
Antiproliferative and Cytotoxic Cytochalasins from Sparticola triseptata Inhibit Actin Polymerization and Aggregation

 , , ,
, , ,  ,
,  ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. General Experimental Procedures
2.2. Fungal Material
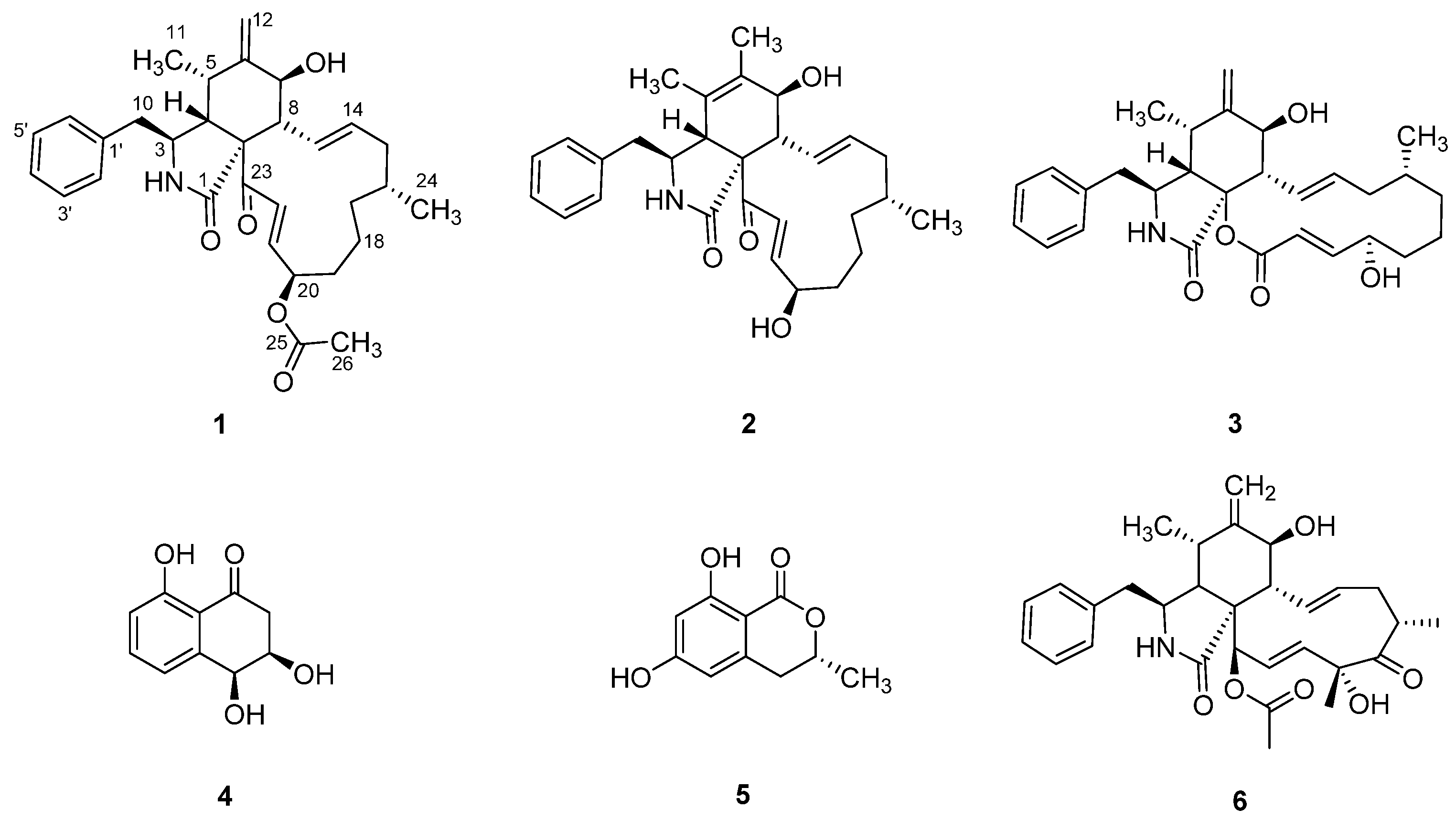
2.3. Production, Extraction, Isolation, and Structural Characterization
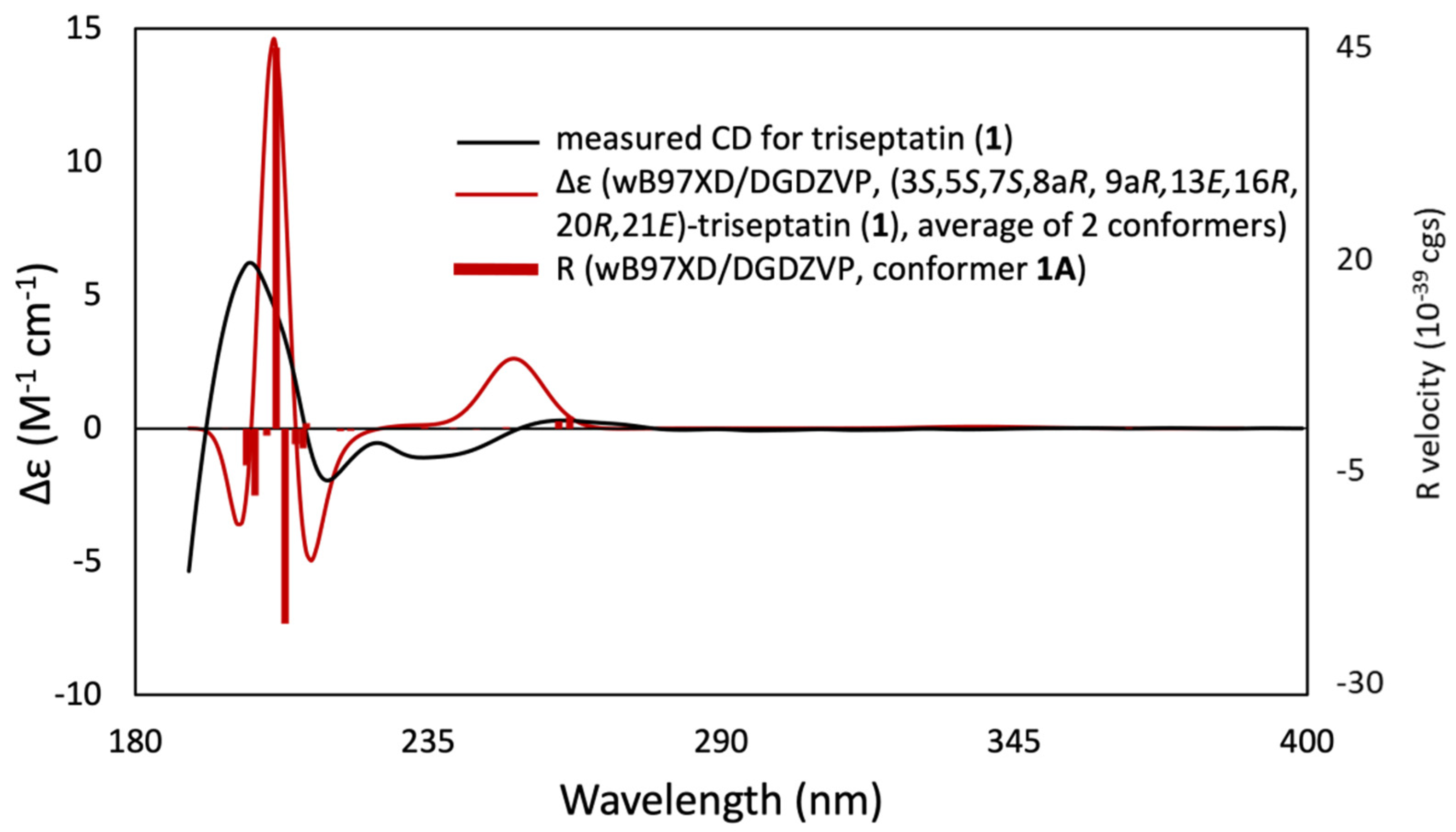
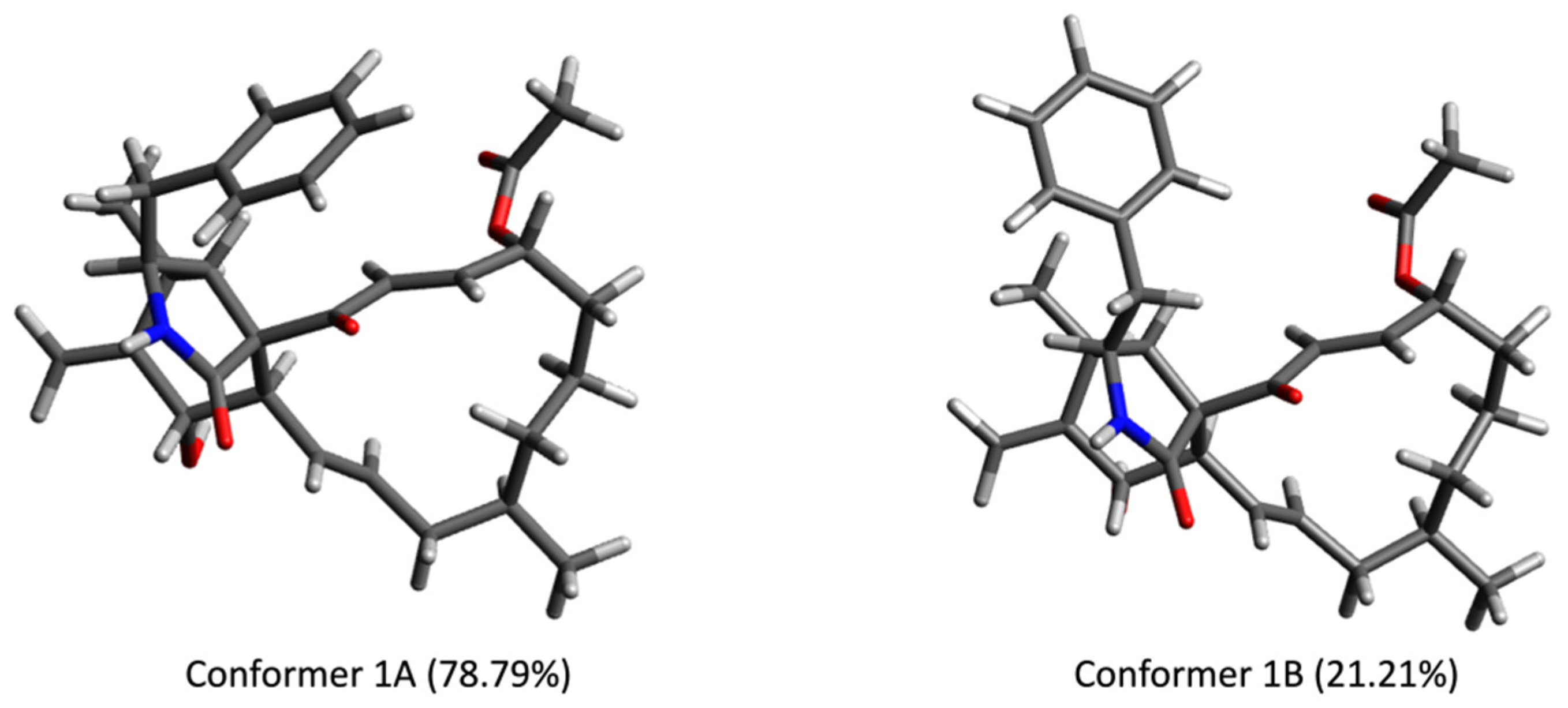
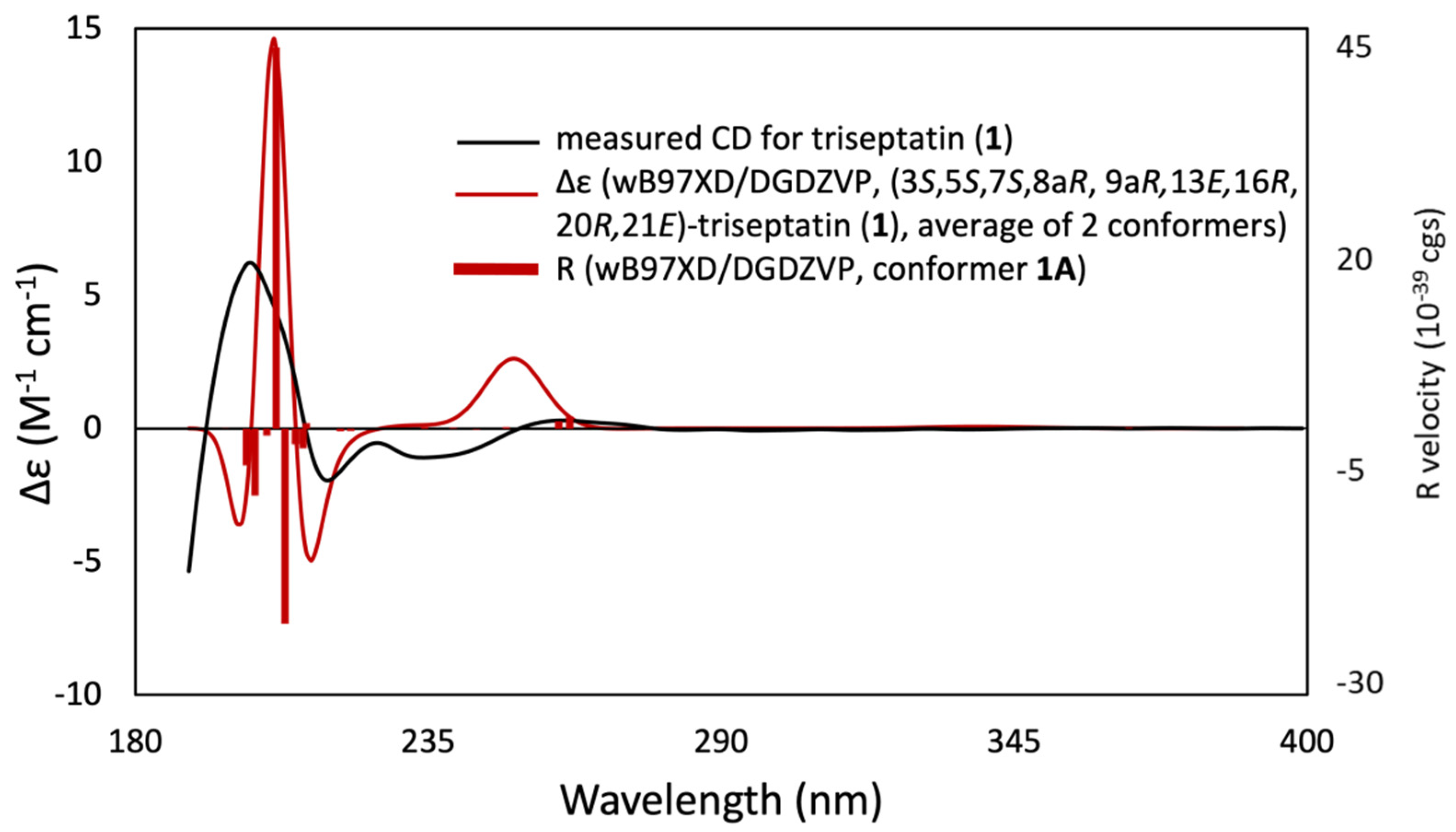
2.4. Computational Calculations
2.5. Antiproliferation and Cytotoxicity Assays
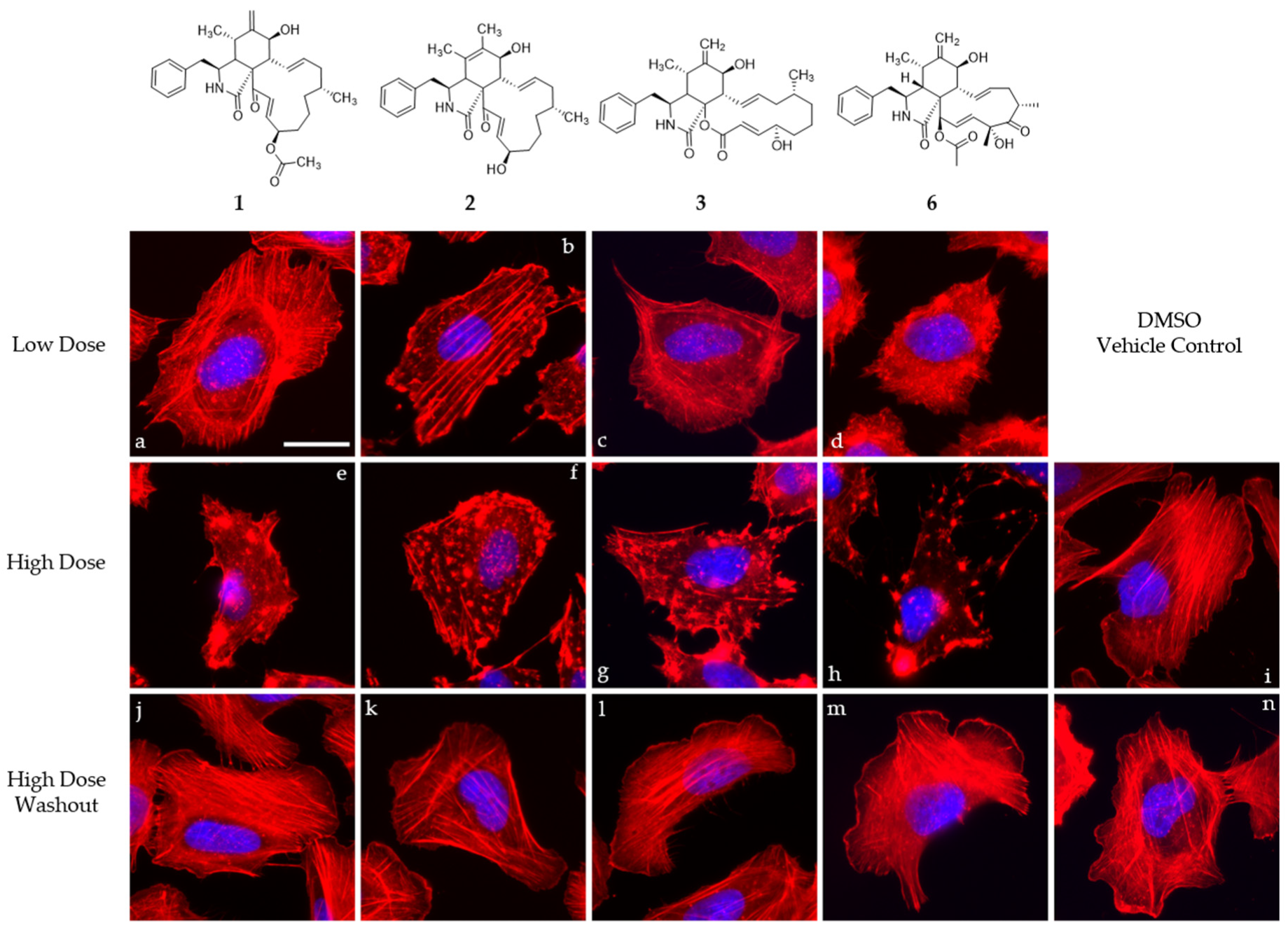
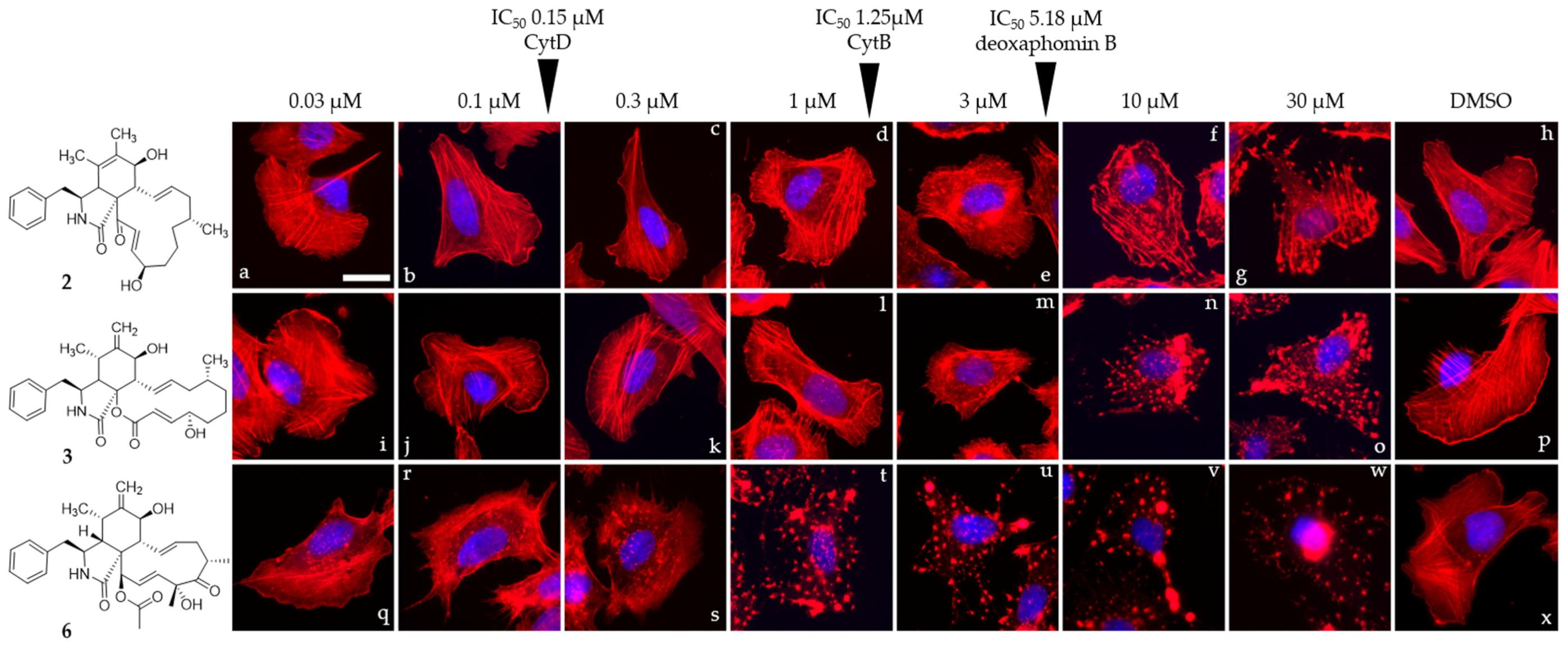
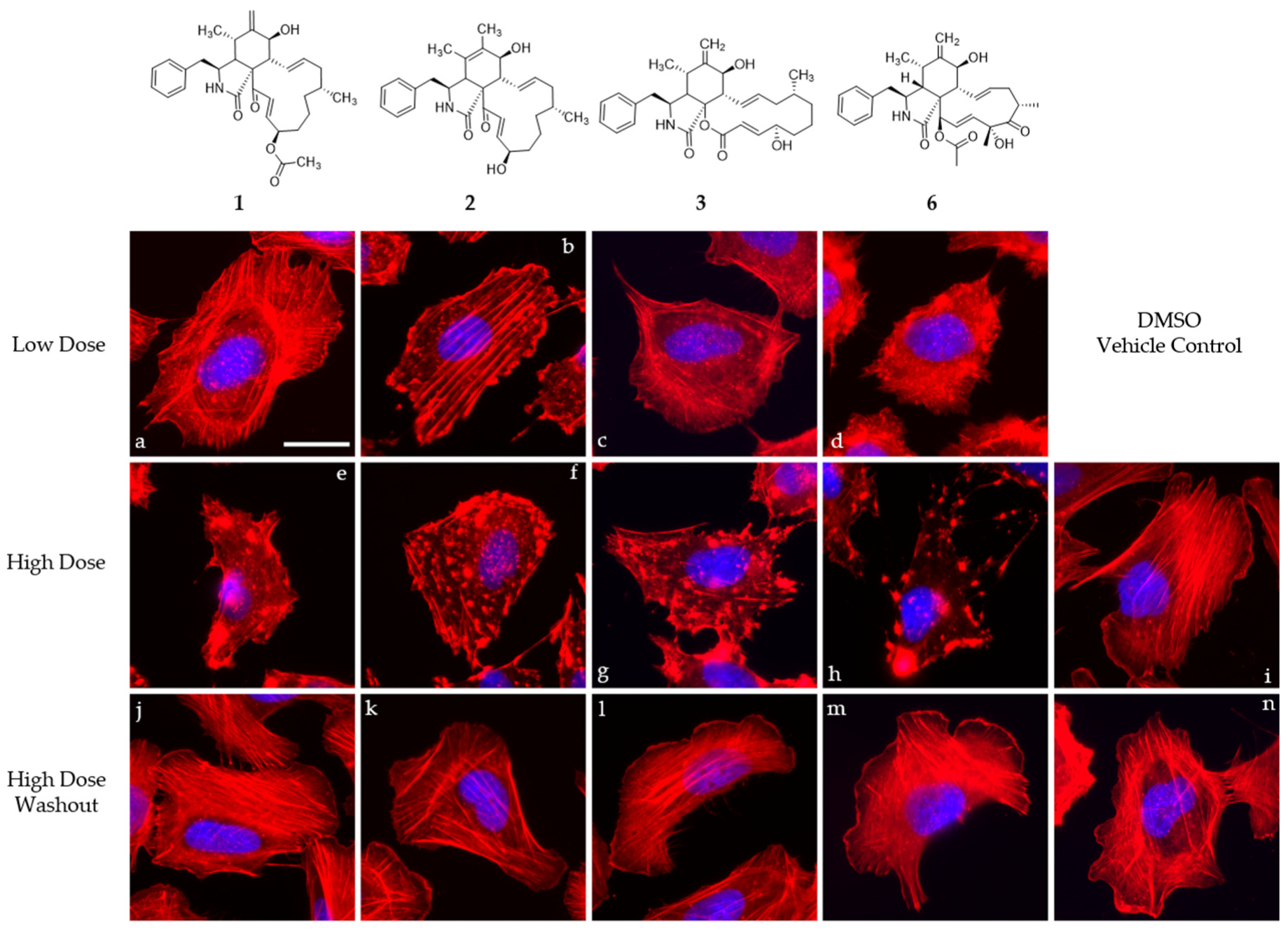
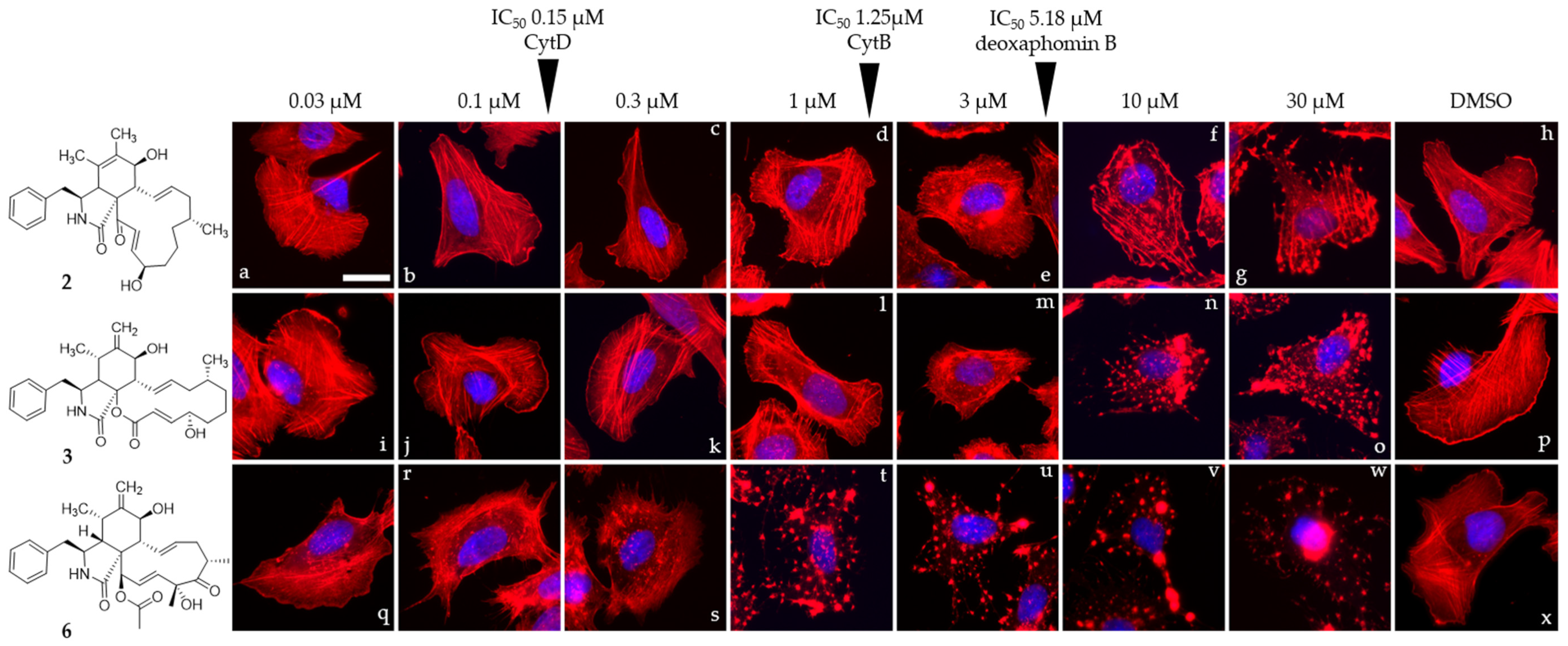
2.6. Cell Culture of U2-OS Cells and Actin Disruption Assay
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Skellam, E. The biosynthesis of cytochalasans. Nat. Prod. Rep. 2017, 34, 1252–1263. [Google Scholar] [CrossRef] [PubMed]
- Rothweiler, W.; Tamm, C. Isolation and structure of phomin. Experentia 1966, 22, 750–752. [Google Scholar] [CrossRef]
- Aldridge, D.C.; Armstrong, J.J.; Speake, R.N.; Turner, W.B. The cytochalasins, a new class of biologically active mould metabolites. Chem. Comm. 1967, 26, 1667–1676. [Google Scholar] [CrossRef]
- Capasso, R.; Evidente, A.; Ritieni, A.; Randazzo, G.; Vurro, M.; Bottalico, A. Ascochalasin, a new cytochalasin from Ascochyta heteromorpha. J. Nat. Prod. 1988, 51, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Scherlach, K.; Boettger, D.; Remme, N.; Hertweck, C. The chemistry and biology of cytochalasans. Nat. Prod. Rep. 2010, 27, 869–886. [Google Scholar] [CrossRef] [PubMed]
- Kretz, R.; Wendt, L.; Wongkanoun, S.; Luangsa-ard, J.J.; Surup, F.; Helaly, S.E.; Noumeur, S.R.; Stadler, M.; Stradal, T.E.B. The effect of cytochalasans on the actin cytoskeleton of eukaryotic cells and preliminary structure–activity relationships. Biomolecules 2019, 9, 73. [Google Scholar] [CrossRef] [Green Version]
- Lambert, C.; Pourmoghaddam, M.J.; Cedeño-Sanchez, M.; Surup, F.; Khodaparast, S.A.; Krisai-Greilhuber, I.; Voglmayr, H.; Stradal, T.E.B.; Stadler, M. Resolution of the Hypoxylon fuscum complex (Hypoxylaceae, Xylariales) and discovery and biological characterization of two of its prominent secondary metabolites. J. Fungi 2021, 7, 131. [Google Scholar] [CrossRef]
- Betina, V.; Micekova, D.; Nemec, P. Antimicrobial properties of cytochalasins and their alteration of fungal morphology. J. Gen. Microbiol. 1972, 71, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Horn, W.S.; Simmonds, M.S.J.; Blaney, W.M. Phomopsichalasin, a novel antimicrobial agent from an endophytic Phomopsis sp. Tetrahedron 1995, 51, 3969–3978. [Google Scholar] [CrossRef]
- Makioka, A.; Kumagai, M.; Kobayashi, S.; Takeuchi, T. Effect of proteasome inhibitors on the growth, encystation, and excystation of Entamoeba histolytica and Entamoeba invadens. Parasitol. Res. 2004, 93, 68–71. [Google Scholar] [CrossRef]
- Jayasuriya, H.; Herath, K.B.; Ondeyka, J.G.; Polishook, J.D.; Bills, G.F.; Dombrowski, A.W.; Springer, M.S.; Siciliano, S.; Malkowitz, L.; Sanchez, M.; et al. Isolation structure of antagonists of chemokine receptor (CCR5). J. Nat. Prod. 2004, 67, 1036–1038. [Google Scholar] [CrossRef] [PubMed]
- Rochfort, S.; Ford, J.; Ovenden, S.; Wan, S.S.; George, S.; Wildman, H.; Tait, R.M.; Meurer-Grimes, B.; Cox, S.; Coates, J.; et al. A novel aspochalasin with HIV-1 integrase inhibitory activity from Aspergillus flavipes. J. Antibiot. 2005, 58, 279–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schofield, J.G. Cytochalasin B and release of growth hormone. Nat. New Biol. 1971, 234, 215–216. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Wolff, J. Cytochalasin B inhibits thyroid secretion. Biochem. Biophys. Res. Commun. 1971, 44, 422–425. [Google Scholar] [CrossRef]
- Crivello, J.F.; Jefcoate, C.R. Intracellular movement of cholesterol in rat adrenal cells. Kinetics and effects of inhibitors. J. Biol. Chem. 1980, 255, 8144–8151. [Google Scholar] [CrossRef]
- Yuyama, K.T.; Wendt, L.; Surup, F.; Chepkirui, C.; Wittstein, K.; Boonlarppradab, C.; Wongkanoun, S.; Luangsa-ard, J.; Stadler, M.; Abraham, W.-R. Cytochalasans act as inhibitors of biofilm formation of Staphylococcus aureus. Biomolecules 2018, 8, 129. [Google Scholar] [CrossRef] [Green Version]
- Rampal, A.L.; Pinkofsky, H.B.; Jung, C.Y. Structure of cytochalasins and cytochalasin B binding sites in human erythrocyte membranes. Biochemistry 1980, 19, 679–683. [Google Scholar] [CrossRef]
- George, T.P.; Cook, H.W.; Byers, D.M.; Palmer, F.B.S.C.; Spence, M.W. Inhibition of phosphatidylcholine and phosphatidylethanolamine biosynthesis by cytochalasin B in cultured glioma cells: Potential regulation of biosynthesis by Ca2+-dependent mechanisms. Biochim. Biophys. Acta. 1991, 1084, 185–193. [Google Scholar] [CrossRef]
- Leuchtmann, A. Phaeosphaera padellana und Massariosphaeria triseptata, zwei neue bitunicate Ascomyceten aus den Alpen. Mycol. Helv. 1987, 2, 183–191. [Google Scholar]
- Phukhamsakda, C.; Ariyansawa, H.A.; Phillips, A.J.L.; Wanasinghe, D.N.; Bhat, D.J.; McKenzie, E.H.C.; Hyde, K.D. Additions to sporormiaceae: Introducing two novel genera, Sparticola and Forliomyces, from Spartium. Cryptogam. Mycol. 2016, 37, 75–97. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeerschd, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminf. 2012, 4, 17–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.M.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Karuth, F.; Dahse, H.-M.; Rüttinger, H.-H.; Frohberg, P. Synthesis and characterization of novel 1,2,4-triazine derivatives with antiproliferative activity. Bioorg. Med. Chem. 2010, 18, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.; Wessel, A.C.; Luangsa-ard, J.J.; Stadler, M. Viridistratins A–C, antimicrobial and cytotoxic benzo[j]fluoranthenes from stromata of Annulohypoxylon viridistratum (Hypoxylaceae, Ascomycota). Biomolecules 2020, 10, 805. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Lambert, C.; Hauser, M.; Deuschmann, A.; Zeilinger, C.; Rottner, K.; Stradal, T.E.B.; Stadler, M.; Skellam, E.J.; Cox, R.J. Diversely Functionalised Cytochalasins via Mutasynthesis and Semi-Synthesis. Chem. Eur. Chem. 2020, 26, 13578–13583. [Google Scholar] [CrossRef]
- Kim, E.L.; Li, J.L.; Dang, H.T.; Hong, J.; Lee, C.O.; Kim, D.K.; Yoon, W.D.; Kim, E.; Liu, Y.; Jung, J.H. Cytotoxic cytochalasins from the endozoic fungus Phoma sp. of the giant jellyfish Nemopilema nomurai. Bioorg. Med. Chem. Lett. 2012, 22, 3126–3129. [Google Scholar] [CrossRef]
- Couché, E.; Fkyerat, A.; Tabacchi, R. Stereoselective Synthesis of cis- and trans-3,4-dihydro-3,4,8-trihydroxynaphthalen-1(2H)-one. Helv. Chim. Acta. 2009, 92, 903–917. [Google Scholar] [CrossRef]
- Islam, M.S.; Ishigami, K.; Watanabe, H. Synthesis of (-)-Mellein (I), (+)-Ramulosin (II), and related natural products. Tetrahedron 2007, 63, 1074–1079. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, H.-P.; Li, Y.; Feng, T.; Liu, J.-K. Cytochalasins from cultures of endophytic fungus Phoma multirostrata EA-12. J. Antibiot. 2014, 68, 23–26. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Wang, J.; Liu, J.; Zhu, H.; Sun, B.; Wang, J.; Zhang, J.; Luo, Z.; Yao, G.; Xue, Y.; et al. Armochaetoglobins A–J: Cytochalasan alkaloids from Chaetomium globosum TW1-1, a fungus derived from the terrestrial arthropod Armadillidium vulgare. J. Nat. Prod. 2015, 78, 1193–1201. [Google Scholar] [CrossRef]
- Evidente, A.; Andolfi, A.; Vurro, M.; Zonno, M.C.; Motta, A. Ascochalasin, a new cytochalasin from Ascochyta heteromorpha. J. Nat. Prod. 2003, 66, 1540–1544. [Google Scholar] [CrossRef]
- Yahara, I.; Harada, F.; Sekita, S.; Yoshihira, K.; Natori, S. Correlation between effects of 24 different cytochalasins on cellular structures and cellular events and those on actin in vitro. J. Cell Biol. 1982, 92, 69–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, J.A. Effects of cytochalasin and phalloidin on actin. J. Cell Biol. 1987, 105, 1473–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | |
|---|---|---|
| δH (mult., J in Hz) | δC, Type | |
| 1 | - | 176.4, C |
| 3 | 3.38 (m) | 54.0, CH |
| 4 | 2.91 (m) | 46.3, CH |
| 5 | 2.72 (m) | 33.2, CH |
| 6 | - | 151.0, C |
| 7 | 3.90 (d, 9.9) | 72.8, CH |
| 8 | 2.50 (t, 9.9) | 51.9, CH |
| 9 | - | 63.9, C |
| 10a | 2.46 (dd, 13.0, 7.3) | 43.5, CH2 |
| 10b | 2.64 (dd, 13.0, 6.4) | |
| 11 | 0.79 (d, 6.8) | 13.3, CH3 |
| 12a | 5.03 (s) | 114.1, CH |
| 12b | 5.17 (s) | |
| 13 | 6.01 (ddd, 15.2, 9.6, 1.9) | 128.3, CH |
| 14 | 5.22 (m) | 137.3, CH |
| 15a | 2.06 (m) | 40.5, CH2 |
| 15b | 1.79 (dd, 11.2, 2.8) | |
| 16 | 1.51 (m) | 34.0, CH |
| 17a | 1.09 (m) | 34.9, CH2 |
| 17b | 1.26 (m) | |
| 18a | 1.15 (m) | 19.7, CH2 |
| 18b | 1.39 (m) | |
| 19a | 1.52 (m) | 32.6, CH2 |
| 19b | 1.92 (m) | |
| 20 | 5.20 (m) | 76.4, CH |
| 21 | 6.48 (dd, 15.4, 8.5) | 144.0, CH |
| 22 | 6.86 (d, 15.4) | 128.0, CH |
| 23 | - | 198.6, C |
| 24 | 0.89 (d, 6.9) | 20.8, CH3 |
| 25 | - | 171.6, C |
| 26 | 2.04 (s) | 21.1, CH3 |
| 1′ | - | 137.9, C |
| 2′/6′ | 7.08 (d, 7.6) | 130.7/131.0, CH |
| 3′/5′ | 7.32 (t, 7.6) | 129.7/129.6, CH |
| 4′ | 7.23 (t, 7.6) | 127.8, CH |
| Cell Line | Compound | Positive Control | |||
|---|---|---|---|---|---|
| 1 | 2 | 3 | Epothilone B | Imatinib | |
| Cytotoxicity a IC50 (μM) | |||||
| Mouse fibroblast L929 | 4.16 | 5.18 | N.D. | 1.4 × 10−3 | N.D. |
| HeLa cells KB3.1 | 1.80 | 1.83 | N.D. | 8.9 × 10−5 | N.D. |
| Human breast adenocarcinoma MCF-7 | 1.86 | 1.79 | N.D. | 2.4 × 10−4 | N.D. |
| Human lung carcinoma A549 | 7.32 | 6.91 | N.D. | 6.9 × 10−5 | N.D. |
| Human prostate cancer PC-3 | 11.28 | 2.81 | N.D. | 1.6 × 10−3 | N.D. |
| Ovarian carcinoma SKOV-3 | 1.84 | 1.55 | N.D. | 2.8 × 10−4 | N.D. |
| Squamous cell carcinoma A431 | 2.17 | 1.60 | N.D. | 7.9 × 10−5 | N.D. |
| Antiproliferative Effect b GI50 (µM) | |||||
| HUVEC | 4.55 | 1.08 | 8.31 | N.D. | 18.5 |
| Myelogenous leukemia K-562 | 8.31 | 3.67 | 3.34 | N.D. | 0.17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia, K.Y.M.; Quimque, M.T.J.; Lambert, C.; Schmidt, K.; Primahana, G.; Stradal, T.E.B.; Ratzenböck, A.; Dahse, H.-M.; Phukhamsakda, C.; Stadler, M.; et al. Antiproliferative and Cytotoxic Cytochalasins from Sparticola triseptata Inhibit Actin Polymerization and Aggregation. J. Fungi 2022, 8, 560. https://doi.org/10.3390/jof8060560
Garcia KYM, Quimque MTJ, Lambert C, Schmidt K, Primahana G, Stradal TEB, Ratzenböck A, Dahse H-M, Phukhamsakda C, Stadler M, et al. Antiproliferative and Cytotoxic Cytochalasins from Sparticola triseptata Inhibit Actin Polymerization and Aggregation. Journal of Fungi. 2022; 8(6):560. https://doi.org/10.3390/jof8060560
Chicago/Turabian StyleGarcia, Katherine Yasmin M., Mark Tristan J. Quimque, Christopher Lambert, Katharina Schmidt, Gian Primahana, Theresia E. B. Stradal, Andreas Ratzenböck, Hans-Martin Dahse, Chayanard Phukhamsakda, Marc Stadler, and et al. 2022. "Antiproliferative and Cytotoxic Cytochalasins from Sparticola triseptata Inhibit Actin Polymerization and Aggregation" Journal of Fungi 8, no. 6: 560. https://doi.org/10.3390/jof8060560
APA StyleGarcia, K. Y. M., Quimque, M. T. J., Lambert, C., Schmidt, K., Primahana, G., Stradal, T. E. B., Ratzenböck, A., Dahse, H.-M., Phukhamsakda, C., Stadler, M., Surup, F., & Macabeo, A. P. G. (2022). Antiproliferative and Cytotoxic Cytochalasins from Sparticola triseptata Inhibit Actin Polymerization and Aggregation. Journal of Fungi, 8(6), 560. https://doi.org/10.3390/jof8060560