Abstract
A new ascomycetous species of Parafenestella was isolated from Acer negundo during the survey of diseased trees in Southern Ontario, Canada. The species is morphologically similar to other taxa of Cucurbitariacea (Pleosporales). The new species is different from the extant species in the morphology of ascospores, culture characteristics and molecular data. The novel species is described as Parafenestella ontariensis sp. nov. based on morphological and multi-gene phylogenetic analyses using a combined set of ITS, LSU, tef1 and tub2 loci. Additionally, the genome of P. ontariensis was sequenced and analyzed. The phylogenomic analysis confirmed the close relationship of the species to the fenestelloid clades of Cucurbitariaceae. The comparative genomics analysis revealed that the species lifestyle appears to be multitrophic (necrotrophic or hemi-biotrophic) with a capability to turn pathogenic on a corresponding plant host.
1. Introduction
The genus Parafenestella typified by P. pseudoplatani was introduced by Jaklitsch et al. [1]. The genus is included in the family of Cucurbitariaceae (Pleosporales) which is characterized by ostiolate ascomata seated on basal stromatic structure, with cylindrical asci and pigmented, muriform ascospores [2]. The recent studies discovered a number of new species of Parafenestella on different plant hosts worldwide [3,4]. There are 15 species epithets in Index Fungorum, but only 14 species of Parafenestella are legitimate. Parafenestella mackenziei was synonymized with P. faberi (www.indexfungorum.org, accessed 23 January 2022). The species of Parafenestella are mainly fungicolous (associated with perithecial ascomycetes), saprobic or necrotrophic on woody plants [1,4].
The species recognition within the genus Parafenestella and other Cucurbitariaceae is mainly based on morphology characteristics accompanied with multi-gene (SSU-ITS-LSU, rpb2, tef1, tub2) phylogenetic analyses. This approach reveals both cryptic and new species.
The rapidly developing new generation sequencing technologies provide high-quality, cost-effective genomic data that can be applied to different analyses. It has been found that phylogenomic approach can improve the resolution of phylogenetic trees used to resolve taxonomic uncertainties or support species reclassification [5]. One such study found out that a polyphagous plant pathogen, Corynespora olivacea, was previously misclassified at the family level [6]. Genomic data are also employed for comparative genomics analysis that may reveal genomic underpins for lifestyle adaptations within the set of close organisms and ultimately help define a species lifestyle [7].
The abilities to successfully colonize plant host tissues and further degrade complex plant carbohydrates are important aspects of fungal lifestyles [8]. In particular, plant-associated fungi require specific sets of genes involved in the production of various secondary metabolites (SM) and enzymes needed for interaction with a host. The genes involved in SM biosynthesis pathways are often clustered in a fungal genome [9]. SMs are bioactive, small molecules that are not essential for organism growth. However, they are involved in producing toxic compounds used by necrotrophic, polyphagous fungi to kill a range of cells in plant hosts [10]. Identifying the pathways involved in the production of SMs via the search for the genes encoding key enzymes (e.g., polyketide synthase (PKS)) is crucial for understanding fungus’ capability to swiftly infect a plant host. The SM complement in fungi was found to be specific to different lifestyles [11].
Carbohydrate-active enzymes (CAZYs) play an important role in the invasion of plant hosts by fungi. Fungal species can produce different kinds of CAZYs involved in the breakdown, biosynthesis or modification of glycoconjugates, oligo- and polysaccharides [12]. Plant pathogenic and endophytic fungi produce various enzymes (e.g., glycoside hydrolase (GH)) to deconstruct cell wall polysaccharide families involved in cellulose and hemicellulose hydrolysis. CAZY content also varies among fungal species and often shows adaptation to nutrition type [13].
It is assumed that a comprehensive comparison of the abundance and distribution of SMs and CAZYs between the Cucurbitariaceae species and the model species with different lifestyles will reveal the Parafenestella ontariensis’ lifestyle and its aptitude to be pathogenic on a related plant host. To date, no draft genome sequences have been published for Parafenestella spp. The availability of a high-quality draft genome sequence of P. ontariensis alongside the genomes of other species of Cucurbitariaceae will also contribute to the understanding additional aspects of plant–fungus interactions (e.g., virulence determinants) for this little-studied family.
This paper describes a new species of Parafenestella collected from Acer negundo in Ontario, Canada. Both morphology characteristics and phylogenetic analysis of four-gene sequence data support the introduction of the new species, P. ontariensis. The new taxon was established based on the recent recommendations for description of novel fungal species [14]. The phylogenomic analysis using the protein sequences of P. ontariensis confirmed the species position in Cucurbitariaceae (Pleosporales) and its close relationship to the fenestelloid taxa of the family. For comparative genomics analysis, P. ontariensis was included in a subset of the Cucurbitariaceae species (Cucurbitaria berberidis and Fenestella fenestrata) for better data representation. The analysis revealed that the lifestyle of the members of the Cucurbitariaceae family could be both necrotrophic and hemi-biotrophic (multitrophic), with a capability to become pathogenic on a corresponding plant host.
2. Materials and Methods
2.1. Sample Collection and Isolation
During the survey of forestry areas in the Niagara region (Ontario, Canada) in April 2020, the branches of A. negundo expressing dieback symptoms were collected for further study. Single spore isolates from the sampled ascomata were obtained following the method described in Phookamsak et al. [15]. The isolates were plated on malt extract agar (MEA) and incubated at room temperature (20–25 °C) in the dark. Germinating ascospores were aseptically transferred onto fresh MEA plates and stored under the same conditions. The purified representative isolate EI-6 was selected for the species description, studying micro-morphology of asexual morph and sequencing. The specimen was deposited in the Herbarium of the Nature Research Centre, Institute of Botany, Vilnius, Lithuania and the Canadian National Mycological Herbarium, Ottawa, ON, Canada.
2.2. Morphological Analysis
The morphological description was performed using microscopy techniques with dissecting (AmScope SE306R-PZ) and compound (AmScope B120C-E5) microscopes. Microscopic observations were made in distilled water. The description and measures of asexual stage were carried out using the pure cultures of the representative isolate. Fruiting structures (10 ascomata with ostioles and 10 pycnidia) were measured for the macromorphological characteristics. Micromorphological structures of sexual (20 asci, 50 ascospores) and asexual (50 conidia) morph were further measured. The obtained measures were recorded as a range of minimum and maximum values in parentheses. Picturing was performed with a 5 MP digital AmScope camera MD500 supplied with AmScopeX software (AmScope, Irvine, CA, USA).
2.3. DNA Extraction, PCR Amplification and Sequencing
Genomic DNA (gDNA) was extracted from pure culture using DNeasy PowerSoil Pro Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. The internal transcribed spacer (ITS) region was amplified with the primer pair ITS1/ITS4 [16]. The primer pair LR0R/LR5 [17] was used to amplify large subunit rRNA (LSU). PCR amplifications were executed using C-1000 thermal cycler (Bio-Rad Laboratories, Hercules, CA, USA) under the conditions described in the references for each region. The quality of PCR products was examined using electrophoresis in 1% agarose gel. All the procedures and Sanger sequencing were carried out at Genome Quebec Innovation Centre (Montreal, QC, Canada).
2.4. Sequence Alignment and Phylogenetic Analysis
The isolate identification was first conducted by standard BLASTn search using the obtained ITS and LSU sequences. Then, the sequences of all available Parafenestella spp. were extracted to perform phylogenetic analysis. The sequence dataset of the fenestelloid species [4] was further employed to build the final sequence matrix. The sequences were initially aligned using CLUSTAL-X2 [18] and manually edited employing MEGA-X [19]. Some characters were trimmed from both ends of the alignments to approximate the length of the obtained sequences to those included in the dataset. Phylogenetic analyses were executed using randomized accelerated maximum likelihood (RAxML) v. 8.0 method [20] for maximum likelihood (ML) analysis, and MEGA-X [19] was employed for maximum parsimony (MP) analysis. Bayesian posterior (BP) probabilities were defined using MrBayes v.3.2.7 [21].
The ML analysis was performed using the transitional unequal (TIM2) substitution model [22] with gamma-distributed rate of heterogeneity and proportion of invariant sites selected with ModelTest-NG v.0.1.7 [23]. The branch support was estimated with bootstrapping of 1000 replicates [24]. To execute the MP analysis, heuristic search with random sequence additions and tree bisection and reconnection algorithm were selected. Max-trees were set to 500, branches of zero length were collapsed and all equally parsimonious trees were saved. The BP values were defined by Markov chain Monte Carlo (MCMC) sampling with the general time reversible (GTR) model. Six simultaneous Markov chains were run for 100,000 generations. The first 1000 trees were discarded and the remaining trees were used to calculate BP in the majority rule consensus tree. The phylograms were visualized using FigTree v. 1.4.4 [25]. The newly generated sequences were deposited in GenBank (Table 1). The alignments used in the analyses were submitted to TreeBase (www.treebase.org; ID: S29577; accessed 14 April 2022).

Table 1.
Strains used in phylogenetic analysis with their GenBank numbers. Strain from this study is in bold. Holo-, neo- and epi-types are marked with *. Sequences retrieved from genome sequence (GCA_021596325) are marked with **.
2.5. DNA Extraction, Library Preparation, Genome Sequencing and Assembly
Highly purified total gDNA was extracted from the fungal mycelia obtained from the 12 d-old culture of the representative isolate EI-6 using DNeasy PowerSoil Pro Kit (Qiagen, Hilden, Germany). gDNA was quantified using the Quant-iT PicoGreen dsDNA Assay Kit (Life Technologies, Carlsbad, CA, USA). Libraries were generated using the NEBNext Ultra II DNA Library Preparation Kit for Illumina (New England Biolabs, Ipswich, MA, USA). The genome was sequenced with the Illumina NovaSeq 6000 platform using 150 bp pair-end sequencing strategy. All the procedures were performed at Genome Quebec Innovation Centre (Montreal, Canada). The raw reads were filtered with Trimmomatic v. 0.38.1 [26] and assembled into scaffolds using SPAdes v. 3.12.1 [27]. The genome assembly characteristics were accessed using QUAST v. 5.0.2 [28]. Repeat sequence content was identified with RepeatMasker v. 4.0.9 [29] using the Repbase library of repeats for fungi [30]. The completeness of assembly was estimated with BUSCO v.4.0.5 based on the dataset ascomycota_odb10 [31].
2.6. Gene Prediction and Functional Annotation
Gene prediction was performed using Augustus v. 3.4.0 [32]. Augustus was trained with the gene structures of the representative genome of C. berberidis (CBS 394.84) which were obtained with the MAKER2 pipeline v. 2.31.11 [33]. The EuKariotic Orthologous Group (KOG) distribution was identified employing the eggNOG 5.0 dataset [34]. The CAZY annotation was performed using dbCAN2 with the HMMER-based classification tool [35]. The SM backbone genes were predicted using the SMIPS tool [36].
2.7. Phylogenomic Analysis and Genome Sequence Alignment
The recently published protein sequence dataset of Pleosporales species [6] with some modifications was employed for the phylogenomic analysis. The proteomes for 18 species were retrieved from the MycoCosm portal [37] and GenBank genome database (Table 2). Orthofinder v. 2.5.4 [38] was used to identify the single-copy orthogroup (SCO) within all species included in the analysis. Multiple sequence alignment was performed using MAFFT v. 7.489 [39]. The ML tree was inferred with FastTree v. 2.1.10 [40]. Phylogram was visualized employing FigTree v.1.4.4 [25]. Genome alignment between genome sequences was performed with MashMap v. 2.0 [41] and displayed as a dot-plot graph using D-Genies [42].
Table 2.
Strains of Pleosporales species used in phylogenomic analyses. Strain from this study is in bold. NA—data not available.
3. Results
3.1. Taxonomy
Cucurbitariaceae G. Winter [as Cucurbitarieae], Rabenh. Krypt. -Fl., Edn 2 (Leipzig) 1.2: 308. 1885.
Parafenestella Jaklitsch & Voglmayr, in Jaklitsch et al., Stud. Mycol. 90: 108. 2018.
Parafenestella ontariensis Ilyukhin & Markovsk., sp. nov. Figure 1.

Figure 1.
(A) Ascomata of P. ontariensis on Acer negundo. (B) Ascus. (C–E) Ascospores. (F) Peridium. (G) Fourteen-day-old culture on MEA. Scale bars: A = 200 μm, B = 20 μm, C–E = 10 μm, F = 5 μm.
MycoBank number: MB 843298.
Etymology—Epithet refers to the geographical region, vicinities of the lake of Ontario (Ontario, Canada), where the species was found.
Saprophytic or pathogenic on branches of Acer negundo. Sexual morph: Ascomata (210–) 265–340(–370) μm (n = 10) diameter, subglobose to globose, black, solitary or aggregated in groups, immersed in bark, some on conidiomata of Diaporthe sp., dark brown, thick-walled and ostiolate. Ostioles 65–130(–150) μm (n = 10) diameter, indistinct, black and rounded. Peridium 15–45 μm thick, pseudoparenchymatous, consisting of isodiametric cells 5–10 μm wide, thick-walled, dark brown and t. angularis to globulosa. Hamathecium consists of numerous branched and anastomosing paraphyses, 2–3.5 μm wide with free ends. Asci (151–)165–195(–215) × (15.5–)17.5–20.5(–23.0) μm (n = 20), cylindrical, bitunicate, with an ocular chamber, short stipe and knob-like base, containing eight ascospores in uniseriate arrangement. Ascospores (22.5–)26.0–32.5(–36.0) × 10.0–12.0(–13.5) μm (n = 50), oblong, fusoid or narrowly ellipsoid with rounded ends, muriform, constricted at the median septum, usually with three to five transverse and one to two longitudinal septa, pale or yellowish brown when young, turning brown to dark brown at maturity, thick-walled and smooth.
Culture characteristics and asexual morph: colony slow-growing (13 mm) on MEA at room temperature in the dark after 7 days, initially white, dense, with thick texture, turning brownish, later grey-brown, darker at the center, covered with brown aerial mycelium; reverse side brown to grey brown. Pycnidia appearing after 12 days, light brown to dark brown (texture angularis), globose, solitary, rare, 65–120 in diameter (n = 10). Conidia (3.2–)3.5–4(–4.5) × (1.0–)1.2–1.5(–1.7) μm (n = 50), 1-celled, oblong or allantoid, sometimes attenuated towards one end, hyaline, with two subterminal drops and smooth.
Notes: Based on morphological characteristics and phylogenetic analysis, P. ontariensis is close to P. alpina sampled from the twigs of Cotoneaster integerrimus in Austria [4]. Both species have similar morphological characteristics but ascospores of P. ontariensis are slightly longer (22.5–36 µm) than those of P. alpina (19–35µm). P. alpina also has wider (10.5–15.5 µm) and more septate ascospores. Lack of brown color of the P. alpina cultures grown on corn malt agar (CMA) also makes these two species different [4]. Culture color of P. alpina is dark grey to olivaceous, meanwhile P. ontariensis is grey brown.
Material examined: CANADA, Ontario, St. Catharines, N43°12′45.3″, W79°14′33.6″, sexual morph (ascomata) was collected from branches of Acer negundo, 29 April 2020, E. Ilyukhin (Holotype: BILAS 51516, ex-type culture BILAS 51517 = EI-6, isotype DAOM 984987).
3.2. Phylogenetic and Phylogenomic Analyses
A combined set of ITS, LSU, tef1 and tub2 sequence data of 29 Cucurbitariaceae species was analyzed to define the taxonomic placement of the isolate EI-6 and infer its relationships at the intrageneric level. The performed RAxML analysis produced the phylogenetic tree with the topology similar to the multi-gene tree obtained in the recent study on fenestelloid clades of Cucurbitariaceae [4]. Individually generated single gene trees did not show conflicts in phylogenetic signals as well (data not shown). The best scoring ML tree with a log likelihood value of −3046.830912 is depicted in Figure 2. The isolate of the novel species P. ontariensis was clustered separately and formed a subclade with P. alpina. The new species was nested within the well-supported clade of Parafenestella spp. (96% ML, 51% MP, 1.00 BP).
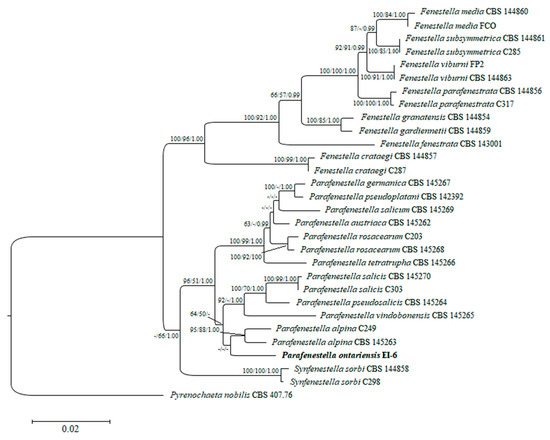
Figure 2.
Phylogram of RAxML tree generated based on the analysis of combined ITS, LSU, tef1 and tub2 sequence data. Bootstrap support values for ML and MP ≥ 50% and BP ≥ 0.90 are defined as ML/MP/BP above and below the nodes. Strain of the new species is in bold. The tree is rooted to Pyrenochaeta nobilis (CBS 407.76).
A total of 4 789 orthogroups present in all species included in the analysis with 3 145 SCOs were uncovered. The phylogenomic tree constructed from the matrix of protein sequence data has shown the strain P. ontariensis EI-6 clustered with F. fenestrata ATCC 66461. This clade was placed within the Cucurbitariaceae family of Pleosporales confirming the family level classification results obtained from the phylogenetic analysis. All the clades were formed with 100% ML support (Figure 3A).
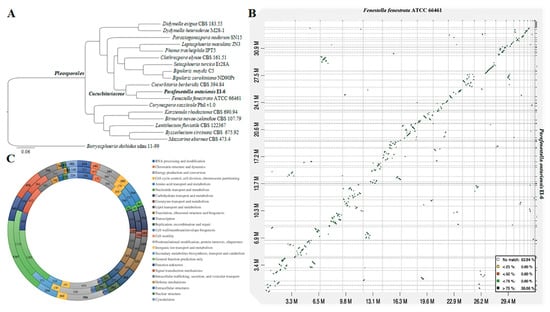
Figure 3.
(A) ML tree generated from the concatenated alignment of 3 145 SCOs. (B) Dot-plot graph showing syntenic blocks between scaffold sequences of F. fenestrata and P. ontariensis. (C) Graph showing the KOG gene annotations of C. berberidis (inner), F. fenestrata (middle) and P. ontariensis (outer layer).
The genome sequence comparison was performed between P. ontariensis and the closely allied F. fenestrata. The analysis showed that 36.1% of syntenic blocks were more than 75% similar with several inversions (Figure 3B).
3.3. Genome Sequencing and Assembly Characteristics of Parafenestella ontariensis and Other Closely Related Cucurbitariaceae Species
The draft genome sequence of P. ontariensis EI-6 was characterized and compared to other publicly available genomes of Cucurbitariaceae (C. berberidis CBS 394.84, F. fenestrata ATCC 66461). The assembly statistics of the species are presented in Table 3. The genome of P. ontariensis was sequenced with higher coverage (267.4×) than the genomes of C. berberidis and F. fenestrata (145.7× and 81.9×, respectively) and assembled into 162 scaffolds (>1000 bp) with the size of 34.4 Mb. That was slightly larger than the genomes of other Cucurbitariaceae species (32.7–32.9 Mb) arranged into 42 (C. berberidis) and 22 (F. fenestrata) scaffolds. The genome of C. berberidis had a lower genomic GC content (49.3%) compared to P. ontariensis (50.79%) and F. fenestrata (51.53). Despite the smaller genome size, 12, 429 gene models were predicted for C. berberidis and 11, 804 for F. fenestrata, which is a larger number than for P. ontariensis (11, 748). The repeat contents of the genomes were found to be relatively low (1.59% for P. ontariensis, 2.24% for C. berberidis and 3.91% for F. fenestrata). The genome assembly completeness assessed by BUSCO indicated that the genomes used in the analysis were nearly complete (97.2–98.1%) which makes the sequences suitable for further analysis.
Table 3.
Assembly statistics of P. ontariensis and other close Cucurbitariaceae species.
Of the total protein sequences analyzed, 7 895 for C. berberidis, 6 473 for F. fenestrata and 6 694 for P. ontariensis were functionally assigned based on the KOG hits. The KOG analysis revealed that the protein allocation pattern of P. ontariensis was very similar to the other two species of Cucurbitariaceae, indicating that the species may have the same ecological niches and lifestyles. It has also been found that C. berberidis had more genes involved in carbohydrate transport and metabolism (424) as well as signal transduction (612). F. fenestrata had more genes related to secondary metabolite biosynthesis, transport and catabolism (371). P. ontariensis contained a similar number of genes related in these important processes (Figure 3C).
3.4. Secondary Metabolites
The genomes of the Cucurbitariaceae species, including P. ontariensis, were searched for the genes encoding the key enzymes (NRPS (non-ribosomal peptide synthetase), PKS (polyketidesynthase), HYBRID (PKS-NRPS) and DMATS (dimethylallyl tryptophane synthase). The identified SMs were compared to the SM content of the model species with different lifestyles (Botryosphaeria dothidea sdau11-99 (B. dothidea), Colletotrichum higginsianum MAff305635-RFP (C. higginsianum), Laccaria bicolor S238N-H28 (L. bicolor), Neurospora crassa 73 trp3 (N. crassa), Peltaster fructicola LNHT1506 (P. fructicola), Pyrenophora tritici-repensis Pt-1C-BFP (P. tritici-repensis), Pyricularia oryzae 70-15 (P. oryzae), Puccinia graminis 21-0 (P. graminis), Rhizopus oryzae GL49 (R. oryzae), Valsa (Cytospora) mali 03-8 (V. mali) and Zymoseptoria tritici IPO323 (Z. tritici)). The members of the Cucurbitariaceae family were found to contain a moderate number of the genes encoding SMs (29 in C. bereberidis, 26 in F. fenestrata and P. ontariensis). The Cucurbitariaceae species had a significantly lower number of SM clusters compared to the hemi-biotrophic C. higginsianum (74) or necrotrophic V. mali (47), B. dothidea (44). At the same time, it was notably higher than those for other lifestyles (e.g., saprotrophic R. oryzae (5) and symbiotic L. laccata (7)) (Figure 4A). Interestingly, C. berberidis had an almost identical complement of SMs (except DMATS) as necrotrophic P. tritici-repensis. The clustering analysis demonstrated that the Cucurbitariaceae species were grouped with hemi-biotrophic P. oryzae. It has also shown that the members of the Cucurbitariaceae family had a relatively expanded set of genes encoding NRPS (Figure 4B).
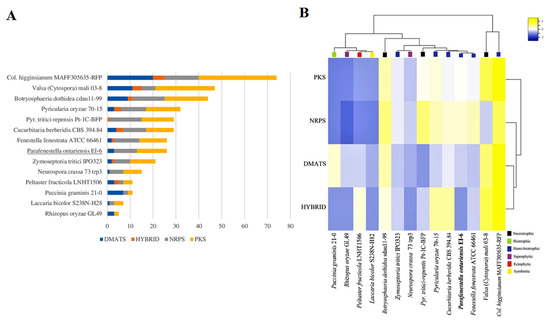
Figure 4.
(A) Overall comparison of SM backbone genes in genomes of Cucurbitariaceae species and 11 other fungi with different lifestyles. (B) Heatmap graph showing SM clusters in Cucurbitariaceae and other fungal species with different lifestyles.
3.5. Carbohydrate Active Enzymes
The CAZY content analysis was performed for the same set of species. It was found that the Cucurbitariaceae species were well equipped with the genes encoding CAZYs. The species contained an extensive set of auxiliary activity (AA) enzymes, carbohydrate esterases (CE), glycoside hydrolases (GH) and polysaccharide lyase (PL), which indicated their capacity to degrade plant cell walls effectively. In particular, the genomes of P. ontarienis were found to have 250 CAZY-related genes, C. berberidis (230) and F. fenestrata (206) (Figure 5A). It has also shown that the CAZY complement of P. ontariensis was very similar to necrotrophic V. mali. However, these two species carried a different numbers of genes involved in cellulose and hemicellulose degradation. Broadly, the species of Cucurbitariaceae had a much more expanded set of CAZYs than ectophytic P. fructucola (92), while it was significantly reduced compared to necrotrophic B. dothidea (372). The cluster analysis further confirmed the high similarity of CAZY content of the Cucurbitariaceae species with V. mali (Figure 5B). It was also demonstrated that the members of Cucurbitariaceae (P. ontariensis, especially) could encode a relatively high number of the genes involved in cellulose degradation. The further analysis of the selected CAZY families [11] has shown that the Cucurbitariaceae species formed a separate cluster with 25 modules (seven hemicellulose, six pectin, five cellulose, four cutin, three chitin) which are relatively expanded in at least two species in the sub-set compared to other species included in the analysis (Figure 6).
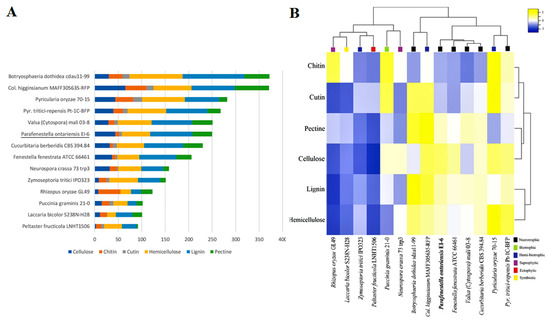
Figure 5.
(A) Overall comparison of CAZY complements in genomes of Cucurbitariaceae species and fungi with different lifestyles. (B) Heatmap graph showing CAZY expansion in Cucurbitariaceae and 11 other fungal species related to different lifestyles.
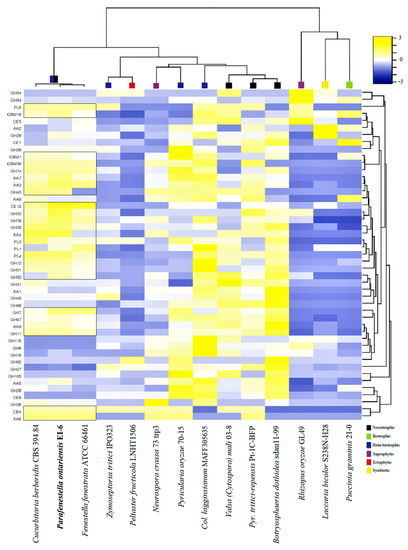
Figure 6.
Distribution of selected plant cell wall degrading enzymes among Cucurbitariaceae species and other fungi with different lifestyles.
4. Discussion
The Cucurbitariaceae family is a heterogeneous group of Ascomycota (Pleosporales) that includes the species with necrotrophic or saprobic lifestyles on woody plants [2,43]. The recently introduced genus Parafenestella was characterized as a phylogenetically close taxon to Fenestella, mainly associated with Diaporthales spp [1]. Morphologically, the new species P. ontariensis meets the genus diagnosis of lacking well-delimited pseudostromata and ascospores that would indicate possible transition from fenestella-like to (neo) cucurbitaria-like species [4].
The genome sequence obtained for the isolate P. ontarienis EI-6 allowed the comparison of the species with other members of the Cucurbitriaceae family and the model fungal species with different lifestyles. The genome size of the Cucurbitariaceae species is similar to the average for Ascomycota (36.9 Mb) [44]. However, it is significantly smaller compared to some pathogenic ascomycetes (e.g., Colletotrichum truncatum of Glomerellaceae 55.4 Mb [45], Phomopsis longicolla of Diaporthaceae 64.7 Mb [46]). Considering relatively low similarity with multiple inversions between the genome sequences of P. ontarienis and F. fenestrata, a mesosynteny-like pattern can be assumed. The pattern was observed for Dothideomycetes, indicating a random reshuffling of gene content with the genes remaining conserved [47].
The fungal lifestyles can be distinguished based on the SM clusters, which were found to be notably expanded in necrotrophic and hemi-biotrophic fungal species compared to biotrophic, saprobic, ectophytic and symbiotic fungi [11]. The SMs (e.g., PKS) are involved in important processes such as producing mycotoxins needed for initial host colonization. It has also been found that PKS and NRPS are rapidly evolving genes in fungi during gene transfer, loss and duplication [48]. P. ontariensis and other Cucurbitariacea species have arsenals of SMs which are similar to both hemi-biotrophic and necrotrophic fungi. For instance, the SM content of the Cucurbitariacea species is close to hemi-biotrophic P. oryzae, a well-known fungal pathogen causing rice blast [49]. It was also observed that C. berberidis had the same number of SM clusters with similar distribution as P. tritici-repensis, a necrotrophic pathogen causing destructive foliar wheat disease [50]. Overall, the members of Cucurbitariaceae were well equipped with the NRPS backbone genes related to virulence processes in fungal pathogens [51].
The CAZY repertoires were also defined and classified for necrotrophic, biotrophic, hemi-biotrophic, saprophytic, ectophytic and symbiotic fungi. The fungal lifestyles were well delimited based on this classification. It has been shown that necrotrophic and hemi-biotrophic fungi have notably more genes related to plant cell wall degrading enzymes (e.g., PL1, GH61) than biotrophic and symbiotic fungi [13]. The members of the Cucurbitariaceae family have a relatively expanded set of CAZYs involved in penetrating plant cell walls needed for the successful colonization of a plant host. Based on the CAZY component, the Cucurbitariaceae species were clustered separately with the strain V. mali 03-8, a highly virulent pathogen causing Valsa canker on apple trees [52]. Another finding was that the Cucurbitariaceae species (P. ontariensis, especially) had a large arsenal of CAZYs involved in cellulose degradation that was previously noticed for saprotrophic fungi [53].
Based on results of the comparative genomics analysis, it can be concluded that P. ontarienis and other close Cucurbitariacea species may have a multitrophic lifestyle. A species can become necrotrophic or hemi-biotrophic under specific circumstances (e.g., host shift). This kind of lifestyle was also assumed for saprotrophic Phomopsis liquidambari [54] and endophytic Sarocladium brachiariae [55]. To confirm lifestyle switch or transition, more species of Cucurbitariaceae with different ecological characteristics should be sequenced and analyzed using the comparative genomics approach. The identified arsenals of SMs and CAZYs can cause the Cucurbitariaceae species to be pathogenic on a related plant host. However, pathogenicity assays should be performed to observe the fungus’ capability to cause a symptomatic disease that eventually leads to plant death.
Author Contributions
Conceptualization, E.I., S.M. and S.S.N.M.; methodology, E.I. and S.M.; formal analysis, E.I., S.M. and S.S.N.M.; writing—original draft preparation, E.I.; writing—review and editing, S.M., S.S.N.M. and A.M.E. and S.S.A.-R.; funding acquisition, A.M.E. and S.S.A.-R. All authors have read and agreed to the published version of the manuscript.
Funding
Researchers supporting project number (RSP-2021/120), King Saud University, Riyadh, Saudi Arabia.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The datasets generated for this study can be found in the GenBank (NCBI), TreeBASE, MycoBank and MycoCosm.
Acknowledgments
We acknowledge Genome Quebec for providing sequencing services. Abdallah M. Elgorban and Salim S. Al-Rejaie are thanked for financial support related to the project.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jaklitsch, W.; Checa, J.; Blanco, M.; Olariaga, I.; Tello, S.; Voglmayr, H. A Preliminary Account of the Cucurbitariaceae. Stud. Mycol. 2018, 90, 71–118. [Google Scholar] [CrossRef] [PubMed]
- Hyde, K.D.; Jones, E.B.G.; Liu, J.-K.; Ariyawansa, H.; Boehm, E.W.A.; Boonmee, S.; Braun, U.; Chomnunti, P.; Crous, P.W.; Dai, D.-Q.; et al. Families of Dothideomycetes. Fungal Divers. 2013, 63, 1–313. [Google Scholar] [CrossRef]
- Crous, P.W.; Wingfield, M.J.; Lombard, L.; Roets, F.; Swart, W.J.; Alvarado, P.; Carnegie, A.J.; Moreno, G.; Luangsa-Ard, J.; Thangavel, R.; et al. Fungal Planet Description Sheets: 951–1041. Persoonia 2019, 43, 223–425. [Google Scholar] [CrossRef] [PubMed]
- Jaklitsch, W.M.; Voglmayr, H. Fenestelloid Clades of the Cucurbitariaceae. Persoonia 2020, 44, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Haridas, S.; Albert, R.; Binder, M.; Bloem, J.; LaButti, K.; Salamov, A.; Andreopoulos, B.; Baker, S.; Barry, K.; Bills, G.; et al. 101 Dothideomycetes Genomes: A Test Case for Predicting Lifestyles and Emergence of Pathogens. Stud. Mycol. 2020, 96, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Dal’Sasso, T.C.D.S.; Rody, H.V.S.; Grijalba, P.E.; Oliveira, L.O. Genome Sequences and In Silico Effector Mining of Corynespora cassiicola CC_29 and Corynespora olivacea CBS 114450. Arch. Microbiol. 2021, 203, 5257–5265. [Google Scholar] [CrossRef]
- Rodrigues, C.M.; Takita, M.A.; Silva, N.V.; Ribeiro-Alves, M.; Machado, M.A. Comparative Genome Analysis of Phyllosticta citricarpa and Phyllosticta capitalensis, Two Fungi Species that Share the Same Host. BMC Genom. 2019, 20, 554. [Google Scholar] [CrossRef]
- Stauber, L.; Prospero, S.; Croll, D. Comparative Genomics Analyses of Lifestyle Transitions at the Origin of an Invasive Fungal Pathogen in the Genus Cryphonectria. Ecol. Evol. Sci. 2020, 5, 00737–20. [Google Scholar] [CrossRef]
- Lind, A.L.; Wisecaver, J.H.; Lameiras, C.; Wiemann, P.; Palmer, J.M.; Keller, N.P.; Rodrigues, F.; Goldman, G.H.; Rokas, A. Drivers of Genetic Diversity in Secondary Metabolic Gene Clusters within a Fungal Species. PLoS Biol. 2017, 15, 2003583. [Google Scholar] [CrossRef] [Green Version]
- Amselem, J.; Cuomo, C.A.; van Kan, J.A.; Viaud, M.; Benito, E.P.; Couloux, A.; Coutinho, P.M.; de Vries, R.P.; Dyer, P.S.; Fillinger, S.; et al. Genomic Analysis of the Necrotrophic Fungal Pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet. 2011, 7, 1002230. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Liang, X.; Gleason, M.L.; Zhang, R.; Sun, G. Comparative Genomics of Botryosphaeria dothidea and B. kuwatsukai, Causal Agents of Apple Ring Rot, Reveals both Species Expansion of Pathogenicity–Related Genes and Variations in Virulence Gene Content during Speciation. IMA Fungus 2018, 9, 243–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombard, V.; Golaconda, R.H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The Carbohydrate–Active Enzymes Database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, 490–495. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Liu, H.; Wang, C.; Xu, R. Comparative Analysis of Fungal Genomes Reveals Different Plant Cell Wall Degrading Capacity in Fungi. BMC Genom. 2013, 14, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aime, M.C.; Miller, A.N.; Aoki, T.; Bensch, K.; Cai, L.; Crous, P.W.; Hawksworth, D.L.; Hyde, K.D.; Kirk, P.M.; Lücking, R.; et al. How to Publish a New Fungal Species, or Name, Version 3.0. IMA Fungus 2021, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Phookamsak, R.; Norphanphoun, C.; Tanaka, K.; Dai, D.-Q.; Luo, Z.-L.; Liu, J.-K.; Su, H.-Y.; Bhat, D.J.; Bahkali, A.H.; Mortimer, P.E.; et al. Towards a Natural Classification of Astrosphaeriella-like Species; Introducing Astrosphaeriellaceae and Pseudoastrosphaeriellaceae fam. nov. and Astrosphaeriellopsis, gen. nov. Fungal Divers. 2015, 74, 143–197. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.D.; Lee, S.; Taylor, J.W. Amplification and Direct Sequencing of Fungal Ribosomal RNA Genes for Phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J., White, T.J., Eds.; Academic Press: New York, NY, USA, 1990; pp. 312–322. [Google Scholar]
- Vilgalys, R.; Hester, M. Rapid Genetic Identification and Mapping of Enzymatically Amplified Ribosomal DNA from Several Cryptococcus Species. J. Bacteriol. 1990, 172, 4238–4246. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position–Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, S. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian Inference of Phylogenetic Trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [Green Version]
- Posada, D.; Buckley, T. Model Selection and Model Averaging in Phylogenetics: Advantages of Akaike Information Criterion and Bayesian Approaches Over Likelihood Ratio Tests. Syst. Biol. 2004, 53, 793–808. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest–NG: A New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillis, D.M.; Bull, J.J. An Empirical Test of Bootstrapping as a Method for Assessing Confidence in Phylogenetic Analysis. Syst. Biol. 1993, 42, 182–192. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree, version 1.4.4; Institute of Evolutionary Biology, University of Edinburgh: Edinburgh, UK, 2018.
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and its Applications to Single–Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Mikheenko, A.; Prjibelski, A.; Saveliev, V.; Antipov, D.; Gurevich, A. Versatile Genome Assembly Evaluation with QUAST–LG. Bioinformatics 2018, 34, 142–150. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Hubley, R.; Green, P. 2013–2015 RepeatMasker Open–4.0. Available online: http://www.repeatmasker.org (accessed on 4 February 2022).
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a Database of Repetitive Elements in Eukaryotic Genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness with Single–Copy Orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A Web Server for Gene Prediction in Eukaryotes that Allows User–Defined Constraints. Nucleic Acids Res. 2005, 33, 465–467. [Google Scholar] [CrossRef] [Green Version]
- Cantarel, B.L.; Korf, I.; Robb, S.M.C.; Parra, G.; Ross, E.; Moore, B.; Holt, C.; Alvarado, A.S.; Yandell, M. MAKER: An Easy-to-Use Annotation Pipeline Designed for Emerging Model Organism Genomes. Genome Res. 2008, 18, 188–196. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.V.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. EggNOG 5.0: A Hierarchical, Functionally and Phylogenetically Annotated Orthology Resource Based on 5090 Organisms and 2502 Viruses. Nucleic Acids Res. 2019, 47, 309–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Mao, X.; Yang, J.; Chen, X.; Mao, F.; Xu, Y. dbCAN: A Web Resource for Automated Carbohydrate-Active Enzyme Annotation. Nucleic Acids Res. 2012, 40, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Wolf, T.; Shelest, V.; Nath, N.; Shelest, E. CASSIS and SMIPS: Promoter–Based Prediction of Secondary Metabolite Gene Clusters in Eukaryotic Genomes. Bioinformatics 2016, 32, 1138–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grigoriev, I.V.; Nikitin, R.; Haridas, S.; Kuo, A.; Ohm, R.A.; Otillar, R.; Riley, R.; Salamov, A.A.; Zhao, X.; Korzeniewski, F.; et al. MycoCosm Portal: Gearing up for 1000 Fungal Genomes. Nucleic Acids Res. 2014, 42, 699–704. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving Fundamental Biases in Whole Genome Comparisons Dramatically Improves Orthogroup Inference Accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2-Approximately Maximum–Likelihood Trees for Large Alignments. PLoS ONE. 2010, 5, e9490. [Google Scholar] [CrossRef]
- Jain, C.; Koren, S.; Dilthey, A.T.; Phillippy, A.M.; Aluru, S. A Fast Adaptive Algorithm for Computing Whole–Genome Homology Maps. Bioinformatics 2018, 34, 748–756. [Google Scholar] [CrossRef] [Green Version]
- Cabanettes, F.; Klopp, C.D. GENIES: Dot Plot Large Genomes in an Interactive, Efficient and Simple Way. PeerJ 2018, 6, 4958. [Google Scholar] [CrossRef]
- Wanasinghe, D.N.; Phookamsak, R.; Jeewon, R.; Li, W.J.; Hyde, K.D.; Gareth Jones, E.B.; Erio, C.; Promputtha, I. A Family Level rDNA Based Phylogeny of Cucurbitariaceae and Fenestellaceae with Descriptions of New Fenestella Species and Neocucurbitaria gen. nov. Mycosphere 2017, 8, 397–414. [Google Scholar] [CrossRef]
- Mohanta, T.K.; Bae, H. The Diversity of Fungal Genome. Biol. Proced. Online 2015, 17, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.; Nandineni, M.R. Genome Sequencing and Comparative Genomics Reveal a Repertoire of Putative Pathogenicity Genes in Chilli Anthracnose Fungus Colletotrichum truncatum. PLoS ONE 2017, 12, 0183567. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Song, Q.; Ji, P.; Cregan, P. Draft Genome Sequence of Phomopsis longicolla Type Strain TWH P74, a Fungus Causing Phomopsis Seed Decay in Soybean. Genome Announc. 2015, 3, e00010-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hane, J.K.; Williams, A.H.; Oliver, R.P. 9 Genomic and Comparative Analysis of the Class Dothideomycetes. In Evolution of Fungi and Fungal–Like Organisms. The Mycota, 14; Pöggeler, S., Wöstemeyer, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 205–229. [Google Scholar]
- Khaldi, N.; Collemare, J.; Lebrun, M.H.; Wolfe, K.H. Evidence for Horizontal Transfer of a Secondary Metabolite Gene Cluster Between Fungi. Genome Biol. 2008, 9, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadeawi, S.; Nasution, A.; Hairmansis, A.; Telebanco-Yanoria, M.J.; Obara, M.; Hayashi, N.; Fukuta, Y. Pathogenicity of Isolates of the Rice Blast Pathogen (Pyricularia oryzae) from Indonesia. Plant Dis. 2021, 105, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Kamel, S.; Cherif, M.; Hafez, M.; Despins, T.; Aboukhaddour, R. Pyrenophora tritici–repentis in Tunisia: Race Structure and Effector Genes. Front. Plant Sci. 2019, 10, 1562. [Google Scholar] [CrossRef] [Green Version]
- Oide, S.; Turgeon, B.G. Natural Roles of Nonribosomal Peptide Metabolites in Fungi. Mycoscience 2020, 61, 101–110. [Google Scholar] [CrossRef]
- Wang, X.; Wei, J.; Huang, L.; Kang, Z. Re-evaluation of Pathogens Causing Valsa Canker on Apple in China. Mycologia 2011, 103, 317–324. [Google Scholar] [CrossRef]
- Ohm, R.A.; Feau, N.; Henrissat, B.; Schoch, C.L.; Horwitz, B.A.; Barry, K.W.; Condon, B.J.; Copeland, A.C.; Dhillon, B.; Glaser, F.; et al. Diverse Lifestyles and Strategies of Plant Pathogenesis Encoded in the Genomes of Eighteen Dothideomycetes fungi. PLoS Pathog. 2012, 8, 1003037. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Li, X.; Huang, P.W.; Dai, C.C. Endophytism or Saprophytism: Decoding the Lifestyle Transition of the Generalist Fungus Phomopsis liquidambari. Microbiol. Res. 2017, 206, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, X.; Cai, J.; Chen, Y. Genomic Characteristics and Comparative Genomics Analysis of the Endophytic Fungus Sarocladium brachiariae. BMC Genom. 2019, 20, 782. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).