

To the Origin of Fungi: Analysis of MFS Transporters of First Assembled Aphelidium Genome Highlights Dissimilarity of Osmotrophic Abilities between Aphelida and Fungi
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Extraction and Genome Sequencing
2.2. Genome Assembly and Annotation
2.3. The Selection and Analysis of MFS-Domain Proteins
3. Results
3.1. Assembly of Aphelidium insulamus Genome
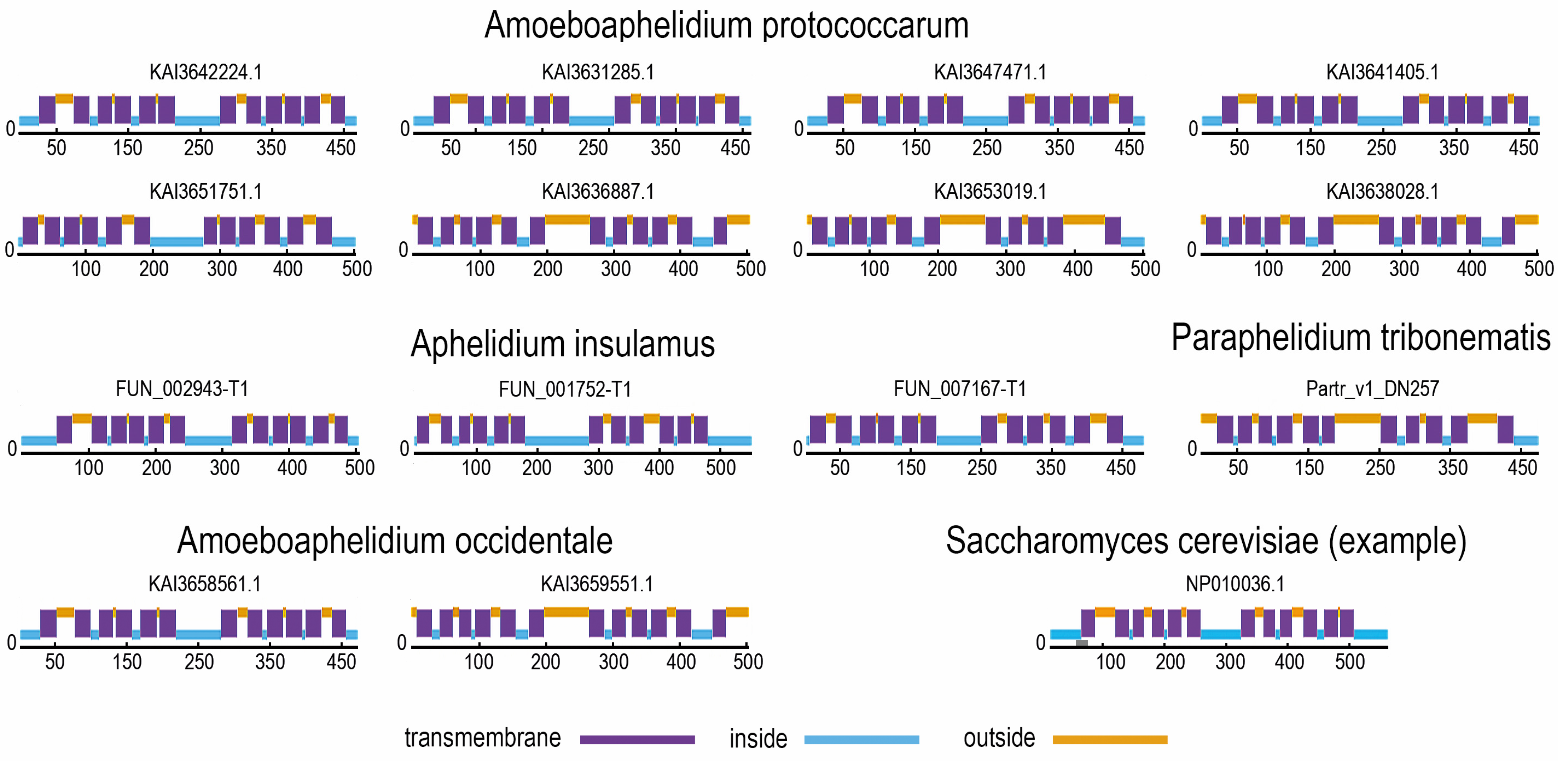
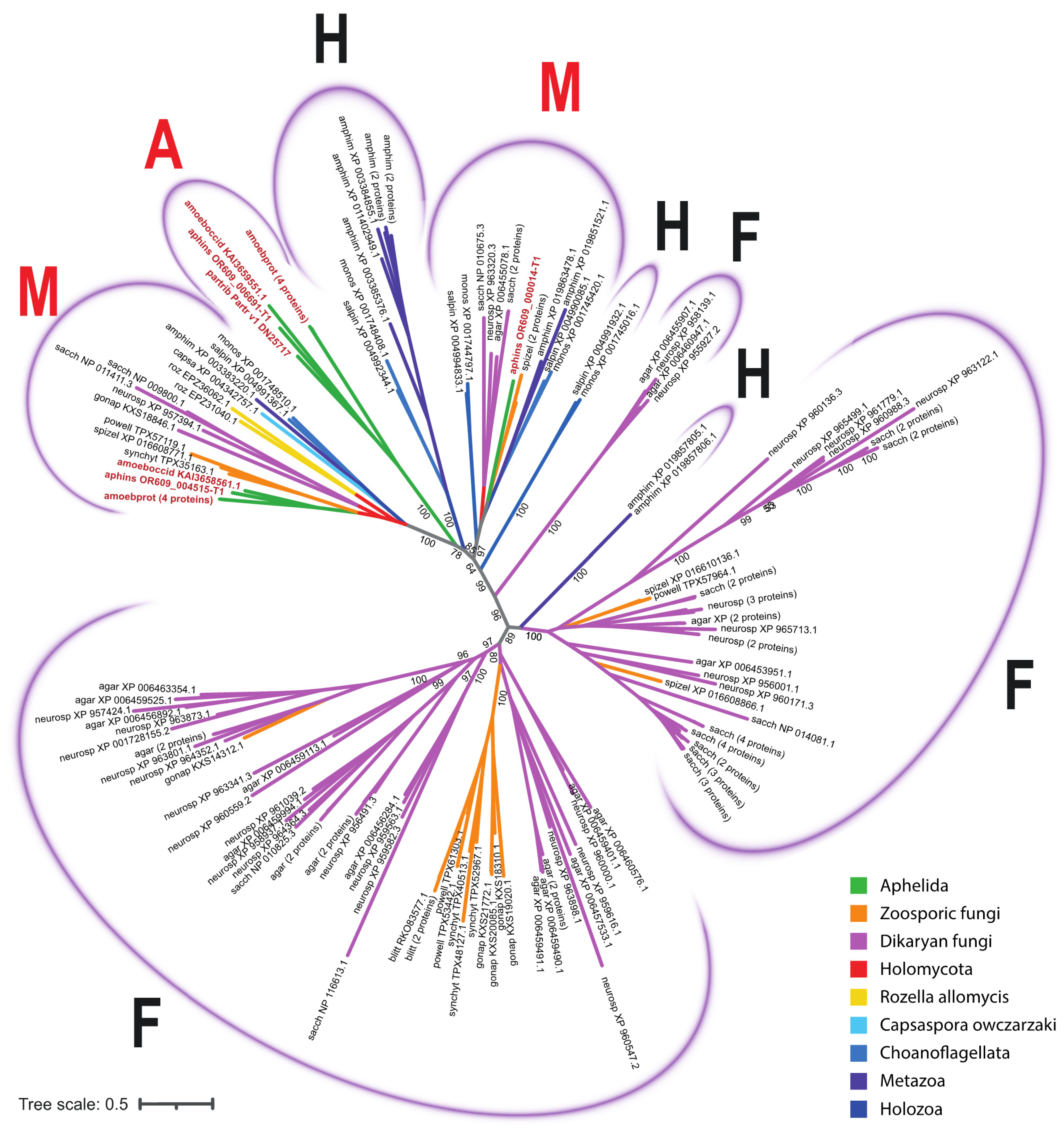
3.2. MFS Protein Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gromov, B.V. Algal parasites of the genera Aphelidium, Amoeboaphelidium and Pseudoaphelidium from the Cienkovski’s “Monadea” group as representatives of new class. Zool. Z. 2000, 79, 517–525. [Google Scholar]
- Karpov, S.A.; Mamkaeva, M.A.; Aleoshin, V.V.; Nassonova, E.; Lilje, O.; Gleason, F.H. Morphology, phylogeny, and ecology of the aphelids (Aphelidea, Opisthokonta) and proposal for the new superphylum Opisthosporidia. Front. Microbiol. 2014, 5, 112. [Google Scholar] [CrossRef] [PubMed]
- Letcher, P.M.; Lopez, S.; Schmieder, R.; Lee, P.A.; Behnke, C.; Powell, M.J.; McBride, R.C. Characterization of Amoeboaphelidium protococcarum, an algal parasite new to the cryptomycota isolated from an outdoor algal pond used for the production of biofuel. PLoS ONE 2013, 8, e56232. [Google Scholar] [CrossRef] [PubMed]
- Adl, S.M.; Bass, D.; Lane, C.E.; Lukes, J.; Schoch, C.L.; Smirnov, A.; Agatha, S.; Berney, C.; Brown, M.W.; Burki, F.; et al. Revisions to the classification, nomenclature, and diversity of eukaryotes. J. Eukaryot. Microbiol. 2019, 66, 4–119. [Google Scholar] [CrossRef] [PubMed]
- Letcher, P.M.; Powell, M.J. A taxonomic summary of Aphelidiaceae. IMA Fungus 2019, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Torruella, G.; Grau-Bové, X.; Moreira, D.; Karpov, S.A.; Burns, J.A.; Sebé-Pedrós, A.; Völcker, E.; López-García, P. Global transcriptome analysis of the aphelid Paraphelidium tribonemae supports the phagotrophic origin of fungi. Commun. Biol. 2018, 1, 231. [Google Scholar] [CrossRef]
- Tedersoo, L.; Sánchez-Ramírez, S.; Kõljalg, U.; Bahram, M.; Döring, M.; Schigel, D.; May, T.; Ryberg, M.; Abarenkov, K. High-level classification of the Fungi and a tool for evolutionary ecological analyses. Fungal Divers. 2018, 90, 135–159. [Google Scholar] [CrossRef]
- Galindo, L.J.; Torruella, G.; López-García, P.; Ciobanu, M.; Gutiérrez-Preciado, A.; Karpov, S.A.; Moreira, D. Phylogenomics supports the monophyly of aphelids and fungi and identifies new molecular synapomorphies. Syst. Biol. 2022, 72, 505–515. [Google Scholar] [CrossRef]
- Mikhailov, K.V.; Karpov, S.A.; Letcher, P.M.; Lee, P.A.; Logacheva, M.D.; Penin, A.A.; Nesterenko, M.A.; Pozdnyakov, I.R.; Potapenko, E.V.; Sherbakov, D.Y.; et al. Genomic analysis reveals cryptic diversity in aphelids and sheds light on the emergence of Fungi. Curr. Biol. 2022, 32, 4607–4619.E7. [Google Scholar] [CrossRef]
- Guarro, J.; Gené, J.; Stchigel, A.M. Developments in fungal taxonomy. Clin. Microbiol. Rev. 1999, 12, 454–500. [Google Scholar] [CrossRef]
- Margulis, L.; Chapman, M.J. (Eds.) Chapter Four—Kingdom Fungi. In Kingdoms and Domains, 4th ed.; Academic Press: Cambridge, MA, USA, 2009; pp. 379–409. [Google Scholar] [CrossRef]
- Naranjo-Ortiz, M.A.; Gabaldón, T. Fungal evolution: Diversity, taxonomy and phylogeny of the Fungi. Biol. Rev. 2019, 94, 2101–2137. [Google Scholar] [CrossRef] [PubMed]
- Richards, T.A.; Leonard, G.; Wideman, J.G. What defines the “Kingdom” Fungi? Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Henderson, P.J. The homologous glucose transport proteins of prokaryotes and eukaryotes. Res. Microbiol. 1990, 141, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Pao, S.S.; Paulsen, I.T.; Saier, M.H., Jr. Major facilitator superfamily. Microbiol. Mol. Biol. Rev. 1998, 62, 1092–2172. [Google Scholar] [CrossRef] [PubMed]
- Yan, N. Structural advances for the major facilitator superfamily (MFS) transporters. Trends Biochem. Sci. 2013, 38, 151–159. [Google Scholar] [CrossRef] [PubMed]
- InterPro—Classification of Protein Families. Major Facilitator Superfamily Domain. Available online: https://www.ebi.ac.uk/interpro/entry/InterPro/IPR020846/ (accessed on 18 July 2023).
- Reddy, V.S.; Shlykov, M.A.; Castillo, R.; Sun, E.I.; Saier, M.H. The major facilitator superfamily (MFS) revisited. FEBS J. 2012, 279, 2022–2035. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, C.; Coelho, M.A.; Salema-Oom, M.; Gonçalves, P. Stepwise functional evolution in a fungal sugar transporter family. Mol. Biol. Evol. 2016, 33, 352–366. [Google Scholar] [CrossRef]
- Merényi, Z.; Krizsán, K.; Sahu, N.; Liu, X.; Bálint, B.; Stajich, J.; Spatafora, J.W.; Nagy, L.G. Taxonomic vs genomic fungi: Contrasting evolutionary loss of protistan genomic heritage and emergence of fungal novelties. bioRxiv 2022. [Google Scholar] [CrossRef]
- Van Dijck, P.; Brown, N.A.; Goldman, G.H.; Rutherford, J.; Xue, C.; Van Zeebroeck, G. Nutrient sensing at the plasma membrane of fungal cells. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Bisson, L.F.; Coons, D.M.; Kruckeberg, A.L.; Lewis, D.A. Yeast sugar transporters. Crit. Rev. Biochem. Mol. Biol. 1993, 28, 259–308. [Google Scholar] [CrossRef]
- Goffeau, A.; Park, J.; Paulsen, I.T.; Jonniaux, J.L.; Dinh, T.; Mordant, P.; Saier, M.H. Multidrug-resistant transport proteins in yeast: Complete inventory and phylogenetic characterization of yeast open reading frames with the major facilitator superfamily. Yeast 1997, 13, 43–54. [Google Scholar] [CrossRef]
- Dias, P.J.; Sá-Correia, I. The drug:H+ antiporters of family 2 (DHA2), siderophore transporters (ARN) and glutathione:H+antiporters (GEX) have a common evolutionary origin in hemiascomycete yeasts. BMC Genom. 2013, 14, 901. [Google Scholar] [CrossRef] [PubMed]
- Karpov, S.A.; Vishnyakov, A.E.; López-García, P.; Zorina, N.A.; Ciobanu, M.; Tcvetkova, V.S.; Moreira, D. Morphology and molecular phylogeny of Aphelidium insulamus sp. nov. (Aphelida, Opisthosporidia). Protistology 2020, 14, 191–203. [Google Scholar] [CrossRef]
- Fahrni, J.F.; Bolivar, I.; Berney, C.; Nassonova, E.; Smirnov, A.; Pawlowski, J. Phylogeny of lobose amoebae based on actin and small-subunit ribosomal RNA genes. Mol. Biol. Evol. 2003, 20, 1881–1886. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P. Assembly of long error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Challis, R.; Richards, E.; Rajan, J.; Cochrane, G.; Blaxter, M. BlobToolKit–interactive quality assessment of genome assemblies. G3-Genes Genom. Genet. 2020, 10, 1361–1374. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.; Stajich, J. Funannotate v1.8.1: Eukaryotic Genome Annotation (v1.8.1). Zenodo 2020. [Google Scholar] [CrossRef]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [PubMed]
- Käll, L.; Krogh, A.; Sonnhammer, E.L. Advantages of combined transmembrane topology and signal peptide prediction—The Phobius web server. Nucleic Acids Res. 2007, 35, W429–W432. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. In Gene Prediction. Methods in Molecular Biology; Kollmar, M., Ed.; Humana: New York, NY, USA, 2019; Volume 1962. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Wheeler, T.J.; Eddy, S.R. nhmmer: DNA homology search with profile HMMs. Bioinformatics 2013, 29, 2487–2489. [Google Scholar] [CrossRef]
- Di Tommaso, P.; Moretti, S.; Xenarios, I.; Orobitg, M.; Montanyola, A.; Chang, J.M.; Taly, J.F.; Notredame, C. T-Coffee: A web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 2011, 39, W13–W17. [Google Scholar] [CrossRef]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Ocaña-Pallarès, E.; Williams, T.A.; López-Escardó, D.; Arroyo, A.S.; Pathmanathan, J.S.; Bapteste, E.; Tikhonenkov, D.V.; Keeling, P.J.; Szöllősi, G.J.; Ruiz-Trillo, I. Divergent genomic trajectories predate the origin of animals and fungi. Nature 2022, 609, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Wang, S.; Sekimoto, S.; Aerts, A.L.; Choi, C.; Clum, A.; LaButti, K.M.; Lindquist, E.A.; Ngan, C.Y.; Ohm, R.A.; et al. Phylogenomic analyses indicate that early fungi evolved digesting cell walls of algal ancestors of land plants. Genome Biol. Evol. 2015, 7, 1590–1601. [Google Scholar] [CrossRef] [PubMed]
- Tikhonenkov, D.V.; Hehenberger, E.; Esaulov, A.S.; Belyakova, O.I.; Mazei, Y.A.; Mylnikov, A.P.; Keeling, P.J. Insights into the origin of metazoan multicellularity from predatory unicellular relatives of animals. BMC Biol. 2020, 18, 39. [Google Scholar] [CrossRef] [PubMed]
- Brun, S.; Silar, P. Convergent evolution of morphogenetic processes in Fungi. In Evolutionary Biology—Concepts, Molecular and Morphological Evolution; Pontarotti, P., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 317–328. [Google Scholar] [CrossRef]
- Kożyczkowska, A.; Najle, S.R.; Ocaña-Pallarès, E.; Aresté, C.; Shabardina, V.; Ara, P.S.; Ruiz-Trillo, I.; Casacuberta, E. Stable transfection in protist Corallochytrium limacisporum identifies novel cellular features among unicellular animals relatives. Curr. Biol. 2021, 31, 4104–4110.e5. [Google Scholar] [CrossRef] [PubMed]
- Torruella, G.; de Mendoza, A.; Grau-Bové, X.; Antó, M.; Chaplin, M.A.; del Campo, J.; Eme, L.; Pérez-Cordón, G.; Whipps, C.M.; Nichols, K.M.; et al. Phylogenomics reveals convergent evolution of lifestyles in close relatives of animals and fungi. Curr. Biol. 2015, 25, 2404–2410. [Google Scholar] [CrossRef] [PubMed]
- Pozdnyakov, I.R.; Zolotarev, A.V.; Karpov, S.A. Comparative analysis of zoosporogenesis’ genes of the bastoclad Blastocladiella emersonii and the aphelid Paraphelidium tribonematis reveals the new directions of evolutionary research. Protistology 2021, 15, 10–23. [Google Scholar] [CrossRef]
- Nagy, L.G.; Kovács, G.M.; Krizsán, K. Complex multicellularity in fungi: Evolutionary convergence, single origin, or both? Biol. Rev. Camb. Philos. Soc. 2018, 93, 1778–1794. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Assembly Size (bp) | Num Scaffolds | Scaffold N50 (bp) | Average Scaffold (bp) | Largest Scaffold (bp) | GC, % | Num Genes | Num Proteins | Single Copy BUSCOs (%) |
|---|---|---|---|---|---|---|---|---|---|
| Aphelidium insulamus | 18,927,283 | 274 | 252,907 | 69,078 | 1,020,338 | 52.05 | 7925 | 7820 | 91.4% |
| Amoeboaphelidium protococcarum | 24,734,778 | 258 | 2,170,272 | 95,871 | 3,250,117 | 40.50 | 13,180 | 13,180 | 92.7% |
| Amoeboaphelidium occidentale | 13,559,732 | 951 | 73,507 | 14,258 | 366,412 | 39.93 | 7568 | 7495 | 91.4% |
| Gonapodya prolifera | 48,794,828 | 352 | 347,324 | 138,622 | 1,572,201 | 51.75 | 13,911 | 13,831 | 93.4% |
| Blyttiomyces helicus | 46,468,912 | 8398 | 6675 | 5533 | 73,981 | 53.75 | 12,446 | 12,167 | 55.4% |
| Powellomyces hirtus | 26,238,698 | 482 | 157,542 | 54,437 | 764,225 | 51.37 | 6536 | 6536 | 96.4% |
| Spizellomyces punctatus | 24,131,112 | 38 | 1,465,700 | 635,029 | 2,242,449 | 47.16 | 9164 | 9422 | 97.4% |
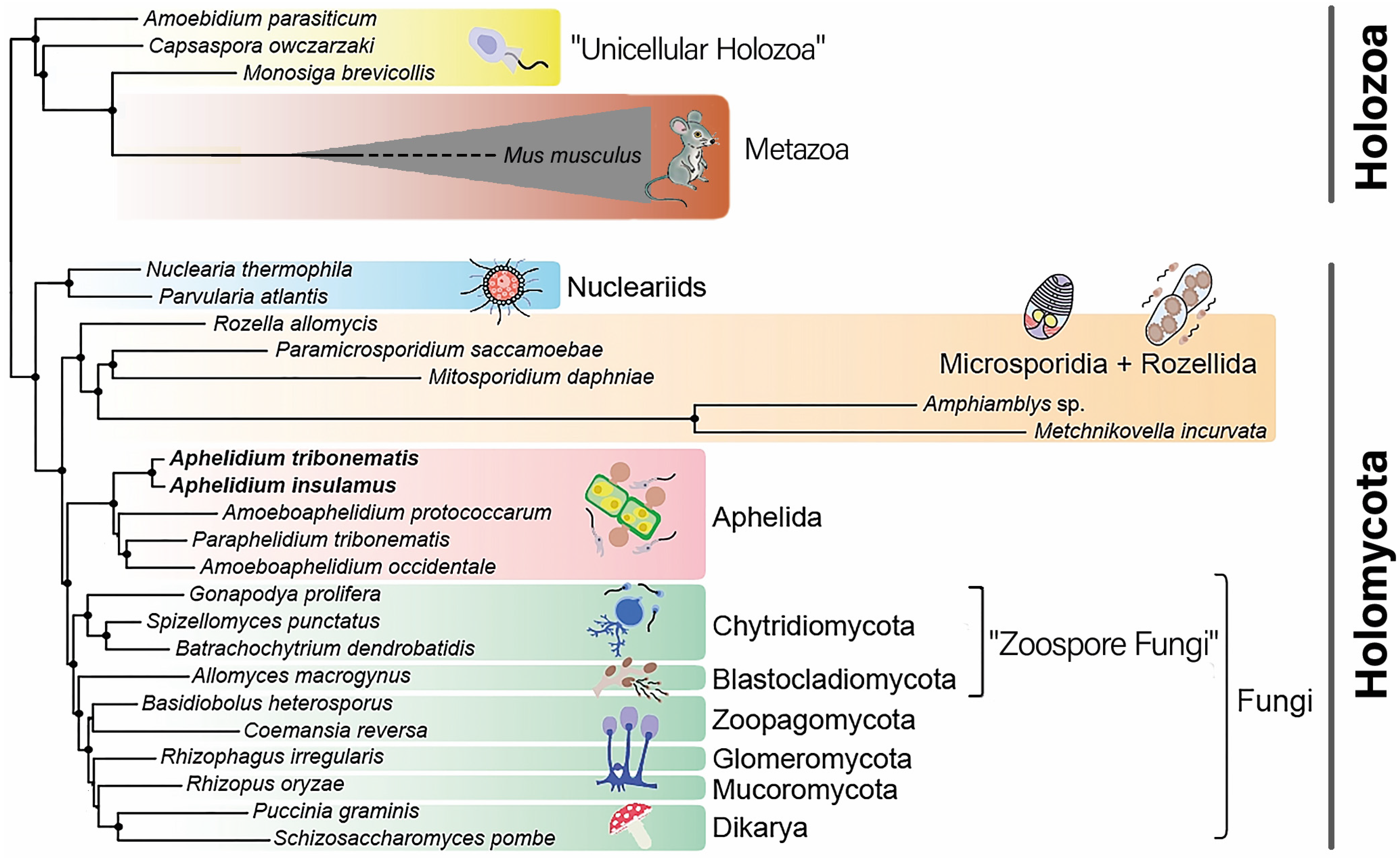
| Aphelida | Zoosporic Fungi | Dikaryan Fungi | Rozellida | Filasterea | Choanoflagellata | Metazoa |
|---|---|---|---|---|---|---|
| Species | Designation in the Phylogenetic Tree | Number of SP Proteins | Accession Numbers | |||
| Aphelidium insulamus | aphins | 3 | OR609_004515-T1 (FUN_002943-T1), OR609_000014-T1 (FUN_001752-T1), OR609_006691-T1 (FUN_007167-T1) | |||
| Paraphelidium tribonematis | partrib | 1 | Partr_v1_DN257 | |||
| Amoeboaphelidium protococcarum | amoebprot | 8 | KAI3642224.1, KAI3631285.1, KAI3647471.1, KAI3641405.1, KAI3636887.1, KAI3651751.1, KAI3653019.1, KAI3638028.1 | |||
| Amoeboaphelidium occidentale | amoeboccid | 2 | KAI3658561.1, KAI3659551.1 | |||
| Blyttiomyces helicus | blitt | 3 | RKO83577.1, RKO87217.1, RKO84726.1 | |||
| Gonapodya prolifera | gonap | 6 | KXS20085.1, KXS21772.1, KXS18310.1, KXS19020.1, KXS14312.1, KXS18846.1 | |||
| Powellomyces hirtus | powell | 4 | TPX57964, TPX61303, TPX53442, TPX57119 | |||
| Spizellomyces punctatus | spizel | 5 | XP_016610136.1, XP_016608866.1, XP_016607884.1, XP_016608771.1, XP_016607634.1 | |||
| Synchytrium endobioticum | synchyt | 4 | TPX52967.1, TPX40513.1, TPX48127.1, TPX35163.1 | |||
| Agaricus bisporus | agar | 26 | XP_006454719.1, XP_006461850.1, XP_006453951.1, XP_006456284.1, XP_006459401.1, XP_006454620.1, XP_006457533.1, XP_006460576.1, XP_006459994.1, XP_006459113.1, XP_006463002.1, XP_006459487.1, XP_006459489.1, XP_006459491.1, XP_006458691.1, XP_006459490.1, XP_006456584.1, XP_006460947.1, XP_006455907.1, XP_006455078.1, XP_006456754.1, XP_006456796.1, XP_006456892.1, XP_006463354.1, XP_006459525.1, XP_006458285.1 | |||
| Neurospora crassa | neurosp | 34 | XP_959573.2, XP_959411.2, XP_965713.1, XP_955977.1, XP_962392.2, XP_958069.2, XP_960171.3, XP_956001.1, XP_959563.1, XP_960000.1, XP_964364.3, XP_958937.1, XP_956491.3, XP_959616.1, XP_959582.3, XP_961039.2, XP_963898.1, XP_963341.3, XP_963320.3, XP_960136.3, XP_960988.3, XP_961779.1, XP_960559.2, XP_958139.1, XP_955927.2, XP_964352.1, XP_963873.1, XP_965499.1, XP_960547.2, XP_963122.1, XP_957424.1, XP_001728155.2, XP_963801.1, XP_957394.1 | |||
| Saccharomyces cerevisiae | sacch | 30 | NP_010087.1, NP_010143.1, NP_011960.2, NP_116644.1, NP_013724.1, NP_013182.1, NP_011962.1, NP_010632.1, NP_012316.1, NP_010629.3, NP_010630.1, NP_011964.1, NP_014486.1, NP_012321.1, NP_014470.1, NP_010845.1, NP_010036.1, NP_012692.3, NP_014081.1, NP_010825.3, NP_010785.1, NP_014538.2, NP_012694.1, NP_010034.1, NP_011805.3, NP_116613.1, NP_009857.1, NP_010675.3, NP_009800.1, NP_011411.3 | |||
| Rozella allomycis | roz | 2 | EPZ36062, EPZ31040 | |||
| Capsaspora owczarzaki | capsa | 1 | XP_004342757.1 | |||
| Monosiga brevicollis | monos | 5 | XP_001748408.1, XP_001745016.1, XP_001745420.1, XP_001748510.1, XP_001744797.1 | |||
| Salpingoeca rosetta | salpin | 5 | XP_004992344.1, XP_004991932.1, XP_004990085.1, XP_004994833.1, XP_004991367.1 | |||
| Amphimedon queenslandica | amphim | 13 | XP_019857806.1, XP_019857805.1, XP_019851521.1, XP_011406421.1, XP_003389392.2, XP_003383220.1, XP_019863478.1, XP_011402949.1, XP_019856859.1, XP_003384062.3, XP_003385376.1, XP_011408594.2, XP_003384855.1 | |||
| Description | Max Score | Total Score | Query Cover | E Value | Per. Ident | Acc. Len | Accession |
|---|---|---|---|---|---|---|---|
| Hypothetical protein MP228_003054 (Amoeboaphelidium protococcarum) | 1009 | 1009 | 100% | 0.0 | 100.00% | 501 | KAI3651751.1 |
| Hypothetical protein MIR68_003639 (Amoeboaphelidium protococcarum) | 882 | 882 | 100% | 0.0 | 92.66% | 503 | KAI3638028.1 |
| Hypothetical protein MP228_002444 (Amoeboaphelidium protococcarum) | 881 | 881 | 100% | 0.0 | 92.64% | 502 | KAI3653019.1 |
| Hypothetical protein MIR68_005154 (Amoeboaphelidium protococcarum) | 863 | 863 | 100% | 0.0 | 90.89% | 504 | KAI3636887.1 |
| Hypothetical protein MP638_005237 (Amoeboaphelidium occidentale) | 196 | 196 | 89% | 2 × 1052 | 30.95% | 483 | KAI3659551.1 |
| Sugar transporter family protein (Tieghemostelium lacteum) | 197 | 197 | 93% | 7 × 1052 | 30.42% | 631 | KYQ90579.1 |
| Sugar transporter family protein (Dictyostelium discoideum AX4) | 190 | 190 | 92% | 4 × 1049 | 30.29% | 630 | XP_642246.1 |
| Sugar porter family MFS transporter (Bacteroidales bacterium) | 186 | 186 | 92% | 1 × 1048 | 28.48% | 495 | MBS3771599.1 |
| Sugar porter family MFS transporter (Bacteroidales bacterium) | 185 | 185 | 92% | 2 × 1048 | 28.14% | 495 | MBS3775699.1 |
| Sugar porter family MFS transporter (Acetilactobacillus jinshanensis) | 181 | 181 | 90% | 6 × 1047 | 31.30% | 467 | WP_133441301.1 |
| Sugar porter family MFS transporter (Bacteroidota bacterium) | 181 | 181 | 92% | 6 × 1047 | 27.35% | 486 | NBC83568.1 |
| Sugar porter family MFS transporter (uncultured bacterium) | 181 | 181 | 90% | 7 × 1047 | 31.30% | 467 | URL60617.1 |
| Sugar porter family MFS transporter (Lentilactobacillus sp. SPB1-3) | 178 | 178 | 90% | 5 × 1046 | 29.00% | 467 | WP_268912157.1 |
| Sugar porter family MFS transporter (Lentilactobacillus curieae) | 177 | 177 | 90% | 6 × 46 | 29.93% | 460 | WP_035166644.1 |
| Sugar porter family MFS transporter (Secundilactobacillus hailunensis) | 176 | 176 | 90% | 4 × 45 | 30.79% | 463 | WP_137631226.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pozdnyakov, I.R.; Potapenko, E.V.; Nassonova, E.S.; Babenko, V.V.; Boldyreva, D.I.; Tcvetkova, V.S.; Karpov, S.A. To the Origin of Fungi: Analysis of MFS Transporters of First Assembled Aphelidium Genome Highlights Dissimilarity of Osmotrophic Abilities between Aphelida and Fungi. J. Fungi 2023, 9, 1021. https://doi.org/10.3390/jof9101021
Pozdnyakov IR, Potapenko EV, Nassonova ES, Babenko VV, Boldyreva DI, Tcvetkova VS, Karpov SA. To the Origin of Fungi: Analysis of MFS Transporters of First Assembled Aphelidium Genome Highlights Dissimilarity of Osmotrophic Abilities between Aphelida and Fungi. Journal of Fungi. 2023; 9(10):1021. https://doi.org/10.3390/jof9101021
Chicago/Turabian StylePozdnyakov, Igor R., Evgeniy V. Potapenko, Elena S. Nassonova, Vladislav V. Babenko, Daria I. Boldyreva, Victoria S. Tcvetkova, and Sergey A. Karpov. 2023. "To the Origin of Fungi: Analysis of MFS Transporters of First Assembled Aphelidium Genome Highlights Dissimilarity of Osmotrophic Abilities between Aphelida and Fungi" Journal of Fungi 9, no. 10: 1021. https://doi.org/10.3390/jof9101021
APA StylePozdnyakov, I. R., Potapenko, E. V., Nassonova, E. S., Babenko, V. V., Boldyreva, D. I., Tcvetkova, V. S., & Karpov, S. A. (2023). To the Origin of Fungi: Analysis of MFS Transporters of First Assembled Aphelidium Genome Highlights Dissimilarity of Osmotrophic Abilities between Aphelida and Fungi. Journal of Fungi, 9(10), 1021. https://doi.org/10.3390/jof9101021