
Evaluation of the Antifungal Activities of Photorhabdus akhurstii and Its Secondary Metabolites against Phytopathogenic Colletotrichum gloeosporioides

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Microorganisms, Culture Media, and Growth Conditions
2.2. Extraction of Active Compounds
2.3. Disk-Diffusion Assay
2.4. Confrontation Assay
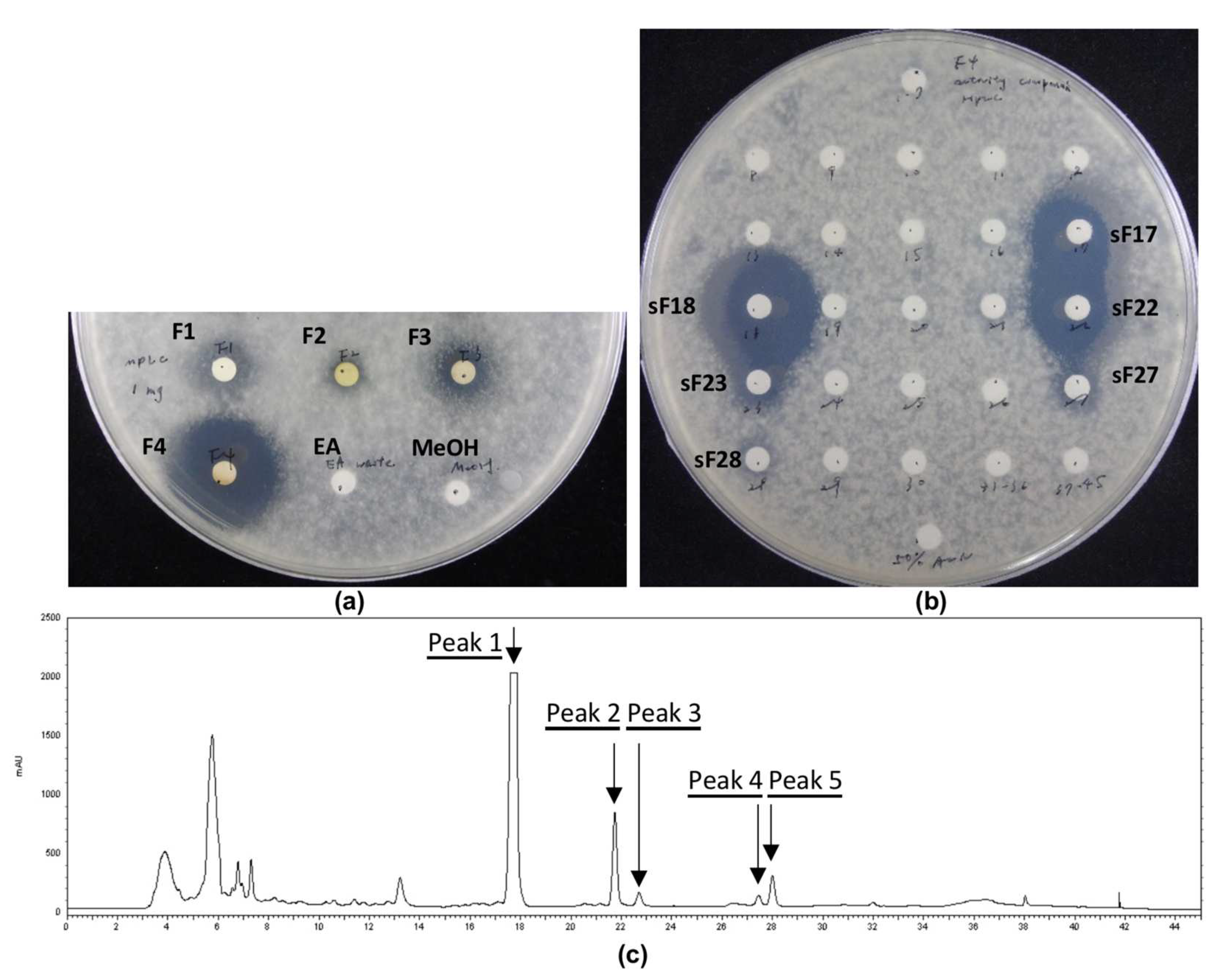
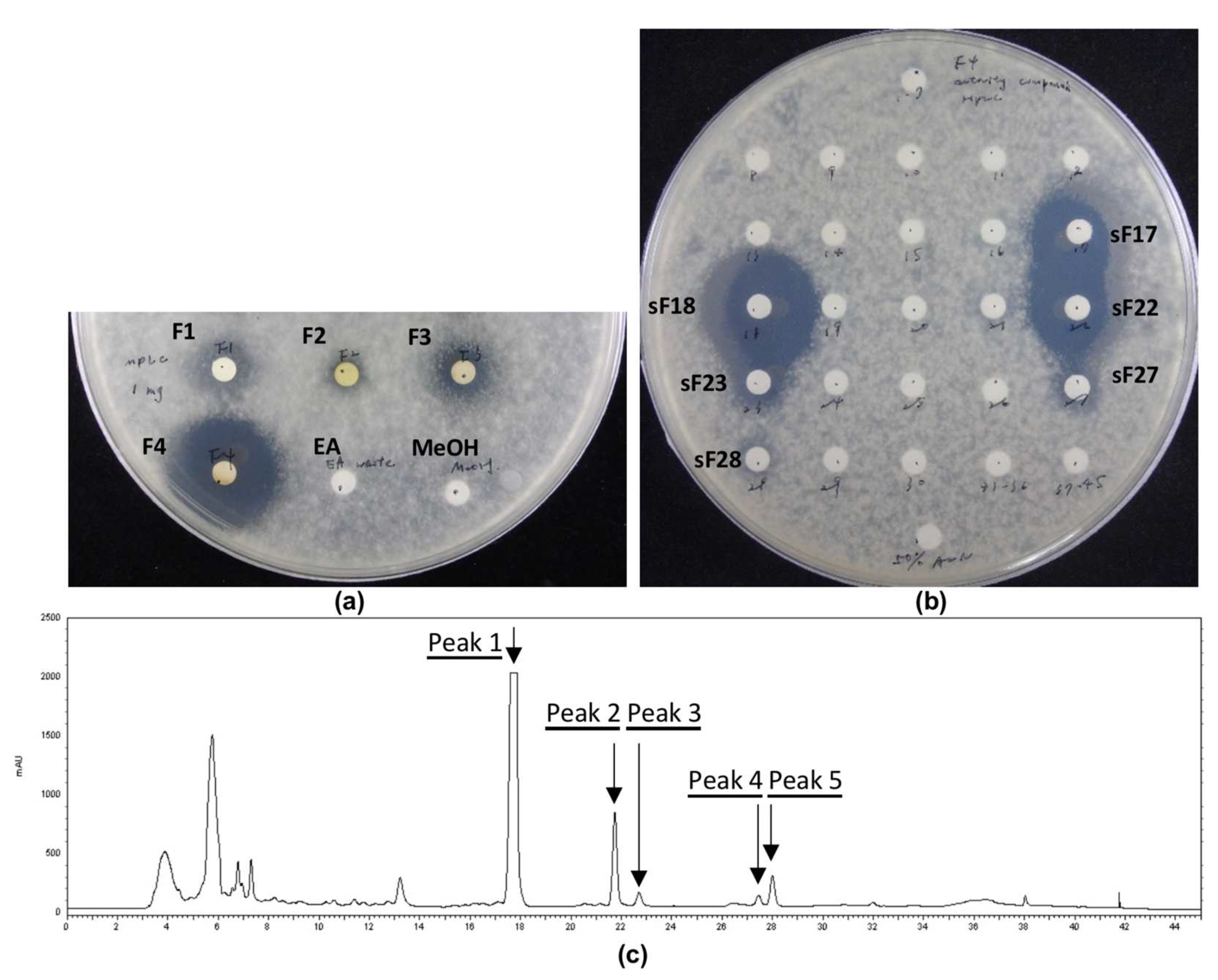
2.5. Medium-Pressure Liquid Chromatography Separation of Crude Extract
2.6. High-Performance Liquid Chromatography Isolation of Medium-Pressure Liquid Chromatography Fractions
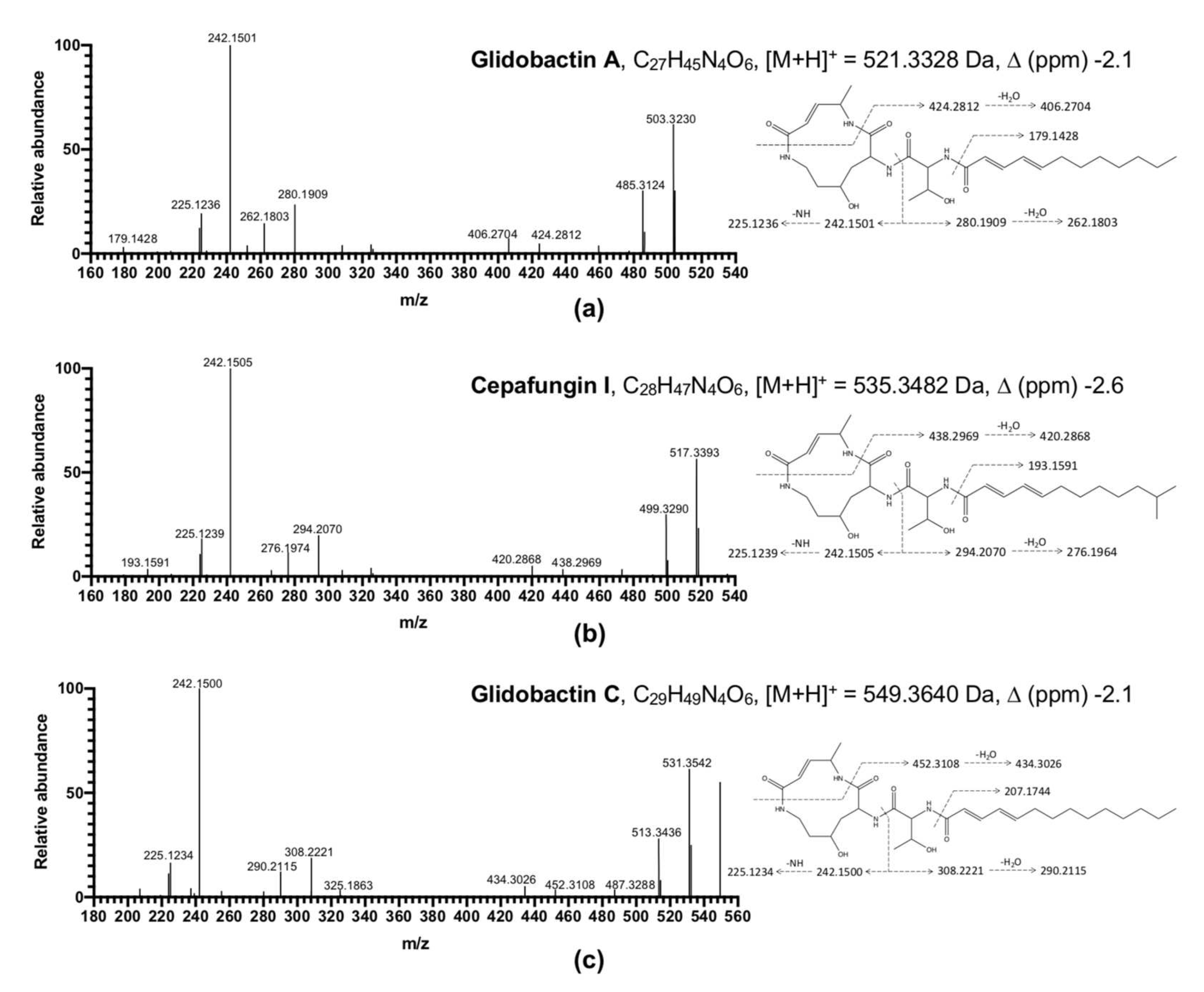
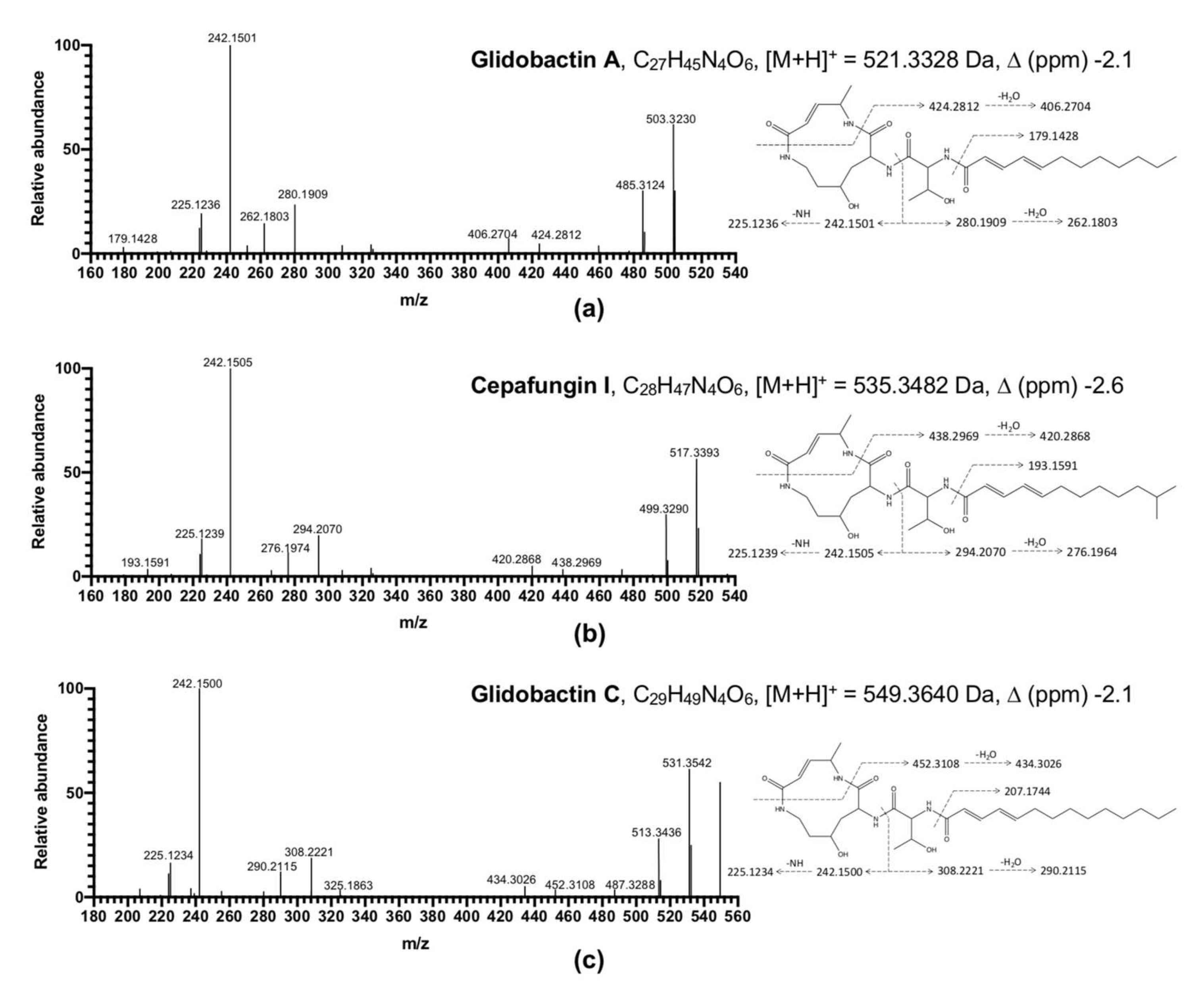
2.7. Compound Prediction Using LC-MS/MS
2.8. NMR Analysis of Isolated Peaks from HPLC
2.9. Determination of Minimum Inhibitory Concentration
2.10. RNA Isolation, Reverse Transcription, and Quantitative Reverse Transcription PCR
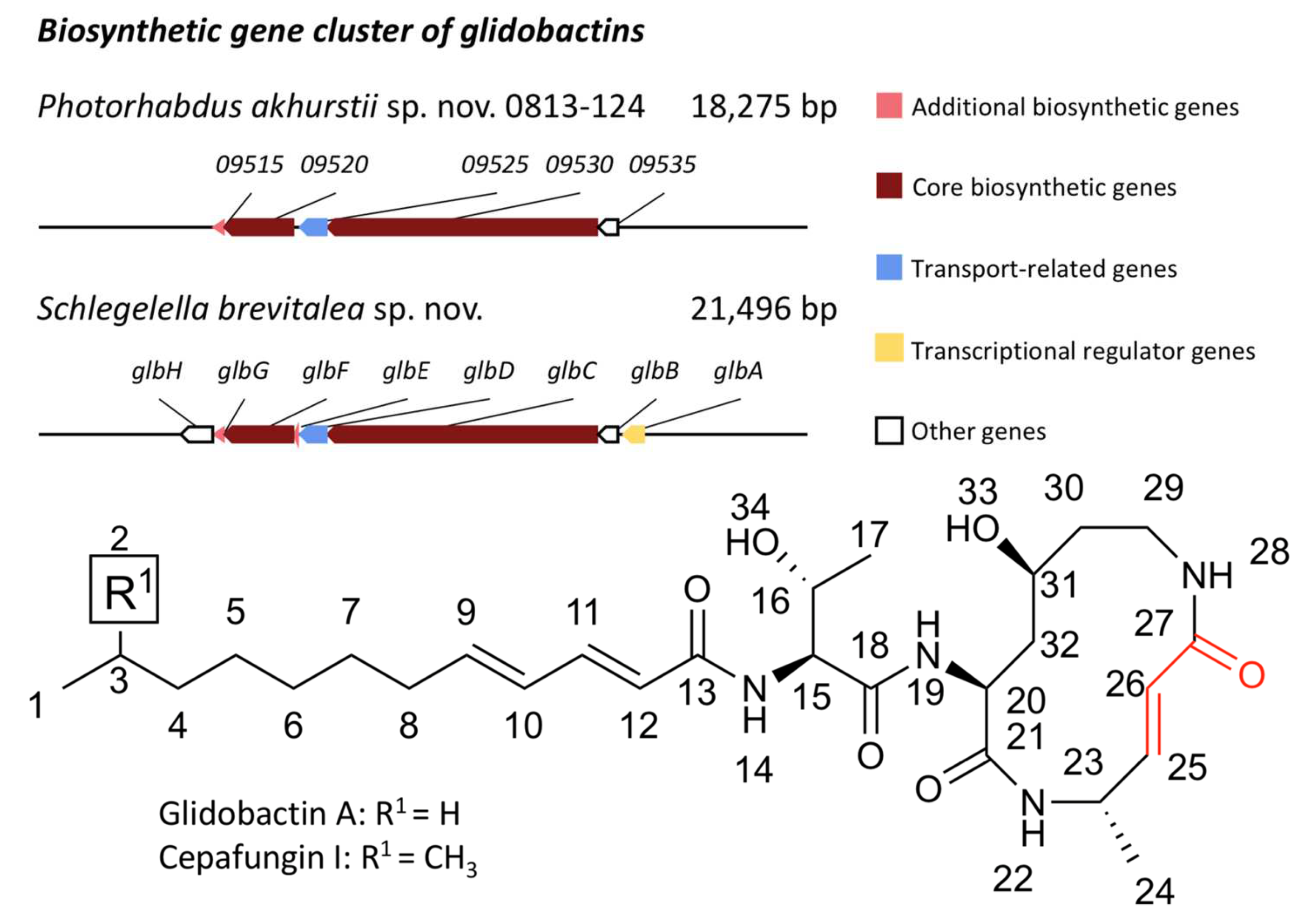
2.11. Biosynthetic Gene Clusters Analysis
3. Results and Discussion
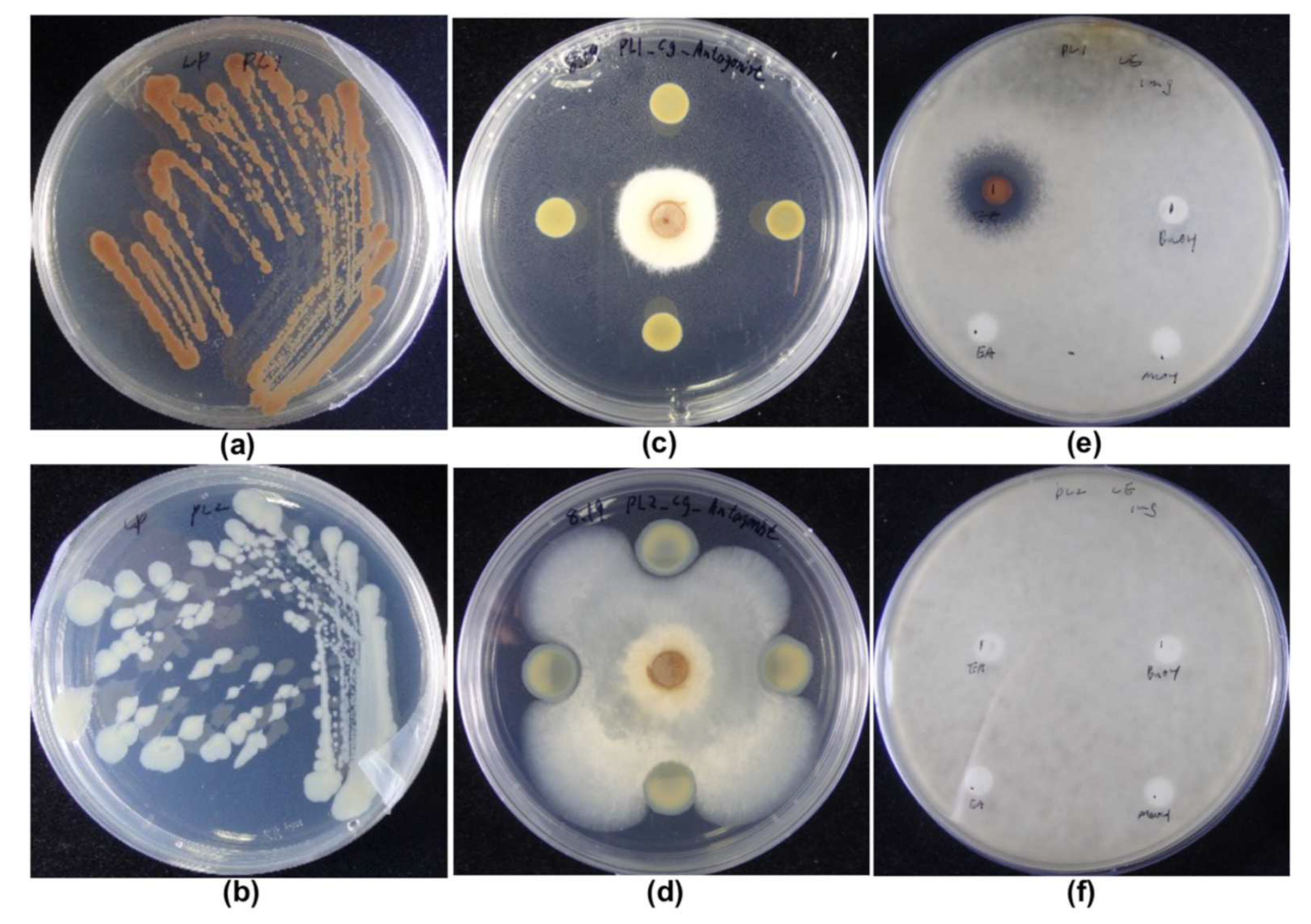
3.1. Phenotypic Comparison of PL1 and PL2; Appearance and Antifungal Activities
3.2. Analysis of Bacterial Extracts
3.3. Minimum Inhibitory Concentration
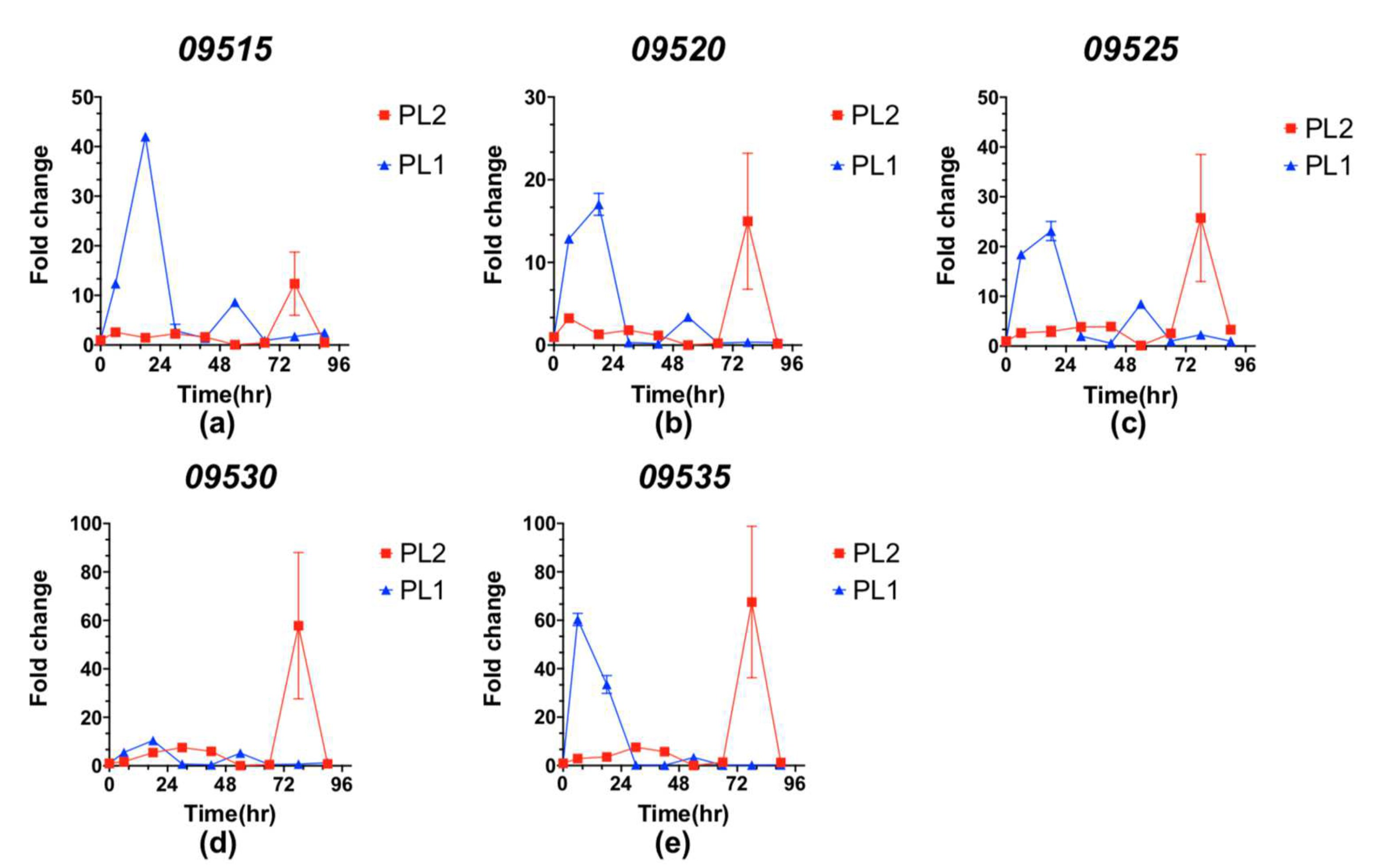
3.4. Genomic Analysis and Expression Level of the Glidobactin BGC in PL1 and PL2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Howard, C.M.; Maas, J.L.; Chandler, C.K.; Albregts, E.E. Anthracnose of strawberry caused by the Colletrotrichum complex in Florida. Plant Dis. 1992, 76, 976–981. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, R.L.; Adhikari, T.B.; Pattison, J.; Yencho, G.C.; Fernandez, G.E.; Louws, F.J. Inheritance of resistance to Colletotrichum gloeosporioides and C. acutatum in strawberry. Phytopathology 2019, 109, 428–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreenivasaprasad, S.; Talhinhas, P. Genotypic and phenotypic diversity in Colletotrichum acutatum, a cosmopolitan pathogen causing anthracnose on a wide range of hosts. Mol. Plant Pathol. 2005, 6, 361–378. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Y.; Hu, X.R.; Zhang, C.Q. Molecular detection of QoI resistance in Colletotrichum gloeosporioides causing strawberry anthracnose based on loop-mediated isothermal amplification assay. Plant Dis. 2019, 103, 1319–1325. [Google Scholar] [CrossRef]
- Mehmood, N.; Yuan, Y.; Ali, M.; Ali, M.; Iftikhar, J.; Cheng, C.; Lyu, M.; Wu, B. Early transcriptional response of terpenoid metabolism to Colletotrichum gloeosporioides in a resistant wild strawberry Fragaria nilgerrensis. Phytochemistry 2021, 181, 112590. [Google Scholar] [CrossRef] [PubMed]
- Schnabel, G.; Tan, Q.; Schneider, V.; Ishii, H. Inherent tolerance of Colletotrichum gloeosporioides to fludioxonil. Pestic. Biochem. Physiol. 2021, 172, 104767. [Google Scholar] [CrossRef]
- Raveau, R.; Fontaine, J.; Sahraoui, A.L.-H. Essential oils as potential alternative biocontrol products against plant pathogens and weeds: A review. Foods 2020, 9, 365. [Google Scholar] [CrossRef] [Green Version]
- Vurukonda, S.S.K.P.; Davide Giovanardi, D.; Stefani, E. Plant growth promoting and biocontrol activity of Streptomyces spp. as Endophytes. Int. J. Mol. Sci. 2018, 19, 952. [Google Scholar] [CrossRef] [Green Version]
- Suárez-Moreno, Z.R.; Vinchira-Villarraga, D.M.; Vergara-Morales, D.I.; Castellanos, L.; Ramos, F.A.; Guarnaccia, C.; Degrassi, G.; Venturi, V.; Moreno-Sarmiento, N. Plant-growth promotion and biocontrol properties of three Streptomyces spp. isolates to control bacterial rice pathogens. Front. Microbiol. 2019, 10, 290. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.; Shah, R.; Joshi, B.; Ramar, K.; Natarajan, A. Molecular identification and biocontrol activity of sugarcane rhizosphere bacteria against red rot pathogen Colletotrichum falcatum. Biotechnol. Rep. 2019, 21, e00317. [Google Scholar] [CrossRef]
- Dutta, T.K.; Santhoshkumar, K.; Mathur, C.; Mandal, A.; Sagar, D. A Photorhabdus akhurstii toxin altered gut homeostasis prior conferring cytotoxicity in Spodoptera frugiperda, S. litura and Helicoverpa armigera. Phytoparasitica 2021, 49, 943–958. [Google Scholar] [CrossRef]
- Bock, C.H.; Shapiro-Ilan, D.I.; Wedge, D.E.; Cantrell, C.L. Identification of the antifungal compound, trans-cinnamic acid, produced by Photorhabdus luminescens, a potential biopesticide against pecan scab. J. Pest Sci. 2013, 87, 155–162. [Google Scholar] [CrossRef]
- Eleftherianos, I.; Boundy, S.; Joyce, S.A.; Aslam, S.; Marshall, J.W.; Cox, R.J.; Simpson, T.J.; Clarke, D.J.; Ffrench-Constant, R.H.; Reynolds, S.E. An antibiotic produced by an insect-pathogenic bacterium suppresses host defenses through phenoloxidase inhibition. Proc. Natl. Acad. Sci. USA 2007, 104, 2419–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, D.J. The regulation of secondary metabolism in Photorhabdus. In The Molecular Biology of Photorhabdus Bacteria; Ffrench-Constant, R.H., Ed.; Springer International Publishing AG: Cham, Switzerland, 2017; Volume 402, pp. 81–102. ISBN 978-3-319-52715-4. [Google Scholar]
- McInerney, B.V.; Taylor, W.C.; Lacey, M.J.; Akhurst, R.J.; Gregson, R.P. Biologically active metabolites from Xenorhabdus spp., Part 2. Benzopyran-1-one derivatives with gastroprotective activity. J. Nat. Prod. 1991, 54, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Muangpat, P.; Yooyangket, T.; Fukruksa, C.; Suwannaroj, M.; Yimthin, T.; Sitthisak, S.; Chantratita, N.; Vitta, A.; Tobias, N.J.; Bode, H.B.; et al. Screening of the antimicrobial activity against drug resistant bacteria of Photorhabdus and Xenorhabdus associated with entomopathogenic nematodes from Mae Wong National Park, Thailand. Front. Microbiol. 2017, 8, 1142. [Google Scholar] [CrossRef]
- Boemare, N.E.; Akhurst, R.J. Biochemical and physiological characterization of colony form variants in Xenorhabdus spp. (Enterobacteriaceae). J. Gen. Microbiol. 1988, 134, 751–761. [Google Scholar] [CrossRef] [Green Version]
- Ffrench-Constant, R.; Waterfield, N.; Daborn, P.; Joyce, S.; Bennett, H.; Au, C.; Dowling, A.; Boundy, S.; Reynolds, S.; Clarke, D. Photorhabdus: Towards a functional genomic analysis of a symbiont and pathogen. FEMS Microbiol. Rev. 2003, 26, 433–456. [Google Scholar] [CrossRef]
- Eckstein, S.; Dominelli, N.; Brachmann, A.; Heermann, R. Phenotypic Heterogeneity of the insect pathogen Photorhabdus luminescens: Insights into the fate of secondary cells. Appl. Environ. Microbiol. 2019, 85, e01910. [Google Scholar] [CrossRef]
- Eckstein, S.; Heermann, R. Regulation of phenotypic switching and heterogeneity in Photorhabdus luminescens cell populations. J. Mol. Biol. 2019, 431, 4559–4568. [Google Scholar] [CrossRef]
- Hurst, S.; Rowedder, H.; Michaels, B.; Bullock, H.; Jackobeck, R.; Abebe-Akele, F.; Durakovic, U.; Gately, J.; Janicki, E.; Tisa Louis, S.; et al. Elucidation of the Photorhabdus temperata genome and generation of a transposon mutant library to identify motility mutants altered in pathogenesis. J. Bacteriol. 2015, 197, 2201–2216. [Google Scholar] [CrossRef]
- Smigielski, A.J.; Akhurst, R.J.; Boemare, N.E. Phase variation in Xenorhabdus nematophilus and Photorhabdus luminescens: Differences in respiratory activity and membrane energization. Appl. Environ. Microbiol. 1994, 60, 120–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turlin, E.; Pascal, G.; Rousselle, J.C.; Lenormand, P.; Ngo, S.; Danchin, A.; Derzelle, S. Proteome analysis of the phenotypic variation process in Photorhabdus luminescens. Proteomics 2006, 6, 2705–2725. [Google Scholar] [CrossRef] [PubMed]
- Machado, R.A.R.; Wuthrich, D.; Kuhnert, P.; Arce, C.C.M.; Thonen, L.; Ruiz, C.; Zhang, X.; Robert, C.A.M.; Karimi, J.; Kamali, S.; et al. Whole-genome-based revisit of Photorhabdus phylogeny: Proposal for the elevation of most Photorhabdus subspecies to the species level and description of one novel species Photorhabdus bodei sp nov., and one novel subspecies Photorhabdus laumondii subsp clarkei subsp nov. Int. J. Syst. Evol. Microbiol. 2018, 68, 2664–2681. [Google Scholar] [CrossRef] [PubMed]
- Muangpat, P.; Suwannaroj, M.; Yimthin, T.; Fukruksa, C.; Sitthisak, S.; Chantratita, N.; Vitta, A.; Thanwisai, A. Antibacterial activity of Xenorhabdus and Photorhabdus isolated from entomopathogenic nematodes against antibiotic-resistant bacteria. PLoS ONE 2020, 15, e0234129. [Google Scholar] [CrossRef] [PubMed]
- Alforja, S.I.R.; Rico, P.M.B.; Caoili, B.L.; Latina, R.A. Two Philippine Photorhabdus luminescens strains inhibit the in vitro growth of Lasiodiplodia theobromae, Fusarium oxysporum f. sp. lycopersici, and Colletotrichum spp. Egypt. J. Biol. Pest Control 2021, 31, 108. [Google Scholar] [CrossRef]
- Hsieh, F.C.; Tzeng, C.Y.; Tseng, J.T.; Tsai, Y.S.; Meng, M.; Kao, S.S. Isolation and characterization of the native entomopathogenic nematode, Heterorhabditis brevicaudis, and its symbiotic bacteria from Taiwan. Curr. Microbiol. 2009, 58, 564–570. [Google Scholar] [CrossRef]
- Chen, W.J.; Kuo, T.Y.; Hsieh, F.C.; Chen, P.Y.; Wang, C.S.; Shih, Y.L.; Lai, Y.M.; Liu, J.R.; Yang, Y.L.; Shih, M.C. Involvement of type VI secretion system in secretion of iron chelator pyoverdine in Pseudomonas taiwanensis. Sci. Rep. 2016, 6, 32950. [Google Scholar] [CrossRef] [Green Version]
- Amatuni, A.; Shuster, A.; Adibekian, A.; Renata, H. Concise chemoenzymatic total synthesis and identification of cellular targets of cepafungin I. Cell Chem. Biol. 2020, 27, 1318–1326. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [Green Version]
- Joyce, S.A.; Watson, R.J.; Clarke, D.J. The regulation of pathogenicity and mutualism in Photorhabdus. Curr. Opin. Microbiol. 2006, 9, 127–132. [Google Scholar] [CrossRef]
- Lulamba, T.E.; Green, E.; Serepa-Dlamini, M.H. Genome assembly and annotation of Photorhabdus heterorhabditis strain ETL reveals genetic features involved in pathogenicity with its associated entomopathogenic nematode and anti-host effectors with biocontrol potential applications. Gene 2021, 795, 145780. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Liang, S.; Cao, L.; Liu, X.; Han, R. Nutritive significance of crystalline inclusion proteins of Photorhabdus luminescens in Steinernema nematodes. FEMS Microbiol. Ecol. 2006, 55, 178–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.M.; Bode, H.B. Chemical language and warfare of bacterial natural products in bacteria–nematode–insect interactions. Nat. Prod. Rep. 2018, 35, 309–335. [Google Scholar] [CrossRef] [PubMed]
- Brachmann, A.O.; Joyce, S.A.; Jenke-Kodama, H.; Schwär, G.; Clarke, D.J.; Bode, H.B. A type II polyketide synthase is responsible for anthraquinone biosynthesis in Photorhabdus luminescens. ChemBioChem 2007, 8, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, A.K.; Glaeser, A.; Tobias, N.J.; Heermann, R.; Bode, H.B. Heterogeneous regulation of bacterial natural product biosynthesis via a novel transcription factor. Heliyon 2016, 2, e00197. [Google Scholar] [CrossRef] [Green Version]
- Cimen, H.; Touray, M.; Gulsen, S.H.; Erincik, O.; Wenski, S.L.; Bode, H.B.; Shapiro-Ilan, D.; Hazir, S. Antifungal activity of different Xenorhabdus and Photorhabdus species against various fungal phytopathogens and identification of the antifungal compounds from X. szentirmaii. Appl. Microbiol. Biotechnol. 2021, 105, 5517–5528. [Google Scholar] [CrossRef]
- Stein, M.L.; Beck, P.; Kaiser, M.; Dudler, R.; Becker, C.F.W.; Groll, M. One-shot NMR analysis of microbial secretions identifies highly potent proteasome inhibitor. Proc. Natl. Acad. Sci. USA 2012, 109, 18367–18371. [Google Scholar] [CrossRef] [Green Version]
- Oka, M.; Nishiyama, Y.; Ohta, S.; Kamei, H.; Konishi, M.; Miyaki, T.; Oki, T.; Kawaguchi, H. Glidobactins A, B and C, new antitumor antibiotics. I. Production, isolation, chemical properties and biological activity. J. Antibiot. 1988, 41, 1331–1337. [Google Scholar] [CrossRef] [Green Version]
- Pierce, M.R.; Robinson, R.M.; Ibarra-Rivera, T.R.; Pirrung, M.C.; Dolloff, N.G.; Bachmann, A.S. Syrbactin proteasome inhibitor TIR-199 overcomes bortezomib chemoresistance and inhibits multiple myeloma tumor growth in vivo. Leuk. Res. 2020, 88, 106271. [Google Scholar] [CrossRef]
- Krahn, D.; Ottmann, C.; Kaiser, M. The chemistry and biology of syringolins, glidobactins and cepafungins (syrbactins). Nat. Prod. Rep. 2011, 28, 1854–1867. [Google Scholar] [CrossRef]
- Wei, D.; Tang, M.; Zhan, C.G. Fundamental reaction pathway and free energy profile of proteasome inhibition by syringolin A (SylA). Org. Biomol. Chem. 2015, 13, 6857–6865. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Le Chapelain, C.; Brachmann, A.O.; Kaiser, M.; Groll, M.; Bode, H.B. Activation, structure, biosynthesis and bioactivity of glidobactin-like proteasome inhibitors from Photorhabdus laumondii. Chembiochem 2021, 22, 1582–1588. [Google Scholar] [CrossRef] [PubMed]
- Ogorevc, E.; Schiffrer, E.S.; Sosič, I.; Gobec, S. A patent review of immunoproteasome inhibitors. Expert Opin. Ther. Pat. 2018, 28, 517–540. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Plaza, A.; Zhang, Y.; Müller, R. Luminmycins A–C, cryptic natural products from Photorhabdus luminescens identified by heterologous expression in Escherichia coli. J. Nat. Prod. 2012, 75, 1652–1655. [Google Scholar] [CrossRef]
- Schellenberg, B.; Bigler, L.; Dudler, R. Identification of genes involved in the biosynthesis of the cytotoxic compound glidobactin from a soil bacterium. Environ. Microbiol. 2007, 9, 1640–1650. [Google Scholar] [CrossRef]
- Wäspi, U.; Blanc, D.; Winkler, T.; Rüedi, P.; Dudler, R. Syringolin, a novel peptide elicitor from Pseudomonas syringae pv. syringae that induces resistance to Pyricularia oryzae in rice. Mol. Plant Microbe Interact. 1998, 11, 727–733. [Google Scholar] [CrossRef] [Green Version]
- Hofstetter, S.S.; Dudnik, A.; Widmer, H.; Dudler, R. Arabidopsis YELLOW STRIPE-LIKE7 (YSL7) and YSL8 transporters mediate uptake of Pseudomonas virulence factor syringolin A into plant cells. Mol. Plant Microbe Interact. 2013, 26, 1302–1311. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zheng, W.; Zhou, H.; Tu, Q.; Tang, Y.-J.; Stewart, A.F.; Zhang, Y.; Bian, X. Improved dsDNA recombineering enables versatile multiplex genome engineering of kilobase-scale sequences in diverse bacteria. Nucleic Acids Res. 2022, 50, e15. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject | MIC (µg/mL) |
|---|---|
| PL1 BuOH crude extract | NS * |
| PL1 EA crude extract | <300 |
| Glidobactin A | ≤1.50 |
| Cepafungin I | ≤2.00 |
| Carbendazim (positive control) | ≤0.32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tu, P.-W.; Chiu, J.-S.; Lin, C.; Chien, C.-C.; Hsieh, F.-C.; Shih, M.-C.; Yang, Y.-L. Evaluation of the Antifungal Activities of Photorhabdus akhurstii and Its Secondary Metabolites against Phytopathogenic Colletotrichum gloeosporioides. J. Fungi 2022, 8, 403. https://doi.org/10.3390/jof8040403
Tu P-W, Chiu J-S, Lin C, Chien C-C, Hsieh F-C, Shih M-C, Yang Y-L. Evaluation of the Antifungal Activities of Photorhabdus akhurstii and Its Secondary Metabolites against Phytopathogenic Colletotrichum gloeosporioides. Journal of Fungi. 2022; 8(4):403. https://doi.org/10.3390/jof8040403
Chicago/Turabian StyleTu, Po-Wen, Jie-Siang Chiu, Chih Lin, Chih-Cheng Chien, Feng-Chia Hsieh, Ming-Che Shih, and Yu-Liang Yang. 2022. "Evaluation of the Antifungal Activities of Photorhabdus akhurstii and Its Secondary Metabolites against Phytopathogenic Colletotrichum gloeosporioides" Journal of Fungi 8, no. 4: 403. https://doi.org/10.3390/jof8040403
APA StyleTu, P.-W., Chiu, J.-S., Lin, C., Chien, C.-C., Hsieh, F.-C., Shih, M.-C., & Yang, Y.-L. (2022). Evaluation of the Antifungal Activities of Photorhabdus akhurstii and Its Secondary Metabolites against Phytopathogenic Colletotrichum gloeosporioides. Journal of Fungi, 8(4), 403. https://doi.org/10.3390/jof8040403