to chromosomes from F. velutipes and A. bisporus.
3.5. ANI Value Analysis
Fungal identification is primarily based on phenotypic and physiological characteristics [39]. Changgengu has been confusedly classified as H. raphanipes, H. furfuracea, or O. radicata [2,5,9] due to a lack of morphologic and genetic information to verify the taxonomy. Many molecular methods have been developed for fungal identification, but the ITS (internal transcribed spacer) remains the key choice [39,40]. However, the ITS sequences cannot distinguish effectively at the species level. On the other hand, ANI values have commonly been used for species determination in prokaryotes on the genome scale [30]. Here, we adopted the ANI values to reflect the genetic correlation between the strains from the genus Oudemansiella and Hymenopellis. Three Hymenopellis strains and one Oudemansiella strain were extracted from the NCBI genome database in order to compare with the two monokaryotic O. raphanipes genomes assembled in this study. The ANI value between strain O. raphanipes CGG-A-s1 and CGG-A-s2 was 98.15%, which displayed the highest similarity. The ANI values between O. raphanipes, H. radicata MG139 and H. chiangmaiae MG56 are all greater than 93.56%, indicating a close taxonomy relationship within species. However, it is interesting to note that the ANI value between the two H. radicata strains themselves is only 85.8%, far below the threshold for intraspecies in prokaryotes, which is 95% (Figure 4a). The O. mucida CBS 558.79 strain displayed the lowest ANI values (~85%) with other species. It is worth noting that the ANI value between O. raphanipes CGG-A-s1 and H. radicata MG139 is 97.5%, suggesting that the two strains are probably from the same species.
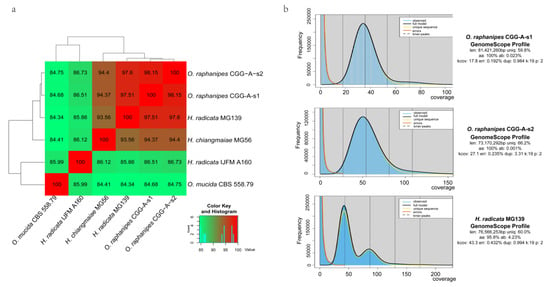
Figure 4.
Genome assembly comparison within O. raphanipes and closely related strains. (a) Cluster heatmap of ANI values between six strains. (b) Histogram of the 19-mer depth distribution of the Illumina sequencing reads of O. raphanipes CGG-A-s1 and H. radicata MG139 plotted by GenomeScope. Blue areas indicate the observed k-mer frequencies and the black line indicates the fitted GenomeScope model.
Although no benchmark analysis has been performed to evaluate the use of fastANI value in the taxonomy of fungi, considering the different genome sizes between fungi and prokaryotes, our results suggested that all six strains should belong to the same genus (Figure 4a) and four strains (CGG-A-s1, CGG-A-s2, MG139, and MG56) should belong to the same species.
Under these circumstances, high-quality genome sequencing and assembly level providing comprehensive instructions are more than necessary. The genome assembly level of strain O. raphanipes CGG-A-s1 was much better than that of strain H. radicata MG139, which was assembled into 31,379 contigs with an N50 of 6.07 Kb and an N90 of 0.92 kb [14]. In a word, further research should be performed to evaluate the digital DNA hybridization analysis methods in the taxonomy research of fungi.
Furthermore, Illumina reads were used for k-mer analysis using GenomeScope to generate a histogram of the depth distribution of the sequencing (k=19) (Figure 4b). A single k-mer coverage peak was observed for O. raphanipes CGG-A-s1 and CGG-A-s2. The heterozygous rate was 0.023% and 0.001%, respectively. Two major peaks between 42 and 85 were observed for H. radicata MG139, and the first peak is higher than the second with high heterozygosity (4.23%). This evidence demonstrates that strain H. radicata MG139 is a dikaryotic strain, while strain O. raphanipes CGG-A-s1 and CGG-A-s2 are monokaryons. However, with the rapid development of sequencing technology, more and more closely related species will be sequenced, which will make the taxonomic classification of Changgengu more accurate.
3.6. Carbohydrate Active Enzymes (CAZymes) in O. raphanipes Genome
CAZyme analysis can provide insight into the metabolism of complex carbohydrates in O. raphanipes. In this study, the dbCAN2 database was used to identify 664 candidate CAZyme genes from the genome of O. raphanipes CGG-A-s1, including 320 glycoside hydrolases (GHs, 48.2%), 82 glycosyl transferases (GTs, 12.3%), 31 polysaccharides lyases (PLs, 4.7%), 44 carbohydrate esterases (CEs, 6.6%), 172 auxiliary activities enzymes (AAs, 25.9%), and 15 carbohydrate-binding modules (CBMs, 2.3%) (Figure 5a). Figure 5b shows the gene numbers of the corresponding CAZyme families. In terms of the genes related to wood degradation, 17 CAZyme families proposed by Floudas [12] were all found in O. raphanipes (Figure 5b).
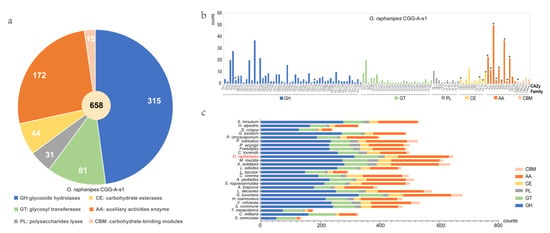
Figure 5.
Distribution and number of carbohydrate-active enzyme (CAZymes) genes in O. raphanipes CGG-A-s1 and 25 other fungi. (a) Distribution of CAZymes in O. raphanipes CGG-A-s1. (b) Gene numbers of CAZyme families in O. raphanipes CGG-A-s1. The asterisks indicate the 17 gene families involved in the wood degradation. (c) Comparison of CAZymes in the 26 fungi. The strain names of these fungi are the same as those in Figure 3.
A comparison of the CAZyme profiles with 25 other fungi was also conducted, as shown in Figure 5c. The result showed that O. raphanipes CGG-A-s1 had the second highest number of CAZyme encoding genes, among which the GHs and AAs families were significantly elevated. Furthermore, the distributions of gene numbers in O. raphanipes CGG-A-s1 were also closely related to that of M. mucida. Genes belonging to the GH family have been observed to play an important role in the degradation of cellulose and hemicellulose, and even in the expansion of the cap and senescence of edible mushrooms [41,42]. The top three GH families (37 GH16 genes, 20 GH3 genes, and 22 GH18) genes identified were involved in hemicellulose digestion, which shows how efficient this process is. The AA-related genes in the O. raphanipes genome include AA1-AA9, AA14, and AA16 (Table S3, Figure 5b). Among them, the AA1 enzymes are multicopper oxidases that are involved in lignin degradation, and the class II lignin-modifying peroxidases (AA2) are also important for lignin degradation as markers to discriminate between white rot fungi and brown rot fungi [43]. A total of 23 AA1-related genes (3 AA1, 15 AA_1, and 5AA_2) and 12 AA2-related genes were found in the O. raphanipes genome, which indicates a prominent lignin degradation ability. In addition, 50 AA3 and 22 AA9-related genes were also identified in O. raphanipes, which are involved in cellulose and hemicellulose degradation [17]. The results are consistent with the strong ability of O. raphanipes to degrade wood.
3.7. Identification of the Mating Genes
Mating type recognition plays a crucial role in the genetics and breeding of mushrooms, influencing the propagating system, fruiting body, gamete quality, etc. [19]. The mating loci in two monokaryotic strains of O. raphanipes were identified by Blastp using mating genes from F. velutips [36,37,38] and P. giganteus [15] as query sequences. In the mating A locus of O. raphanipes, there are two clusters of homeodomain (HD) genes (Figure 6a). In one cluster, two HD genes (including one HD1 and one HD2 genes) were transcribed in opposite directions, as in many other mushrooms [15,37,44]. Both the mitochondrial intermediate peptidase (Mip) and the beta flanking gene (Bfg) were located in the two ends of the mating A locus of O. raphanipes.
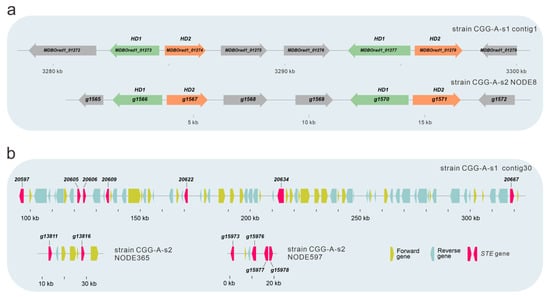
Figure 6.
Gene structure of the mating type locus of in O. raphanipes CGG-A-s1 and CGG-A-s2. (a) Structure of A mating type locus in O. raphanipes. (b) Structure of B mating type locus in O. raphanipes.
Sequence alignments showed that the mating B locus of O. raphanipes strain CGG-A-s1 was located in contig 30 (Figure 6b). Seven proteins with sequence similarity to the pheromone receptor (STE) genes from F. velutips [36,37,38] and P. giganteus [15] were identified in contig 30. Different from the mating A locus, the gene organization of the mating B locus in strain CGG-A-s2 was different from that in strain CGG-A-s1. Identified STE genes are located in different contigs. Two contigs containing more than one STE gene are shown in Figure 6b.
4. Discussion
Changgengu or Heipijizong is one of the most important commercial edible mushrooms in China, which has a long history in agricultural production, but a dispute over its scientific name. Though it has been assigned to the genus Oudemansiella according to the combined ITS and nrLSU [2], it was also classified as a species of other genera, mostly Hymenopellis [1,11]. Currently, a whole-genome similarity analysis has been one of the best ways to figure out the strain taxonomy and get genetic information on the molecular mechanisms of fungal growth and breeding [18,30,45]. However, few studies have investigated the genome sequence and genetic structure of O. raphanipes. Consequently, in this study, monokaryon genome sequencing was performed to explore the evolutionary status and genetic information of related functional genes of O. raphanipes CGG-1. With a combination of Nanopore and Illumina sequencing platforms, we assembled the genome of one of the monokaryons, O. raphanipes CGG-A-s1, into 37 contigs, which was much higher-quality sequencing data than its closely related reported strains, e.g., H. radicata MG139 and H. radicata IJFM A160. However, the ANI values indicated that our O. raphanipes strains and the other four strains should be interspecies or even intraspecies (ANI > 95) [30,46,47], which suggests that some of these strains are probably misnamed. Therefore, high-quality genome sequencing of more species within the genus is required in order to classify their taxonomic status. In order to comprehensively analyze the relationship between O. raphanipes and related species from other genera, 23 fungal species from Basidiomycota and 2 from Ascomycota were used for the phylogenetic analysis. O. raphanipes CGG-A-s1 was closely related to M. mucid (formerly also called Oudemansiella mucida), Cylindrobasidium torrendii, and Flammulina velutipes (one of the most widely cultivated mushroom species in China [48]). All these Agaricomycetes commonly have a small fruiting body, which are good resources for studying the underlying genomic changes in complexity level in mushroom fruiting bodies. Understanding the genetic bases of fruiting body evolution may directly contribute to the improvement of commercial mushroom production [49]. Furthermore, the synteny analysis showed high collinearity between the genomes of O. raphanipes CGG-A-s1 and F. velutipes, which suggests that F. velutipe probably served as a reference mode for cultivation and breeding of O. raphanipes during large-scale industrialization.
This study has identified many essential genes related to secondary metabolites, which endow O. raphanipes with biological activities that promote its survival in a given environment and defense responses to pathogens. Genes involved in the regulation of terpenes and NRPSs were mostly found in the genome of O. raphanipes. Terpenes are one of the largest groups of the bioactive natural products identified, which have been found in wild edible mushrooms and play an important role in biological functions [50,51,52]. For example, triterpenes from Ganoderma lucidum have been proven to have anti-cancer effects and can be used as a potential drug for cancer treatment [53]. It is worth noting that compared with other reported fungi, O. raphanipes is also predominate in the number of terpene synthesis genes [18,51]. NRPSs in fungi are considered to be corresponding to the virulence determinants during host-pathogen interactions, yet the exact role they play remains elusive in many cases [54,55]. To date, the function of NPRSs in Basidiomycetes has not been reported [19]. The expansion of terpenes and NRPSs in O. raphanipes suggests that it has significant potential for medicinal development.
CAZymes are important for the growth and development of wood-rotting mushrooms to thrive in environments rich in carbohydrates, especially lignocellulose and cellulose [18,56,57,58]. In the O. raphanipes genome, the GHs, GTs, and AAs were the main CAZymes, while PLs, CEs, and CMBs were in the minority. GH genes, the most highly elevated family in the genome of O. raphanipes, were four times more elevated than GT genes. This may be due to the lignocellulose degradation capacity that is necessary for the survival of O. raphanipes. Most GH-related genes are involved in starch degradation [41]. These evidences revealed that the enrichment of GH genes contributed to the diversification of nutrient substrate utilization for O. raphanipes. Moreover, compared with other fungi, O. raphanipes has the most AAs genes, which are important for lignin degradation [57,59].
O. raphanipes has the same gene organization structure as the mating A locus in other mushrooms [15,37,44]. Different mushrooms contain a different number of HD genes. P. giganteus and P. ostreatus PC9 only contain one pair of HD genes, while some contain three HD genes, such as those from P. eryngii [44], Pleurotus diamor [60], and F. velutipes [37]. Different monokaryotic strains from the same species may also contain a different number of HD genes. The strains of Hypsizygus marmoreus may contain three, four, or five HD genes [61]. As shown in Figure 6b, the two mating-compatible strains of O. raphanipes contain the same mating A structure with the same number of HD genes (Figure 6a). It is clear that the gene organization of STE genes on NODE365 and NODE597 are different from those in contig 30. Similar results have also been reported in other mushrooms, such as F. velutipes [15]. The gene organizations of STE genes in different mushrooms are quite different from one another [15,44,61]. All of the identified mating loci in O. raphanipes are based on sequence alignments. Nevertheless, the function of these genes in mating and clamp connection formation is still unknown. Further research should be conducted to determine the functions of these mating-related genes.
5. Conclusions
In summary, the whole-genome map of O. raphanipes was depicted for the first time in this study. Two monokaryons of O. raphanipes were isolated and used for sequencing by Nanopore and Illumina platforms. O. raphanipes CGG-A-s1 was sequenced with Nanopore and Illumina sequencing, generating 37 contigs with an N50 length of 2.55 Mb. O. raphanipes CGG-A-s2 was sequenced using the Illumina platform, obtaining 3154 contigs with an N50 of 77.4 Kb. The O. raphanipes CGG-A-s1 genome was functionally annotated using proprietary databases. The genome comparative analysis, genome evolutionary analysis, and CAZymes comparison expand our understanding of the taxonomic status, survival mechanisms, and capacity of O. raphanipes. Mating A and Mating B loci in both mating-compatible monokaryotic strains were identified, which provides useful information for the development of molecular markers for cross-breeding. The genome elucidation in this study provides foundational information and genetic resources for the exploration of metabolic compounds, industrial breeding, and genetic applications of O. raphanipes.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/jof9020266/s1, Figure S1. BUSCO summary of O. raphanipes CGG monokaryotic strain CGG-A-s1. The completeness of CGG-A-s1 genome was evaluated using different databases as indicated on the y-axis. The blue area represents the number of complete (C) and single-copy genes (S), the dark blue area represents the number of complete and duplicated genes (D), and the yellow and red indicate the number of fragmented genes (F) and missing genes (M), respectively; and n indicates the number of all genes used; Table S1: Genome annotation of O. raphanipes CGG-A-s1 by EggNOGmapper, SwissProt, and Pfam databases; Table S2:Prediction of gene clusters for secondary metabolites in O. raphanipes CGG-A-s1; Table S3: Gene lists of CAZymes in O. raphanipes CGG-A-s1.
Author Contributions
Conceptualization, H.Y. (Hailong Yu) and H.Y. (Hao Yu); Data curation, L.Z.; Formal analysis, W.Y. and H.Y. (Hao Yu); Funding acquisition, L.G. and H.Y. (Hao Yu); Investigation, M.Z. and W.Y.; Methodology, L.Z.; Supervision, S.Y., H.Y. (Hailong Yu) and H.Y. (Hao Yu); Validation, M.Z. and C.H.; Writing—original draft, L.Z.; Writing—review & editing, X.G. and H.Y. (Hao Yu). All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by Key R&D Program of Shandong Province, China (2022TZXD0024, 2022LZGC023, 2021ZDSYS28).
Data Availability Statement
The whole genome sequence data reported in this paper have been deposited in the Genome Warehouse in National Genomics Data Center [62,63] under accession number GWHBRAH00000000 and GWHBRAI00000000, respectively, that is publicly accessible at https://ngdc.cncb.ac.cn/gwh, accessed on 1 December 22. The genome information of H. radicata MG139 and H. chiangmaiae MG56 can be acquired from NCBI database with Genbank Accession of GCA_003314005.1 and QLPD00000000, respectively. In addition, the data used to support the findings of this study are available from the corresponding author upon request (ftp://www.mushroomlab.cn, accessed on 1 December 22).
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Zeng, X.Y.; Yuan, X.X.; Peng, K.Q.; Pan, Y.T.; Tan, T.J.; Wu, N.; Tian, F.H. Taxonomy and control of Trichoderma hymenopellicola sp. nov. responsible for the first green mold disease on Hymenopellis raphanipes. Front. Microbiol. 2022, 13, 991987. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.J.; Zhao, Q.; Wang, S.X.; Yang, Z.L. What is the radicate Oudemansiella cultivated in China? Phytotaxa 2016, 286, 10–11646. [Google Scholar] [CrossRef]
- Shim, J.O.; Chang, K.C.; Kim, T.H.; Lee, Y.S.; Lee, Y.S.; Lee, U.Y.; Lee, T.S.; Lee, M.W. The fruiting body formation of Oudemansiella radicata in the sawdust of oak (Quercus variabilis) mixed with rice bran. Mycobiology 2006, 34, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Liu, X.C.; Wang, W.S.; Yang, Q.H.; Dong, Y.H.; Xu, N.; Zhang, C.; Song, X.L.; Ren, Z.Z.; Zhao, F.L.; et al. Characteristic anti-inflammatory and antioxidative effects of enzymatic- and acidic- hydrolysed mycelium polysaccharides by Oudemansiella radicata on LPS-induced lung injury. Carbohydr. Polym. 2019, 204, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; Jia, J.X.; Ren, X.J.; Li, B.H.; Zhang, Q. Extraction, preliminary characterization and in vitro antioxidant activity of polysaccharides from Oudemansiella radicata mushroom. Int. J. Biol. Macromol. 2018, 120, 1760–1769. [Google Scholar] [CrossRef]
- Geng, X.; Lei, J.; Bau, T.; Guo, D.; Chang, M.; Feng, C.; Xu, L.; Cheng, Y.; Zuo, N.; Meng, J. Purification, characterization, and immobilization of a novel protease-resistant alpha-galactosidase from Oudemansiella radicata and its application in degradation of raffinose family oligosaccharides from soymilk. Foods 2022, 11, 3091. [Google Scholar] [CrossRef]
- Liu, Q.; Zhu, M.; Geng, X.; Wang, H.; Ng, T.B. Characterization of polysaccharides with antioxidant and hepatoprotective activities from the edible mushroom Oudemansiella radicata. Molecules 2017, 22, 234. [Google Scholar] [CrossRef]
- Liu, Q.; Chen, H.; Wang, H.; Ng, T.B. Isolation and characterization of a ubiquitin-like ribonuclease from the cultured deep root mushroom, Oudemansiella radicata (higher Basidiomycetes). Int. J. Med. Mushrooms 2015, 17, 1037–1045. [Google Scholar] [CrossRef]
- Wei, Q.; Zhong, X.; Haruna, M.H.; Liu, S.; Zhou, F.; Chen, M. Evaluation of different agricultural wastes for the production of polysaccharides from Oudemansiella raphanipes and its antioxidant properties. Food Sci. Nutr. 2022, 10, 3444–3452. [Google Scholar] [CrossRef]
- Zhou, X.; Yang, C.; Meng, Q.; Cui, Y.; Wang, Y.; Chen, X.; Fu, S. Investigation of chemical compounds and DPPH radical scavenging activity of Oudemansiella raphanipes (Agaricomycetes) based on fermentation. Int. J. Med. Mushrooms 2020, 22, 299–304. [Google Scholar] [CrossRef]
- Niego, A.G.; Raspe, O.; Thongklang, N.; Charoensup, R.; Lumyong, S.; Stadler, M.; Hyde, K.D. Taxonomy, diversity and cultivation of the Oudemansielloid/Xeruloid Taxa Hymenopellis, Mucidula, Oudemansiella, and Xerula with respect to their bioactivities: A Review. J. Fungi. 2021, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Floudas, D.; Binder, M.; Riley, R.; Barry, K.; Blanchette, R.A.; Henrissat, B.; Martinez, A.T.; Otillar, R.; Spatafora, J.W.; Yadav, J.S.; et al. The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 2012, 336, 1715–1719. [Google Scholar] [CrossRef] [PubMed]
- Nagy, L.G.; Riley, R.; Tritt, A.; Adam, C.; Daum, C.; Floudas, D.; Sun, H.; Yadav, J.S.; Pangilinan, J.; Larsson, K.H.; et al. Comparative genomics of early-diverging mushroom-forming fungi provides insights into the origins of lignocellulose decay capabilities. Mol. Biol. Evol. 2016, 33, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wu, S.; Ma, X.; Chen, W.; Zhang, J.; Duan, S.; Gao, Y.; Kui, L.; Huang, W.; Wu, P.; et al. The genome sequences of 90 mushrooms. Sci. Rep. 2018, 8, 9982. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, M.; Sun, Y.; Li, Q.; Liu, J.; Song, C.; Shang, X.; Tan, Q.; Zhang, L.; Yu, H. Whole-genome sequence of a high-temperature edible mushroom Pleurotus giganteus (zhudugu). Front. Microbiol. 2022, 13, 941889. [Google Scholar] [CrossRef]
- Schafhauser, T.; Wibberg, D.; Binder, A.; Ruckert, C.; Busche, T.; Wohlleben, W.; Kalinowski, J. Genome assembly and genetic traits of the pleuromutilin-producer Clitopilus passeckerianus DSM1602. J. Fungi. 2022, 8, 862. [Google Scholar] [CrossRef]
- Li, S.; Zhao, S.; Hu, C.; Mao, C.; Guo, L.; Yu, H.; Yu, H. Whole genome sequence of an edible mushroom Stropharia rugosoannulata (Daqiugaigu). J. Fungi. 2022, 8, 99. [Google Scholar] [CrossRef]
- Geng, Y.; Zhang, S.; Yang, N.; Qin, L. Whole-genome sequencing and comparative genomics analysis of the wild edible mushroom (Gomphus purpuraceus) provide insights into its potential food application and artificial domestication. Genes 2022, 13, 1628. [Google Scholar] [CrossRef]
- Duan, Y.; Han, H.; Qi, J.; Gao, J.M.; Xu, Z.; Wang, P.; Zhang, J.; Liu, C. Genome sequencing of Inonotus obliquus reveals insights into candidate genes involved in secondary metabolite biosynthesis. BMC Genom. 2022, 23, 314. [Google Scholar] [CrossRef]
- Zhang, R.Q.; Feng, X.L.; Wang, Z.X.; Xie, T.C.; Duan, Y.; Liu, C.; Gao, J.M.; Qi, J.Z. Genomic and metabolomic analyses of the medicinal fungus Inonotus hispidus for its metabolite’s biosynthesis and medicinal application. J. Fungi 2022, 8, 1245. [Google Scholar] [CrossRef]
- Dong, W.G.; Wang, Z.X.; Feng, X.L.; Zhang, R.Q.; Shen, D.Y.; Du, S.; Gao, J.M.; Qi, J.Z. Chromosome-level genome sequences, comparative genomic analyses, and secondary-metabolite biosynthesis evaluation of the medicinal edible mushroom Laetiporus sulphureus. Microbiol. Spectr. 2022, 10, e0243922. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Nie, F.; Xie, S.Q.; Zheng, Y.F.; Dai, Q.; Bray, T.; Wang, Y.X.; Xing, J.F.; Huang, Z.J.; Wang, D.P.; et al. Efficient assembly of nanopore reads via highly accurate and intact error correction. Nat. Commun. 2021, 12, 60. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Mikheenko, A.; Prjibelski, A.; Saveliev, V.; Antipov, D.; Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 2018, 34, i142–i150. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 2003, 19, ii215–ii225. [Google Scholar] [CrossRef] [PubMed]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.; V.Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Cantalapiedra, C.P.; Hernandez-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez, R.L.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2004, 5, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef]
- van Peer, A.F.; Park, S.Y.; Shin, P.G.; Jang, K.Y.; Yoo, Y.B.; Park, Y.J.; Lee, B.M.; Sung, G.H.; James, T.Y.; Kong, W.S. Comparative genomics of the mating-type loci of the mushroom Flammulina velutipes reveals widespread synteny and recent inversions. PLoS ONE 2011, 6, e22249. [Google Scholar] [CrossRef]
- Wang, W.; Mukhtar, I.; Chou, T.; Jiang, S.; Liu, X.; van Peer, A.F.; Xie, B. A second HD mating type sublocus of Flammulina velutipes is at least di-allelic and active: New primers for identification of HD-a and HD-b subloci. PeerJ 2019, 7, e6292. [Google Scholar] [CrossRef]
- Wang, W.; Lian, L.; Xu, P.; Chou, T.; Mukhtar, I.; Osakina, A.; Waqas, M.; Chen, B.; Liu, X.; Liu, F.; et al. Advances in understanding mating type gene organization in the mushroom-forming fungus Flammulina velutipes. G3 2016, 6, 3635–3645. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Fungal Barcoding Consortium; Fungal Barcoding Consortium Author List. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef]
- Bhambri, A.; Srivastava, M.; Mahale, V.G.; Mahale, S.; Karn, S.K. Mushrooms as potential sources of active metabolites and medicines. Front. Microbiol. 2022, 13, 837266. [Google Scholar] [CrossRef]
- Tao, Y.; Xie, B.; Yang, Z.; Chen, Z.; Chen, B.; Deng, Y.; Jiang, Y.; van Peer, A.F. Identification and expression analysis of a new glycoside hydrolase family 55 exo-beta-1,3-glucanase-encoding gene in Volvariella volvacea suggests a role in fruiting body development. Gene 2013, 527, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Ge, W.; Buswell, J.A. Molecular cloning and transcriptional expression analysis of an intracellular beta-glucosidase, a family 3 glycosyl hydrolase, from the edible straw mushroom, Volvariella volvacea. FEMS Microbiol. Lett. 2007, 267, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Jin, L.; Lei, P.; Shao, C.; Wu, S.; Yang, Y.; He, Y.; Ren, R.; Xu, J. Whole-genome assembly and analysis of a medicinal fungus: Inonotus hispidus. Front. Microbiol. 2022, 13, 967135. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.; Kim, S.; Kim, M.; Ryu, J.S.; Ro, H.S. Structure analysis of A and B mating type loci in a representative commercial strain of Pleurotus eryngii. Sci. Hortic. 2020, 274, 109686. [Google Scholar] [CrossRef]
- Prasad Singh, P.; Srivastava, D.; Jaiswar, A.; Adholeya, A. Effector proteins of Rhizophagus proliferus: Conserved protein domains may play a role in host-specific interaction with different plant species. Braz. J. Microbiol. 2019, 50, 593–601. [Google Scholar] [CrossRef]
- Vincent, A.T.; Schiettekatte, O.; Goarant, C.; Neela, V.K.; Bernet, E.; Thibeaux, R.; Ismail, N.; Mohd Khalid, M.K.N.; Amran, F.; Masuzawa, T.; et al. Revisiting the taxonomy and evolution of pathogenicity of the genus Leptospira through the prism of genomics. PLoS Negl. Trop. Dis. 2019, 13, e0007270. [Google Scholar] [CrossRef]
- Lee, I.; Ouk Kim, Y.; Park, S.C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef]
- Liu, X.B.; Feng, B.; Li, J.; Yan, C.; Yang, Z.L. Genetic diversity and breeding history of Winter Mushroom (Flammulina velutipes) in China uncovered by genomic SSR markers. Gene 2016, 591, 227–235. [Google Scholar] [CrossRef]
- Floudas, D.; Held, B.W.; Riley, R.; Nagy, L.G.; Koehler, G.; Ransdell, A.S.; Younus, H.; Chow, J.; Chiniquy, J.; Lipzen, A.; et al. Evolution of novel wood decay mechanisms in Agaricales revealed by the genome sequences of Fistulina hepatica and Cylindrobasidium torrendii. Fungal Genet. Biol. 2015, 76, 78–92. [Google Scholar] [CrossRef]
- Quin, M.B.; Flynn, C.M.; Schmidt-Dannert, C. Traversing the fungal terpenome. Nat. Prod. Rep. 2014, 31, 1449–1473. [Google Scholar] [CrossRef]
- Min, B.; Kim, S.; Oh, Y.L.; Kong, W.S.; Park, H.; Cho, H.; Jang, K.Y.; Kim, J.G.; Choi, I.G. Genomic discovery of the hypsin gene and biosynthetic pathways for terpenoids in Hypsizygus marmoreus. BMC Genom. 2018, 19, 789. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Lu, D.; Wang, S.; Zhao, Y.; Gao, S.; Han, R.; Yu, J.; Zheng, W.; Geng, J.; Hu, S. Genome assembly and pathway analysis of edible mushroom Agrocybe cylindracea. Genom. Proteom. Bioinform. 2020, 18, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Liu, L.; Huang, S.; Zhang, Y.; Xu, J.; Zhang, Z. Anti-cancer Effects of a Neutral triterpene fraction from Ganoderma lucidum and its active constituents on SW620 human colorectal cancer cells. Anticancer. Agents Med. Chem. 2020, 20, 237–244. [Google Scholar] [CrossRef]
- Oide, S.; Turgeon, B.G. Natural roles of nonribosomal peptide metabolites in fungi. Mycoscience 2020, 61, 101–110. [Google Scholar] [CrossRef]
- Ilyukhin, E.; Markovskaja, S.; Elgorban, A.M.; Al-Rejaie, S.S.; Maharachchikumbura, S.S.N. Genomic characteristics and comparative genomics analysis of Parafenestella ontariensis sp. nov. J. Fungi 2022, 8, 732. [Google Scholar] [CrossRef]
- Sharma, G.; Kaur, B.; Raheja, Y.; Agrawal, D.; Basotra, N.; Di Falco, M.; Tsang, A.; Singh Chadha, B. Lignocellulolytic enzymes from Aspergillus allahabadii for efficient bioconversion of rice straw into fermentable sugars and biogas. Bioresour. Technol. 2022, 360, 127507. [Google Scholar] [CrossRef]
- Zhu, L.; Su, Y.; Ma, Z.; Guo, L.; Yang, S.; Yu, H. Comparative proteomic analysis reveals differential protein expression of Hypsizygus marmoreus in response to different light qualities. Int. J. Biol. Macromol. 2022, 223, 1320–1334. [Google Scholar] [CrossRef]
- Zheng, J.; Hu, B.; Zhang, X.; Ge, Q.; Yan, Y.; Akresi, J.; Piyush, V.; Huang, L.; Yin, Y. dbCAN-seq update: CAZyme gene clusters and substrates in microbiomes. Nucleic Acids Res. 2023, 51, D557–D563. [Google Scholar] [CrossRef]
- Tian, F.; Li, C.; Li, Y. Genomic analysis of Sarcomyxa edulis reveals the basis of its medicinal properties and evolutionary relationships. Front. Microbiol. 2021, 12, 652324. [Google Scholar] [CrossRef]
- James, T.Y.; Liou, S.R.; Vilgalys, R. The genetic structure and diversity of the A and B mating-type genes from the tropical oyster mushroom, Pleurotus djamor. Fungal Genet. Biol. 2004, 41, 813–825. [Google Scholar] [CrossRef]
- Wang, G.; Wang, Y.; Chen, L.; Wang, H.; Guo, L.; Zhou, X.; Dou, M.; Wang, B.; Lin, J.; Liu, L.; et al. Genetic structure and evolutionary diversity of mating-type (MAT) loci in Hypsizygus marmoreus. IMA Fungus 2021, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Ma, Y.; Wu, S.; Zheng, X.; Kang, H.; Sang, J.; Xu, X.; Hao, L.; Li, Z.; Gong, Z.; et al. Genome Warehouse: A public repository housing genome-scale data. Genom. Proteom. Bioinform. 2021, 19, 584–589. [Google Scholar] [CrossRef] [PubMed]
- CNCB-NGDC Members and Partners. Database resources of the national genomics data center, China National Center for Bioinformation in 2022. Nucleic Acids Res. 2022, 50, D27–D38. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).