
Genome Analyses of Two Blueberry Pathogens: Diaportheamygdali CAA958 and Diaporthe eres CBS 160.32





Abstract
1. Introduction
2. Materials and Methods
2.1. Fungal Material and Culture Conditions
2.2. DNA Extraction
2.3. Genome Sequencing, Assembly, and Prediction
2.4. Dispersed Repeat Sequences and Noncoding tRNA Annotation
2.5. Gene Annotation and Functional Analyses
2.6. Comparative Analyses
3. Results
3.1. Genome Assembly and Genomic Characteristics
3.2. Gene Prediction and Functional Annotation
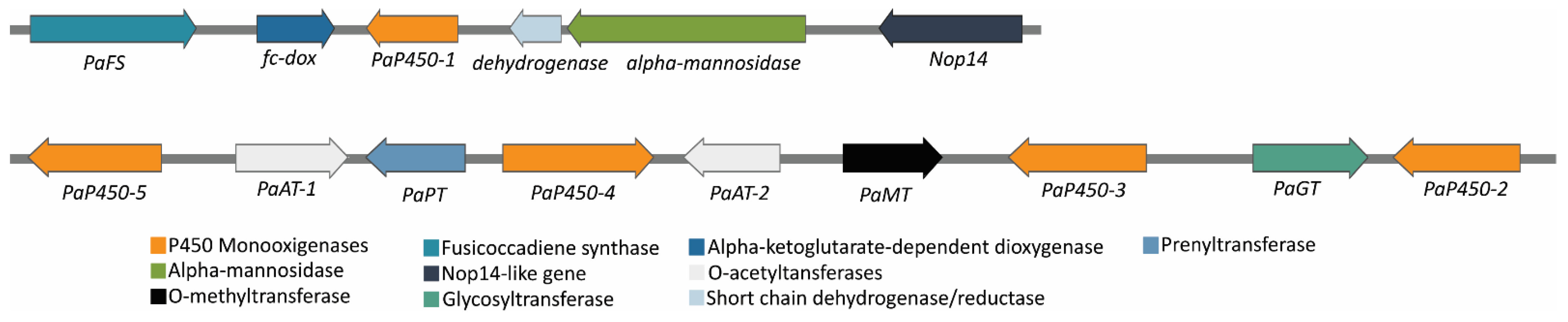
3.3. Fusicoccin A Biosynthesis
3.4. Virulence Factors, Effectors, and Strategies to Overcome Host Responses
3.5. Cellular Transporters
3.6. Comparative Analyses
3.6.1. Predicted Genes and Genome Statistics
3.6.2. CAZymes
3.6.3. BGCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Möller, M.; Stukenbrock, E.H. Evolution and genome architecture in fungal plant pathogens. Nat. Rev. Microbiol. 2017, 15, 756–771. [Google Scholar] [CrossRef]
- Aylward, J.; Steenkamp, E.T.; Dreyer, L.L.; Roets, F.; Wingfield, B.D.; Wingfield, M.J. A plant pathology perspective of fungal genome sequencing. IMA Fungus 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Ellwood, S.R.; Liu, Z.; Syme, R.A.; Lai, Z.; Hane, J.K.; Keiper, F.; Moffat, C.S.; Oliver, R.P.; Friesen, T.L. A first genome assembly of the barley fungal pathogen Pyrenophora teres f. teres. Genome Biol. 2010, 11, R109. [Google Scholar] [CrossRef] [PubMed]
- Gomes, R.R.; Glienke, C.; Videira, S.I.R.; Lombard, L.; Groenewald, J.Z.; Crous, P.W. Diaporthe: A genus of endophytic, saprobic and plant pathogenic fungi. Persoonia 2013, 31, 1–41. [Google Scholar] [CrossRef] [PubMed]
- Bhunjun, C.S.; Niskanen, T.; Suwannarach, N.; Wannathes, N.; Chen, Y.J.; McKenzie, E.H.; Maharachchikumbura, S.S.; Buyck, B.; Zhao, C.L.; Fan, Y.G.; et al. The numbers of fungi: Are the most speciose genera truly diverse? Fungal Divers. 2022, 114, 387–462. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, F.; Duan, W.; Crous, P.W.; Cai, L. Diaporthe is paraphyletic. IMA Fungus 2017, 8, 153–187. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.S.; Crous, P.W.; Bai, Q.; Fu, M.; Yang, M.M.; Wang, X.H.; Du, Y.M.; Hong, N.; Xu, W.X.; Wang, G.P. High diversity of Diaporthe species associated with pear shoot canker in China. Persoonia 2020, 45, 132–162. [Google Scholar]
- Udayanga, D.; Liu, X.; Crous, P.W.; McKenzie, E.H.; Chukeatirote, E.; Hyde, K.D. A multi-locus phylogenetic evaluation of Diaporthe (Phomopsis). Fungal Divers. 2012, 56, 157–171. [Google Scholar] [CrossRef]
- Farr, D.F.; Castlebury, L.A.; Rossman, A.Y. Morphological and molecular characterization of Phomopsis vaccinii and additional isolates of Phomopsis from blueberry and cranberry in the eastern United States. Mycologia 2002, 94, 494–504. [Google Scholar] [CrossRef]
- Varjas, V.; Vajna, L.; Izsépi, F.; Nagy, G.; Pájtli, É. First report of Phomopsis amygdali causing twig canker on almond in Hungary. Plant Dis. 2017, 101, 1674. [Google Scholar] [CrossRef]
- Marra, A.; Camoni, A.L.; Visconti, A.; Fiorillo, A.; Evident, A. The Surprising Story of Fusicoccin: A Wilt-Inducing Phytotoxin, a Tool in Plant Physiology and a 14-3-3-Targeted Drug. Biomolecules 2021, 11, 1393. [Google Scholar] [CrossRef] [PubMed]
- Guarnaccia, V.; Groenewald, J.Z.; Woodhall, J.; Armengol, J.; Cinelli, T.; Eichmeier, A.; Ezra, D.; Fontaine, F.; Gramaje, D.; Gutierrez-Aguirregabiria, A.; et al. Diaporthe diversity and pathogenicity revealed from a broad survey of grapevine diseases in Europe. Persoonia 2018, 40, 135–153. [Google Scholar] [CrossRef] [PubMed]
- Hilário, S.; Amaral, A.I.; Gonçalves, M.F.M.; Lopes, A.; Santos, L.; Alves, A. Diversity and pathogenicity of Diaporthe species on blueberry plants in Portugal, with description of 4 new species. Mycologia 2020, 112, 293–308. [Google Scholar] [CrossRef]
- Singh, S.K.; Strobel, G.A.; Knighton, B.; Geary, B.; Sears, J.; Ezra, D. An endophytic Phomopsis sp. possessing bioactivity and fuel potential with its volatile organic compounds. Microb. Ecol. 2011, 61, 729–739. [Google Scholar] [CrossRef]
- Tanney, J.B.; McMullin, D.R.; Green, B.D.; Miller, J.D.; Seifert, K.A. Production of antifungal and antiinsectan metabolites by the Picea endophyte Diaporthe maritima sp. nov. Fungal Biol. 2016, 120, 1448–1457. [Google Scholar] [CrossRef]
- Mena, E.; Garaycochea, S.; Stewart, S.; Montesano, M.; De León, I.P. Comparative genomics of plant pathogenic Diaporthe species and transcriptomics of Diaporthe caulivora during host infection reveal insights into pathogenic strategies of the genus. BMC Genom. 2022, 23, 175. [Google Scholar] [CrossRef]
- Gai, Y.; Xiong, T.; Xiao, X.; Li, P.; Zeng, Y.; Li, L.; Riely, B.K.; Li, H. The Genome Sequence of the Citrus Melanose Pathogen Diaporthe citri and Two Citrus-Related Diaporthe Species. Phytopathology 2021, 111, 779–783. [Google Scholar] [CrossRef]
- Hilário, S.; Santos, L.; Alves, A. Diversity and pathogenicity of Diaporthe species revealed from a survey of blueberry orchards in Portugal. Agriculture 2021, 11, 1271. [Google Scholar] [CrossRef]
- Hilário, S.; Gonçalves, F.M.; Alves, A. Using genealogical concordance and coalescent-based species delimitation to assess species boundaries in the Diaporthe eres complex. J. Fungi 2021, 7, 507. [Google Scholar] [CrossRef] [PubMed]
- Baroncelli, R.; Scala, F.; Vergara, M.; Thon, M.R.; Ruocco, M. Draft whole-genome sequence of the Diaporthe helianthi 7/96 strain, causal agent of sunflower stem canker. Genom. Data 2016, 10, 151–152. [Google Scholar] [CrossRef]
- Li, S.; Darwish, O.; Alkharouf, N.W.; Musungu, B.; Matthews, B.F. Analysis of the genome sequence of Phomopsis longicolla: A fungal pathogen causing Phomopsis seed decay in soybean. BMC Genom. 2017, 18, 688. [Google Scholar] [CrossRef]
- Morales-Cruz, A.; Amrine, K.C.H.; Blanco-Ulate, B.; Lawrence, D.P.; Travadon, R.; Rolshausen, P.E.; Baumgartner, K.; Cantu, D. Distinctive expansion of gene families associated with plant cell wall degradation, secondary metabolism, and nutrient uptake in the genomes of grapevine trunk pathogens. BMC Genom. 2015, 16, 469. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yao, X.; Xhang, X.; Zou, H.; Chen, J.; Fang, B.; Huang, L. Draft genome sequence of Diaporthe batatatis causing dry rot disease in sweet potato. Plant Dis. 2022, 106, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Qin, K.; Li, S.; Han, S.; Zhu, T. Whole genome sequence of Diaporthe capsici, a new pathogen of walnut blight. Genomics 2020, 112, 3751–3761. [Google Scholar] [CrossRef]
- Hilário, S.; Santos, L.; Alves, A. Diaporthe amygdali, a species complex or a complex species? Fungal Biol. 2021, 125, 505–518. [Google Scholar] [CrossRef]
- Pitcher, D.G.; Saunders, N.A.; Owen, R.J. Rapid extraction of bacterial genomic DNA with guanidium thiocyanate. Lett. Appl. Microbiol. 1989, 8, 151–156. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Babraham Bioinformatics. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2006. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 15 December 2020).
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Stanke, M.; Steinkamp, R.; Waack, S.; Morgenstern, B. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res. 2004, 32, 309–312. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2018. Institute for Systems Biology. 2015. Available online: http://www.repeat-masker.org (accessed on 10 February 2021).
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Gelfand, Y.; Rodriguez, A.; Benson, G. TRDB—The tandem repeats database. Nucleic Acids Res. 2007, 35, D80–D87. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, F.M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Mulder, N.; Apweiler, R. InterPro and InterProScan: Tools for protein sequence classification and comparison. Methods Mol. Biol. 2007, 396, 59–70. [Google Scholar] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef]
- Armenteros, J.J.A.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Hallgren, J.; Tsirigos, K.D.; Pedersen, M.D.; Armenteros, J.J.A.; Marcatili, P.; Nielsen, H.; Krogh, A.; Winther, O. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. bioRxiv 2022. [Google Scholar] [CrossRef]
- Sperschneider, J.; Dodds, P.N. EffectorP 3.0: Prediction of apoplastic and cytoplasmic effectors in fungi and oomycetes. Mol. Plant Microbe Interact. 2022, 35, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Urban, M.; Cuzick, A.; Seager, J.; Wood, V.; Rutherford, K.; Venkatesh, S.Y.; De Silva, N.; Martinez, M.C.; Pedro, H.; Yates, A.D.; et al. PHI-base: The pathogen–host interactions database. Nucleic Acids Res. 2020, 48, D613–D620. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. AntiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef]
- Yin, Y.; Mao, X.; Yang, J.; Chen, X.; Mao, F.; Xu, Y. dbCAN: A web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012, 40, W445–W451. [Google Scholar] [CrossRef] [PubMed]
- Saier, M.H., Jr.; Reddy, V.S.; Tsu, B.V.; Ahmed, M.S.; Li, C.; Moreno-Hagelsieb, G. The transporter classification database (TCDB): Recent advances. Nucleic Acids Res. 2016, 44, D372–D379. [Google Scholar] [CrossRef]
- Figueiredo, L.; Santos, R.B.; Figueiredo, A. Defense and offense strategies: The role of aspartic proteases in plant-pathogen interactions. Biology 2021, 2, 75. [Google Scholar] [CrossRef] [PubMed]
- Félix, C.; Meneses, R.; Gonçalves, M.F.; Tilleman, L.; Duarte, A.S.; Jorrín-Novo, J.V.; Van de Peer, Y.; Deforce, D.; Van Nieuwerburgh, F.; Esteves, A.C.; et al. A multi-omics analysis of the grapevine pathogen Lasiodiplodia theobromae reveals that temperature affects the expression of virulence-and pathogenicity-related genes. Sci. Rep. 2019, 9, 13144. [Google Scholar] [CrossRef] [PubMed]
- Madhu, S.N.; Sharma, S.; Gajjar, D.U. Identification of Proteases: Carboxypeptidase and Aminopeptidase as Putative Virulence Factors of Species Complex. Open Microbiol. J. 2020, 14, 266–277. [Google Scholar] [CrossRef]
- Bettini, P.P.; Frascella, A.; Kolařík, M.; Comparini, C.; Pepori, A.L.; Santini, A.; Scala, F.; Scala, A. Widespread horizontal transfer of the cerato-ulmin gene between Ophiostoma novo-ulmi and Geosmithia species. Fungal Biol. 2014, 118, 663–674. [Google Scholar] [CrossRef][Green Version]
- Temple, B.; Horgen, P.A. Biological roles for cerato-ulmin, a hydrophobin secreted by the elm pathogens, Ophiostoma ulmi and O. novo-ulmi. Mycologia 2000, 92, 1–9. [Google Scholar] [CrossRef]
- Kong, L.A.; Yang, J.; Li, G.T.; Qi, L.L.; Zhang, Y.J.; Wang, C.F.; Zhao, W.S.; Xu, J.R.; Peng, Y.L. Different chitin synthase genes are required for various developmental and plant infection processes in the rice blast fungus Magnaporthe oryzae. PLoS Pathog. 2012, 8, e1002526. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Lu, M.; Ling, E.; Li, P.; Wang, C. A M35 family metalloprotease is required for fungal virulence against insects by inactivating host prophenoloxidases and beyond. Virulence 2020, 11, 222–237. [Google Scholar] [CrossRef]
- Dong, S.; Wang, Y. Nudix Effectors: A common weapon in the arsenal of plant pathogens. PLoS Pathog. 2016, 12, e1005704. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.G.; de Curcio, J.S.; Silva-Bailão, M.G.; Lima, R.M.; Tomazett, M.V.; de Souza, A.F.; Cruz-Leite, V.R.M.; Sbaraini, N.; Bailão, A.M.; Rodrigues, F.; et al. Molecular characterization of siderophore biosynthesis in Paracoccidioides brasiliensis. IMA Fungus 2020, 11, 11. [Google Scholar] [CrossRef]
- Figueiredo, J.; Sousa Silva, M.; Figueiredo, A. Subtilisin-like proteases in plant defence: The past, the present and beyond. Mol. Plant Pathol. 2018, 19, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Reichard, U.; Léchenne, B.; Asif, A.R.; Streit, F.; Grouzmann, E.; Jousson, O.; Monod, M. Sedolisins, a new class of secreted proteases from Aspergillus fumigatus with endoprotease or tripeptidyl-peptidase activity at acidic pHs. Appl. Environ. Microbiol. 2006, 72, 1739–1748. [Google Scholar] [CrossRef] [PubMed]
- López-Berges, M.S.; Hera, C.; Sulyok, M.; Schäfer, K.; Capilla, J.; Guarro, J.; Di Pietro, A. The velvet complex governs mycotoxin production and virulence of Fusarium oxysporum on plant and mammalian hosts. Mol. Microbiol. 2013, 87, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Yamada, K.; Yabumoto, K.; Fujii, S.; Huser, A.; Tsuji, G.; Koga, H.; Dohi, K.; Mori, M.; Shiraishi, T.; et al. Saccharomyces cerevisiae SSD1 orthologues are essential for host infection by the ascomycete plant pathogens Colletotrichum lagenarium and Magnaporthe grisea. Mol. Microbiol. 2007, 64, 1332–1349. [Google Scholar] [CrossRef]
- Thammahong, A.; Dhingra, S.; Bultman, K.M.; Kerkaert, J.D.; Cramer, R.A. An Ssd1 homolog impacts trehalose and chitin biosynthesis and contributes to virulence in Aspergillus fumigatus. mSphere 2019, 4, e00244-19. [Google Scholar] [CrossRef] [PubMed]
- Palmer, G.E. Vacuolar trafficking and Candida albicans pathogenesis. Commun. Integr. Biol. 2011, 4, 240–242. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yan, J.Y.; Zhao, W.S.; Chen, Z.; Xing, Q.K.; Zhang, W.; Chethana, K.T.; Xue, M.F.; Xu, J.P.; Phillips, A.J.L.; Wang, Y.; et al. Comparative genome and transcriptome analyses reveal adaptations to opportunistic infections in woody plant degrading pathogens of Botryosphaeriaceae. DNA Res. 2018, 25, 87–102. [Google Scholar] [CrossRef]
- Nagel, J.H.; Wingfield, M.J.; Slippers, B. Increased abundance of secreted hydrolytic enzymes and secondary metabolite gene clusters define the genomes of latent plant pathogens in the Botryosphaeriaceae. BMC Genom 2021, 22, 589. [Google Scholar] [CrossRef]
- Eschenbrenner, C.J.; Feurtey, A.; Stukenbrock, E.H. Population genomics of fungal plant pathogens and the analyses of rapidly evolving genome compartments. In Statistical Population Genomics; Dutheil, J.Y., Ed.; Humana: New York, NY, USA, 2020; pp. 337–355. [Google Scholar]
- Paietta, J.V. Regulation of sulfur metabolism in mycelial fungi. In The Mycota III: Biochemistry Molecular Biology, 2nd ed.; Brambl, R., Marzluf, G.A., Eds.; Springer: Berlin/Heidelberg, Germany, 2004; pp. 369–383. [Google Scholar]
- Traynor, A.M.; Sheridan, K.J.; Jones, G.W.; Calera, J.A.; Doyle, S. Involvement of sulfur in the biosynthesis of essential metabolites in pathogenic fungi of animals, particularly Aspergillus spp.: Molecular and therapeutic implications. Front. Microbiol. 2019, 10, 2859. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Gao, Y.; Yang, A. Sulfur Homeostasis in Plants. Int. J. Mol. Sci. 2019, 21, 8926. [Google Scholar] [CrossRef] [PubMed]
- Cregut, M.; Piutti, S.; Slezack-Deschaumes, S.; Benizri, E. Compartmentalization and regulation of arylsulfatase activities in Streptomyces sp., Microbacterium sp. and Rhodococcus sp. soil isolates in response to inorganic sulfate limitation. Microbiol. Res. 2013, 168, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Paolinelli-Alfonso, M.; Villalobos-Escobedo, J.M.; Rolshausen, P.; Herrera-Estrella, A.; Galindo-Sánchez, C.; López-Hernández, J.F.; Hernandez-Martinez, R. Global transcriptional analysis suggests Lasiodiplodia theobromae pathogenicity factors involved in modulation of grapevine defensive response. BMC Genom. 2016, 17, 615. [Google Scholar] [CrossRef]
- Gonçalves, M.F.; Nunes, R.B.; Tilleman, L.; Van de Peer, Y.; Deforce, D.; Van Nieuwerburgh, F.; Esteves, A.C.; Alves, A. Dual RNA sequencing of Vitis vinifera during Lasiodiplodia theobromae infection unveils host–pathogen interactions. Int. J. Mol. Sci. 2019, 20, 6083. [Google Scholar] [CrossRef]
- Murry, R.; Traxler, L.; Pötschner, J.; Krüger, T.; Kniemeyer, O.; Krause, K.; Kothe, E. Inositol Signaling in the Basidiomycete Fungus Schizophyllum commune. J. Fungi 2021, 7, 470. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, T.B. Strategies for acquiring the phospholipid metabolite inositol in pathogenic bacteria, fungi and protozoa: Making it and taking it. Microbiology 2009, 155, 1386–1396. [Google Scholar] [CrossRef]
- Meena, M.; Prasad, V.; Zehra, A.; Gupta, V.K.; Upadhyay, R.S. Mannitol metabolism during pathogenic fungal–host interactions under stressed conditions. Front. Microbiol. 2015, 6, 1019. [Google Scholar] [CrossRef]
- Stergiopoulos, I.; Zwiers, L.H.; De Waard, M.A. Secretion of natural and synthetic toxic compounds from filamentous fungi by membrane transporters of the ATP-binding cassette and major facilitator superfamily. Eur. J. Plant Pathol. 2002, 108, 719–734. [Google Scholar] [CrossRef]
- Nogueira, K.M.; de Paula, R.G.; Antoniêto, A.C.C.; Dos Reis, T.F.; Carraro, C.B.; Silva, A.C.; Almeida, F.; Rechia, C.G.V.; Goldman, G.H.; Silva, R.N. Characterization of a novel sugar transporter involved in sugarcane bagasse degradation in Trichoderma reesei. Biotechnol. Biofuels 2018, 11, 84. [Google Scholar] [CrossRef] [PubMed]
- Colabardini, A.C.; Nicolas, L.; Ries, A.; Brown, N.A.; Fernanda, T.; Savoldi, M.; Goldman, M.H.S.; Menino, J.F.; Rodrigues, F.; Goldman, G.H. Functional characterization of a xylose transporter in Aspergillus nidulans. Biotechnol. Biofuels 2014, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.F.; Lawrence, D.P.; Morales-Cruz, A.; Travadon, R.; Minio, A.; Hernandez-Martinez, R.; Rolshausen, P.E.; Baumgartner, K.; Cantu, D. Phylogenomics of plant-associated Botryosphaeriaceae species. Front. Microbiol. 2021, 12, 652802. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.C.; de Godoy, K.F.; Bannitz-Fernandes, R.; Fabri, J.H.M.; Barbosa, M.M.F.; de Castro, P.A.; Almeida, F.; Goldman, G.H.; da Cunha, A.F.; Netto, L.E.; et al. Analyses of the three 1-Cys peroxiredoxins from Aspergillus fumigatus reveal that cytosolic Prx1 is central to H2O2 metabolism and virulence. Sci. Rep. 2018, 8, 12314. [Google Scholar] [CrossRef]
- Jimenez-Jimenez, S.; Hashimoto, K.; Santana, O.; Aguirre, J.; Kuchitsu, K.; Cárdenas, L. Emerging roles of tetraspanins in plant inter-cellular and inter-kingdom communication. Plant Signal. Behav. 2019, 14, e1581559. [Google Scholar] [CrossRef] [PubMed]
- Noike, M.; Liu, C.; Ono, Y.; Hamano, Y.; Toyomasu, T.; Sassa, T.; Kato, N.; Dairi, T. An enzyme catalyzing O-Prenylation of the glucose moiety of Fusicoccin A, a diterpene glucoside produced by the fungus Phomopsis amygdali. ChembBioChem 2012, 13, 566–573. [Google Scholar] [CrossRef]
- Ohkanda, J. Fusicoccin: A chemical modulator for 14-3-3 proteins. Chem. Lett. 2021, 50, 57–67. [Google Scholar] [CrossRef]
- Noike, M.; Ono, Y.; Araki, Y.; Tanio, R.; Higuchi, Y.; Nitta, H.; Hamano, Y.; Toyomasu, T.; Sassa, T.; Kato, N.; et al. Molecular breeding of a fungus producing a precursor diterpene suitable for semi-synthesis by dissection of the biosynthetic machinery. PLoS ONE 2012, 7, e42090. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Graniti, A. Fusicoccin: A fungal toxin that opens stomata. Nature 1969, 223, 1070–1071. [Google Scholar] [CrossRef]
- Feldman, A.W.; Graniti, A.; Sparapano, L. Effect of fusicoccin on abscission, cellulase activity and ethylene production in citrus leaf explants. Physiol. Plant Pathol. 1971, 1, 115–122. [Google Scholar] [CrossRef]
- Lanfermeijer, F.C.; Prins, H. Modulation of H+ -ATPase activity by fusicoccin in plasma membrane vesicles from oat (Avena sativa L.) roots (A comparison of modulation by fusicoccin, trypsin, and lysophosphatidylcholine). Plant Physiol. 1994, 104, 1277–1285. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ballio, A.; Michelis, M.I.; Lado, P.; Randazzo, G. Fusicoccin structure-activity relationships: Stimulation of growth by cell enlargement and promotion of seed germination. Physiol. Plant 1981, 52, 471–475. [Google Scholar] [CrossRef]
- Chen, H.; Singh, H.; Bhardwaj, N.; Bhardwaj, S.K.; Khatri, M.; Kim, K.H.; Peng, W. An exploration on the toxicity mechanisms of phytotoxins and their potential utilities. Crit. Rev. Environ. Sci. Technol. 2022, 52, 395–435. [Google Scholar] [CrossRef]
- Ohm, R.A.; Feau, N.; Henrissat, B.; Schoch, C.L.; Horwitz, B.A.; Barry, K.W.; Condon, B.J.; Copeland, A.C.; Dhillon, B.; Glaser, F.; et al. Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes fungi. PLoS Pathog 2012, 8, e1003037. [Google Scholar] [CrossRef]
- Baroncelli, R.; Amby, D.B.; Zapparata, A.; Sarrocco, S.; Vannacci, G.; Le Floch, G.; Harrison, R.J.; Holub, E.; Sukno, S.A.; Sreenivasaprasad, A.; et al. Gene family expansions and contractions are associated with host range in plant pathogens of the genus Colletotrichum. BMC Genom. 2016, 17, 555. [Google Scholar] [CrossRef] [PubMed]
- Rafiei, V.; Vélëz, H.; Tzelepis, G. The role of glycoside hydrolases in phytopathogenic fungi and oomycetes virulence. Int. J. Mol. Sci. 2021, 22, 9359. [Google Scholar] [CrossRef]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef]
- Lyu, X.; Shen, C.; Fu, Y.; Xie, J.; Jiang, D.; Li, G.; Cheng, J. Comparative genomic and transcriptional analyses of the carbohydrate-active enzymes and secretomes of phytopathogenic fungi reveal their significant roles during infection and development. Sci. Rep. 2015, 5, 15565. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, J.; Yan, J.; Guo, M.; Xu, L.; Hou, L.; Zou, Q. Comparative genome analysis of plant ascomycete fungal pathogens with different lifestyles reveals distinctive virulence strategies. BMC Genom. 2022, 23, 34. [Google Scholar] [CrossRef]
- Udayanga, D.; Liu, X.; McKenzie, E.H.C.; Chukeatirote, E.; Bahkali, A.H.A.; Hyde, K.D. The genus Phomopsis: Biology, applications, species concepts and names of common phytopathogens. Fungal Divers. 2011, 50, 189–225. [Google Scholar] [CrossRef]
- Cardinaals, J.; Wenneker, M.; Voogd, B.; Van Leeuwen, M. Pathogenicity of Diaporthe spp. on two blueberry cultivars (Vaccinium corymbosum). EPPO Bull. 2018, 48, 128–134. [Google Scholar] [CrossRef]
- Howlett, B.J. Secondary metabolite toxins and nutrition of plant pathogenic fungi. Curr. Opin. Plant Biol. 2006, 9, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Tsuge, T.; Harimoto, Y.; Akimitsu, K.; Ohtani, K.; Kodama, M.; Akagi, Y.; Egusa, M.; Yamamoto, M.; Otani, H. Host-selective toxins produced by the plant pathogenic fungus Alternaria alternata. FEMS Microbiol. Rev. 2013, 37, 44–66. [Google Scholar] [CrossRef]
- Wang, M.; Fu, H.; Ruan, R. A small horizontally transferred gene cluster contributes to the sporulation of Alternaria alternata. Genome Biol. Evol. 2019, 12, 3436–3444. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Features | D. amygdali | D. eres |
|---|---|---|
| Genome assembled | 51.5 Mbp | 60.8 Mbp |
| Number of contigs (>500 bp) | 267 | 2524 |
| Largest contig length | 4,327,563 bp | 1,105,552 bp |
| N50 contig length | 1,008,325 bp | 169,851 bp |
| N75 contig length | 622,097 bp | 74,774 bp |
| GC content | 52.1% | 47.6% |
| BUSCO * completeness | 98.3% | 98.4% |
| Predicted genes | 15,818 | 16,499 |
| Predicted proteins with signal peptides | 1874 | 1806 |
| Secreted proteins | 1562 | 1616 |
| Candidate effectors | 109 | 98 |
| Total length of coding genes | 23,649,268 bp | 24,024,391 |
| Average length of predicted genes | 1495 bp | 1456 bp |
| Total length of predicted genes/Genome assembled | 45.9% | 39.5% |
| Average number of exons per gene | 3 | 3 |
| Average number of introns per gene | 2 | 2 |
| Type a | D. amygdali CAA958 | D. eres CBS 160.32 | |||||
|---|---|---|---|---|---|---|---|
| Number | Total Length (bp) | Genome Content (%) | Number | Total Length (bp) | Genome Content (%) | ||
| Interspersed and terminal repeats | LTRs | 131 | 12,096 | 0.0235 | 140 | 12,642 | 0.0208 |
| DNA transposons | 174 | 9942 | 0.0193 | 143 | 11,693 | 0.0192 | |
| LINEs | 18 | 1207 | 0.0023 | 21 | 1611 | 0.0027 | |
| SINEs | 0 | 0 | 0 | 0 | 0 | 0 | |
| Rolling circles | 4 | 243 | 0.0005 | 0 | 0 | 0 | |
| Small RNA | 55 | 8582 | 0.0167 | 52 | 8523 | 0.0140 | |
| Satellites | 25 | 1927 | 0.0037 | 18 | 1434 | 0.0024 | |
| Simple repeats | 11,895 | 486,262 | 0.9445 | 15,264 | 686,233 | 1.1289 | |
| Low complexity | 1067 | 51,681 | 0.1004 | 1571 | 77,250 | 0.1271 | |
| TOTAL | 13,369 | 571,940 | 1.1109 | 17,209 | 799,386 | 1.3151 | |
| Tandem repeats | 8639 | 478,007 | 0.9459 | 33,522 | 2,237,060 | 3.6802 | |
| tRNAs | 162 | 15,038 | 0.0292 | 177 | 17,216 | 0.0283 | |
| Putative Protein | D. amygdali CAA958 | D. eres CBS 160.32 | Function | References |
|---|---|---|---|---|
| Acid aspartase | √ | × | Role in the mechanisms of virulence during fungal infection, participating in the degradation of the host’s physical barriers | [48] |
| Aminobutyrate aminotransferase | √ | √ | Metabolization of γ-aminobutyric acid, providing pathogen nitrogen requirements during infection | [49] |
| Aminopeptidase, carboxypeptidase | √ | √ | Protease required by fungi for host peptide degradation during pathogenesis | [50] |
| Cerato-ulmi | √ | × | Hydrophobic proteins secreted by filamentous fungi (Ophiostoma species). It possesses properties of a wilt toxin in susceptible elms, such as Ulmus americana | [51,52] |
| Chitin synthases | √ | √ | Enzymes that serve as a pathogen-associated molecular pattern (PAMP), triggering immune responses in host plants. Reported in Magnaporthe oryzae, Botrytis cinerea, Fusarium graminearum, and F. verticillioides | [53] |
| Metalloprotease | √ | √ | Zinc-chelating protease that plays an essential role in microbial pathogenesis. In M. oryzae, it is an effector that triggers host defense response | [54] |
| Nudix proteins | √ | √ | Important virulence components manipulating host defense mechanisms | [55] |
| Siderophores | √ | √ | Chelators synthesized to be involved in iron uptake, intracellular transport, and storage. Essential virulence factors allow the fungus to overcome severe iron limitation imposed by the host | [56] |
| Subtilisin-like serine protease | √ | √ | Proteases that are released in infected plant host to degrade pathogenesis-related proteins and disrupt host cell membranes | [57] |
| Tripeptidyl-peptidase | √ | × | Acidification of the microenvironment in the host facilitates the proliferation of the pathogen | [58] |
| Velvet proteins | √ | √ | Promotion of chromatin accessibility and expression of biosynthetic gene clusters involved in pathogenicity as mycotoxins, pigments, and hormones | [59] |
| Virulence protein SSD1 | √ | √ | Important for M. grisea to colonize rice leaves, leading to evasion and tolerance of the host immune response | [60,61] |
| Vacuole protein sorting | √ | √ | Proteins involved in the delivery of soluble vacuolar compounds, metabolite storage, and osmoregulation. Essential for fungal growth and pathogenesis | [62] |
| Transporter Class | D. amygdali CAA958 | D. eres CBS 160.32 |
|---|---|---|
| Channels and pores (TC 1) | 348 | 348 |
| Electrochemical potential-driven transporters (TC 2) | 973 | 911 |
| Primary active transporters (TC 3) | 366 | 371 |
| Group translocators (TC 4) | 48 | 39 |
| Transmembrane electron carriers (TC 5) | 14 | 13 |
| Accessory factors involved in transport (TC 8) | 270 | 266 |
| Incompletely characterized transport systems (TC 9) | 306 | 290 |
| TOTAL | 2325 | 2238 |
| Species | Strain | Host | BUSCO * Completeness % | Genome Size (Mb) | GC Content % | Predicted Genes | Secreted Proteins | CAZymes | BGCs | GenBank Accession Number |
|---|---|---|---|---|---|---|---|---|---|---|
| Diaporthe ampelina | DA912 | Grapevine | 98.7 | 53.4 | 52.8 | 10,704 | ND | 696 | 105 | LWAD01000000 |
| Diaporthe amygdali | CAA958 | Blueberry | 98.3 | 51.5 | 52.1 | 15,818 | 1562 | 856 | 86 | This study |
| Diaporthe batatas | CRI 302-4 | Sweet potato | 97.9 | 54.4 | 50.6 | 13,037 | 1224 | 941 | 91 | JAHWGW000000000 |
| Diaporthe capsici | GY-Z16 | Walnut | 98.4 | 57.6 | 51.3 | 14,425 | 1488 | 843 | 103 | WNXA00000000 |
| Diaporthe caulivora | D57 | Soybean | 97.8 | 57.8 | 52.9 | 18,385 | 1501 | ND | ND | ND |
| Diaporthe citri | ZJUD2 | Citrus | 98.5 | 59.6 | 47.9 | 15,218 | 1860 | 847 | 98 | JADAZQ000000000 |
| Diaporthe citriasiana | ZJUD30 | Citrus | 99.2 | 52.4 | 52.0 | 13,839 | 1643 | 796 | 89 | JADWDH000000000 |
| Diaporthe citrichinensis | ZJUD34 | Citrus | 98.3 | 54.5 | 54.1 | 15,928 | 2043 | 925 | 110 | JADAZR000000000 |
| Diaporthe eres (syn. D. vaccinii) | CBS 160.32 | Blueberry | 98.4 | 60.8 | 47.6 | 16,499 | 1616 | 859 | 88 | This study |
| Diaporthe helianthi | DHEL01 | Sunflower | 98.3 | 63.6 | 43.9 | 13,139 | 1433 | 764 | 67 | MAVT02000001 |
| Diaporthe longicolla | MSPL 10–6 | Soybean | 98.2 | 62.0 | 48.6 | 16,597 | 1535 | 1221 | 174 | AYRD00000000 |
| Classes | Total Number of Genes | Secreted CAZymes | ||
|---|---|---|---|---|
| D. amygdali | D. eres | D. amygdali | D. eres | |
| GT | 107 | 108 | 3 | 10 |
| GH | 404 | 398 | 235 | 40 |
| CBM | 20 | 25 | 11 | 4 |
| AA | 230 | 225 | 131 | 26 |
| CE | 63 | 66 | 44 | 5 |
| PL | 33 | 37 | 30 | 3 |
| TOTAL | 857 | 859 | 454 | 88 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hilário, S.; Gonçalves, M.F.M.; Fidalgo, C.; Tacão, M.; Alves, A. Genome Analyses of Two Blueberry Pathogens: Diaportheamygdali CAA958 and Diaporthe eres CBS 160.32. J. Fungi 2022, 8, 804. https://doi.org/10.3390/jof8080804
Hilário S, Gonçalves MFM, Fidalgo C, Tacão M, Alves A. Genome Analyses of Two Blueberry Pathogens: Diaportheamygdali CAA958 and Diaporthe eres CBS 160.32. Journal of Fungi. 2022; 8(8):804. https://doi.org/10.3390/jof8080804
Chicago/Turabian StyleHilário, Sandra, Micael F. M. Gonçalves, Cátia Fidalgo, Marta Tacão, and Artur Alves. 2022. "Genome Analyses of Two Blueberry Pathogens: Diaportheamygdali CAA958 and Diaporthe eres CBS 160.32" Journal of Fungi 8, no. 8: 804. https://doi.org/10.3390/jof8080804
APA StyleHilário, S., Gonçalves, M. F. M., Fidalgo, C., Tacão, M., & Alves, A. (2022). Genome Analyses of Two Blueberry Pathogens: Diaportheamygdali CAA958 and Diaporthe eres CBS 160.32. Journal of Fungi, 8(8), 804. https://doi.org/10.3390/jof8080804