
Genome Sequencing and Analysis of Trichoderma (Hypocreaceae) Isolates Exhibiting Antagonistic Activity against the Papaya Dieback Pathogen, Erwinia mallotivora

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of Pathogen
2.2. Isolation of Antagonist Fungi
2.3. Antagonistic Activity of Fungal Isolates against E. mallotivora Strain BT-MARDI
2.4. Genomic DNA Extraction
2.5. Identification of Fungi Based on Mycelia Morphology, PCR Amplification, and Sanger Sequencing
2.6. DNA Library Preparation and Sequencing through Oxford Nanopore Technology (ONT)
2.7. Genome Assembly and Error Corrections
2.8. Genome Annotations
2.9. Comparative Genomics and Phylogenomic Analysis
3. Results
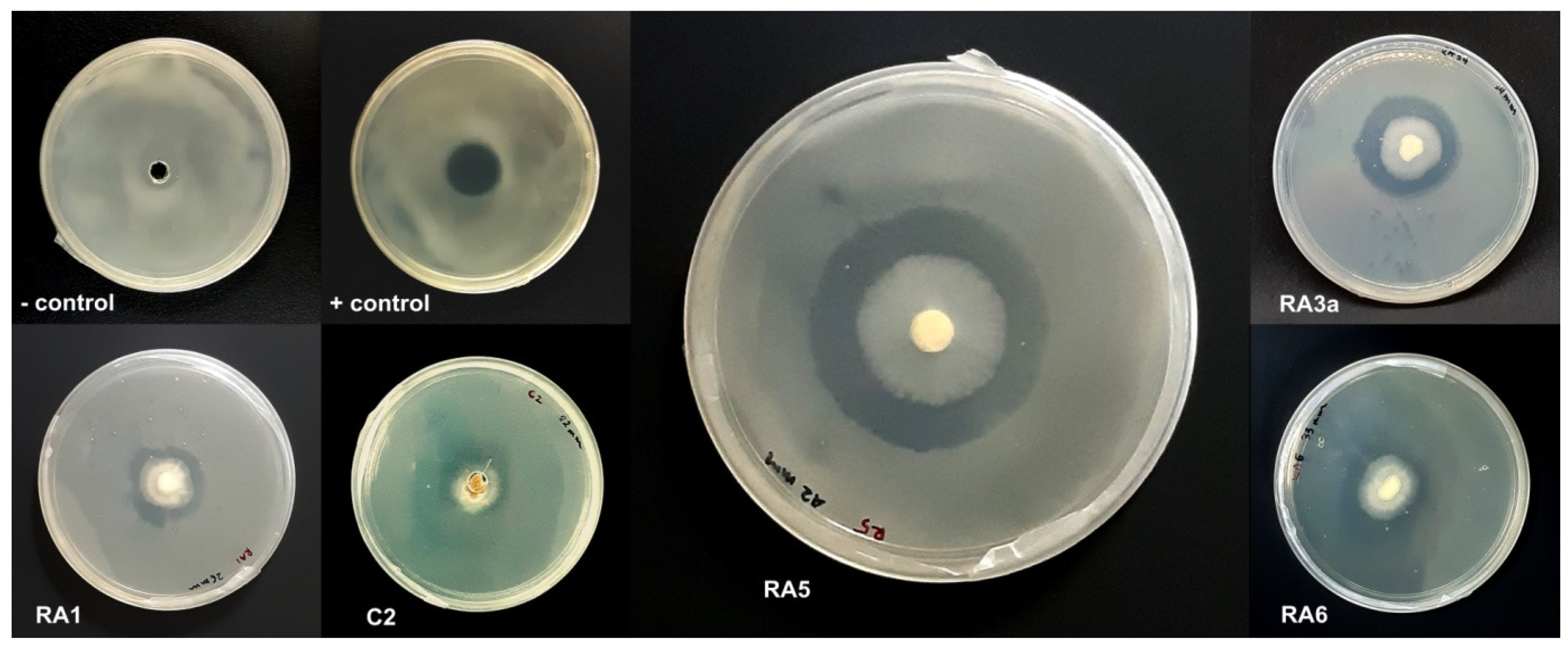
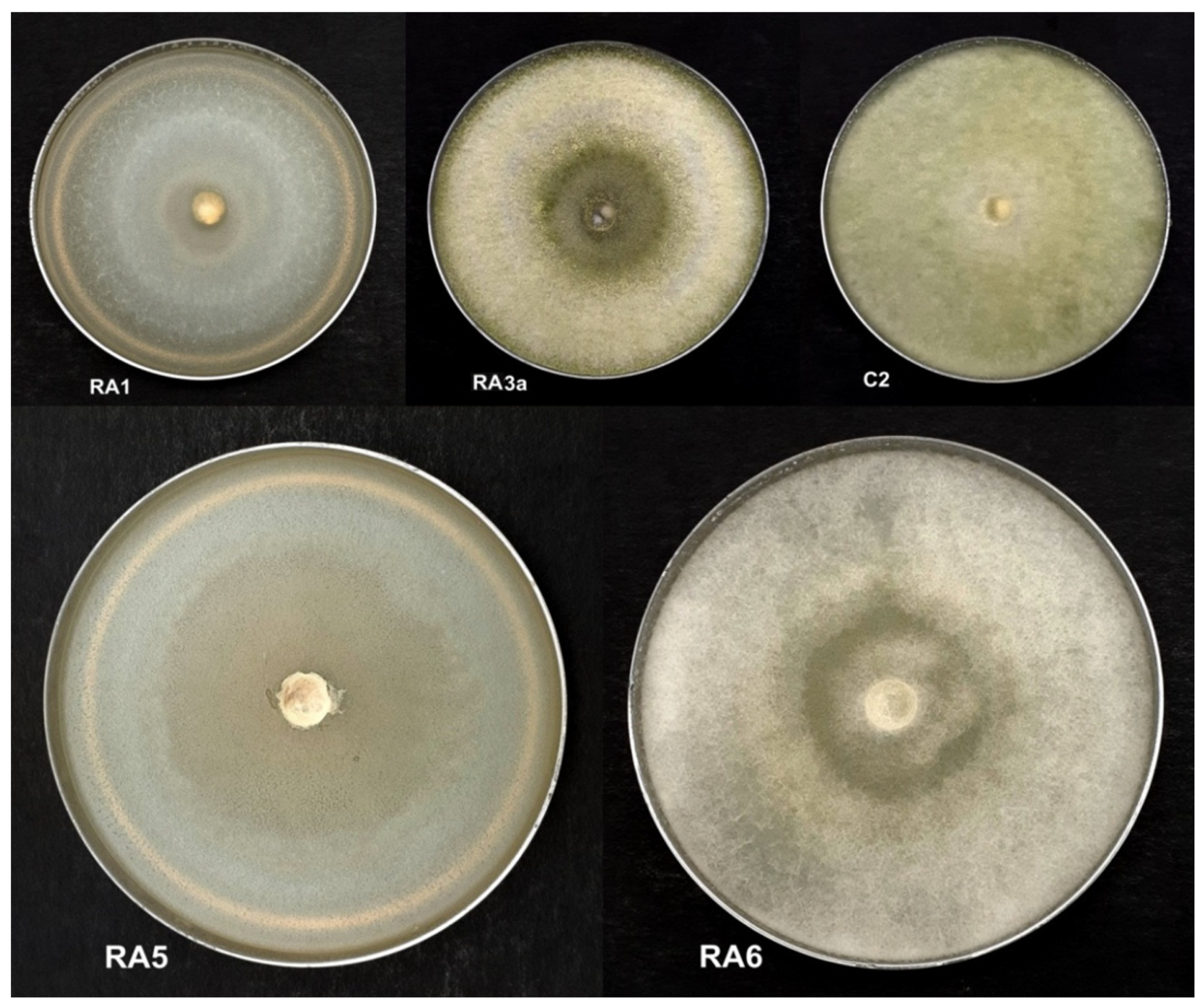
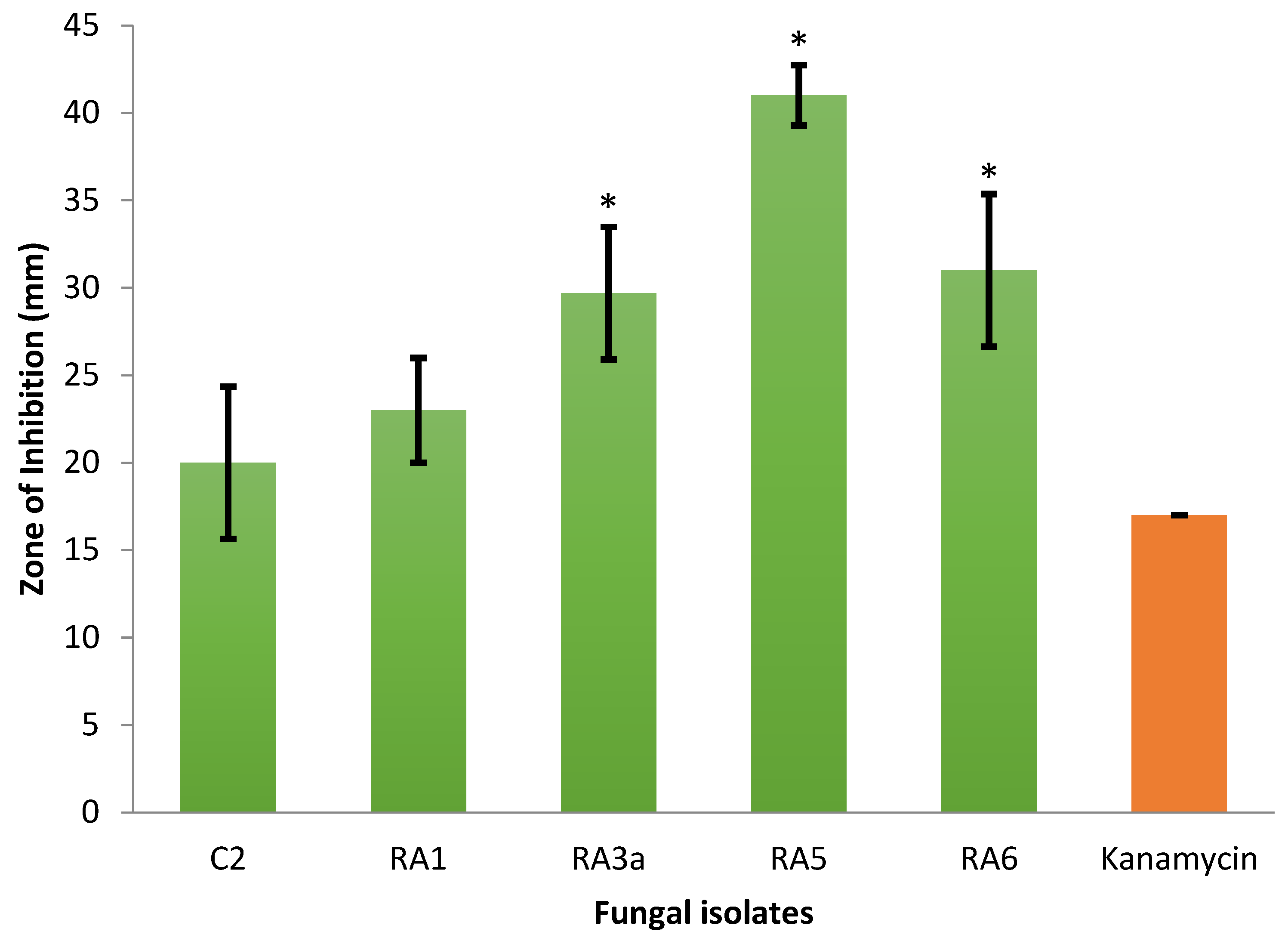
3.1. Antagonism of Fungal Isolates against E. mallotivora Strain BT-MARDI
3.2. Genomic DNA Extraction
3.3. Molecular Identification of Strains
3.4. Genome Sequencing
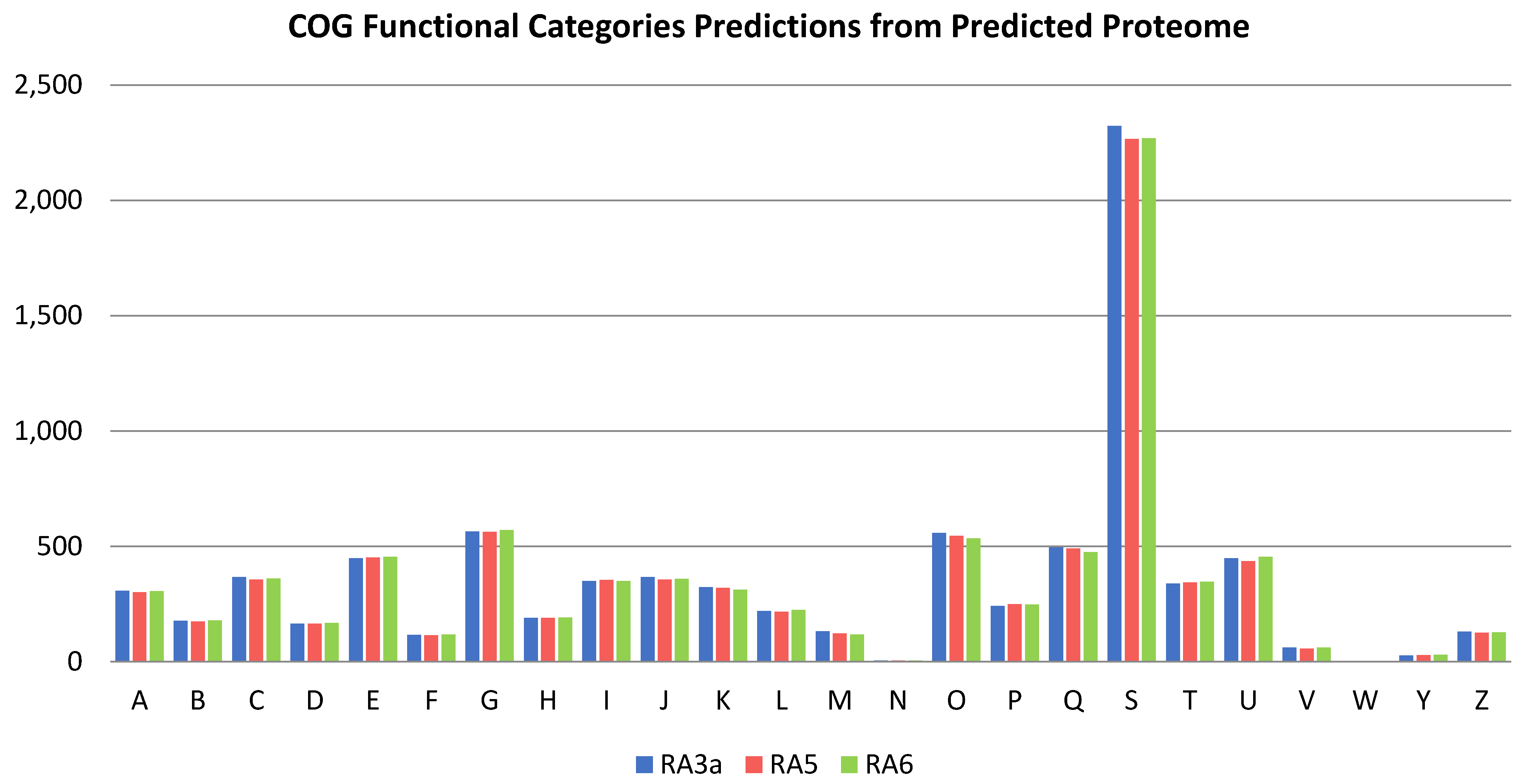
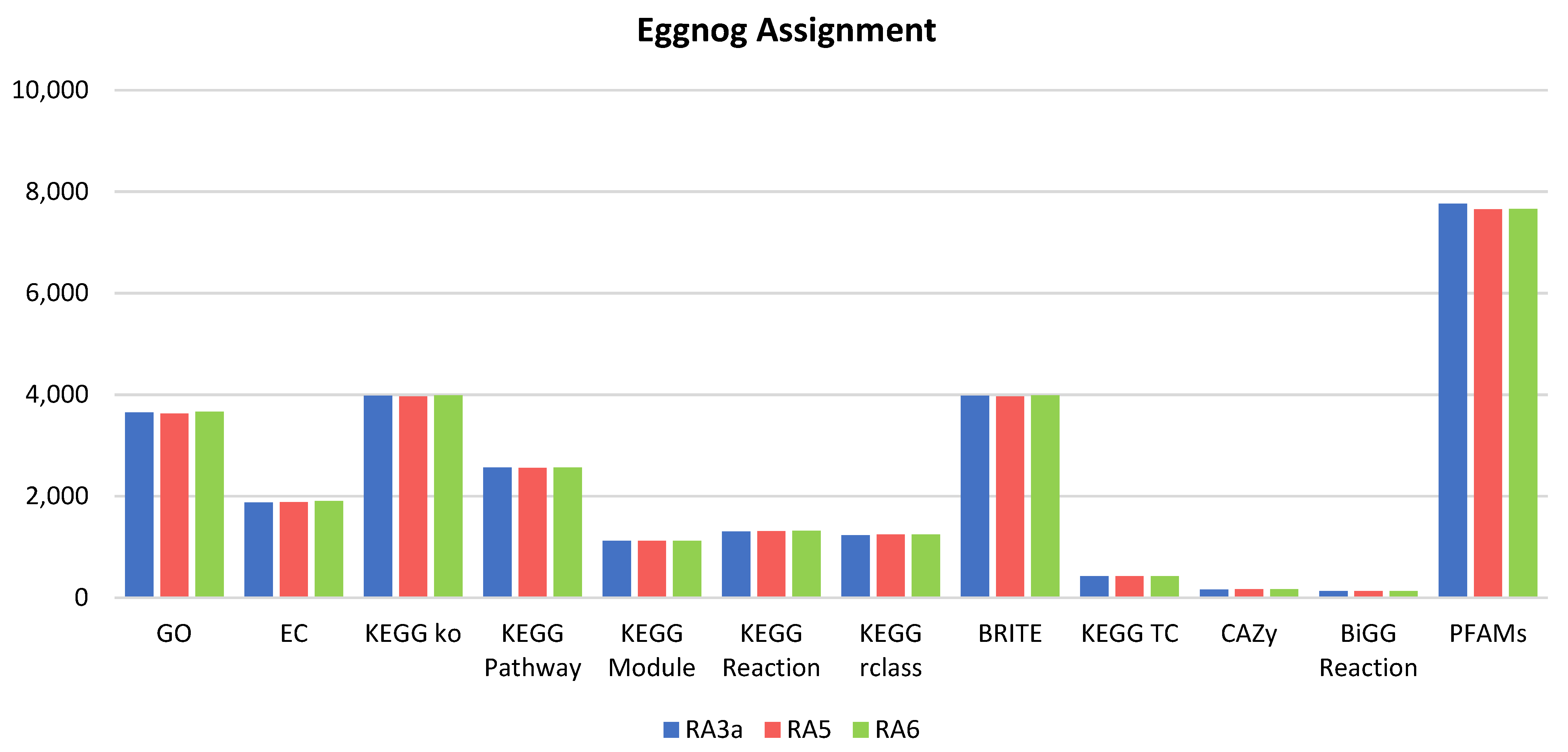
3.5. Genome Analysis
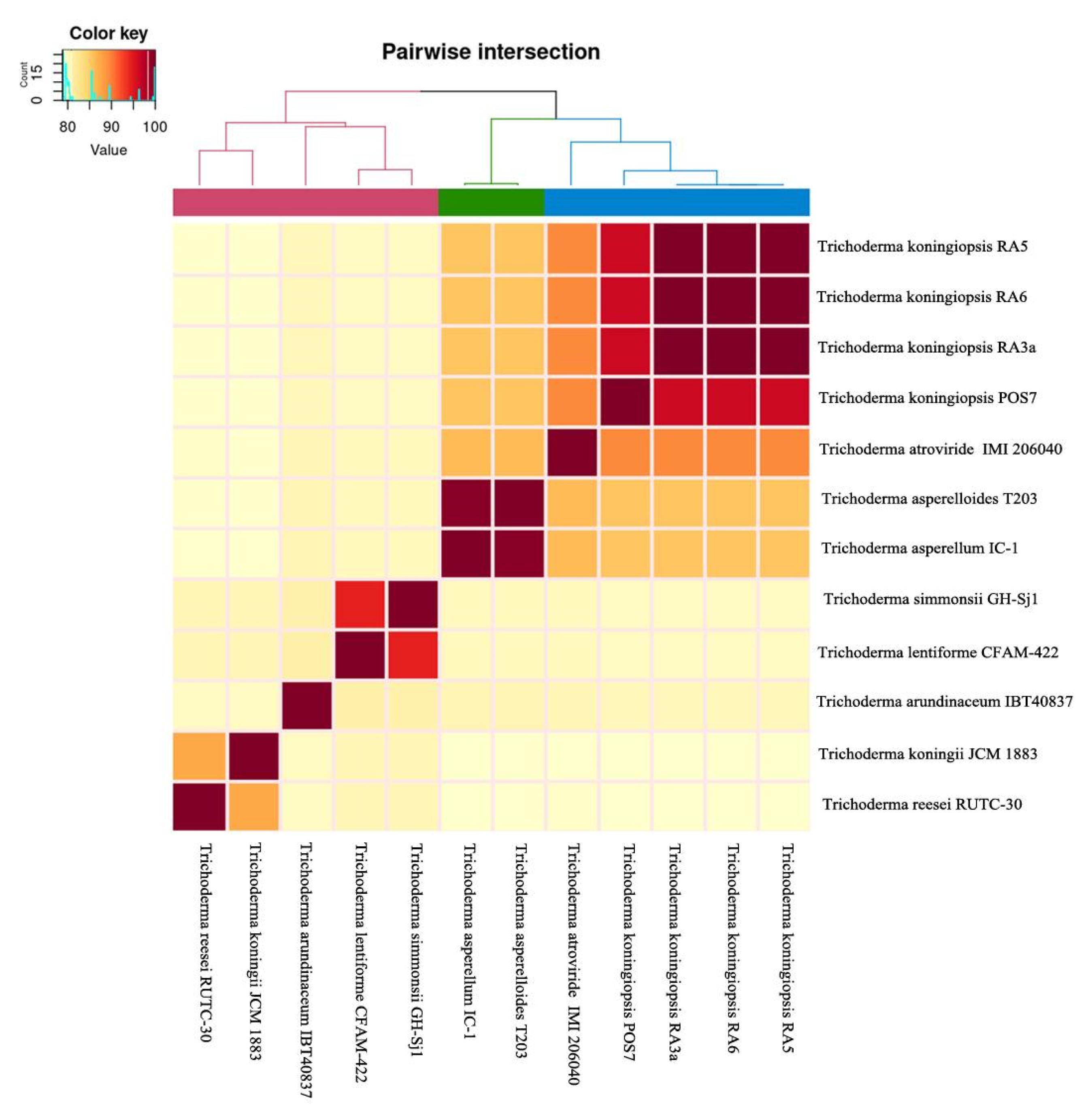
3.6. Comparative Genomics and Phylogenomic Analysis
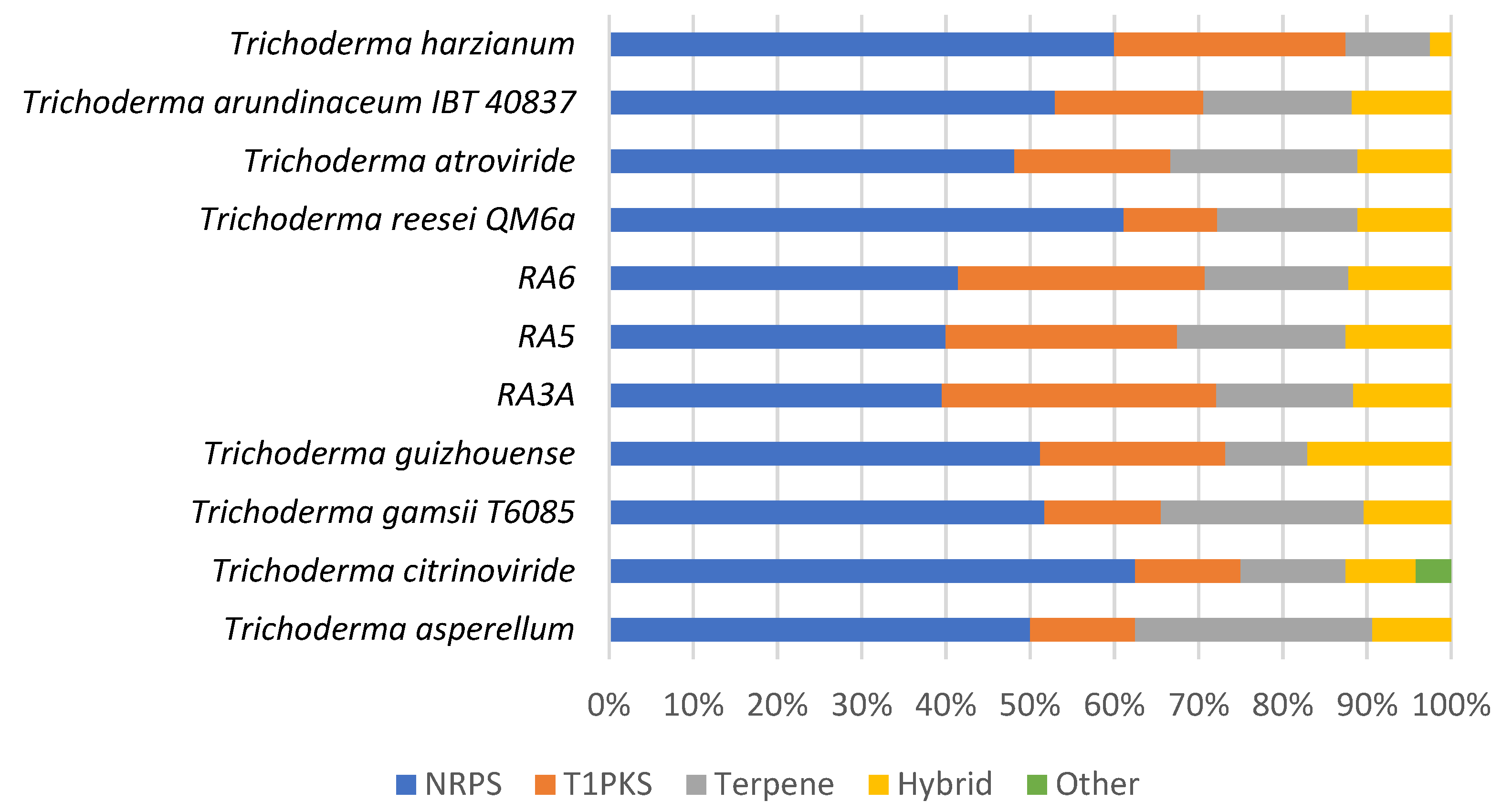
3.7. Secondary Metabolite Clusters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FAOSTAT (Food and Agriculture Organisation of the United Nations Statistics Division). Available online: https://www.fao.org/3/ca5688en/CA5688EN.pdf (accessed on 20 July 2021).
- Maktar, N.H.; Kamis, S.; Mohd Yusof, F.Z.; Hussain, N.H. Erwinia papayae causing papaya dieback in Malaysia. Plant Pathol. 2008, 57, 774. [Google Scholar] [CrossRef]
- Mat Amin, N.; Bunawan, H.; Ahmad Redzuan, R.; Jaganath, I.B.S. Erwinia Mallotivora sp., a new pathogen of papaya (Carica papaya) in Peninsular Malaysia. Int. J. Mol. Sci. 2010, 12, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Ahmad Redzuan, R.; Abu Bakar, N.; Rozano, L.; Badrun, R.; Mat Amin, N.; Mohd Raih, M.F. Draft genome sequence of Erwinia mallotivora BT-MARDI, causative agent of papaya dieback disease. Genome Announc. 2014, 2, e00375-14. [Google Scholar] [CrossRef]
- Supian, S.; Saidi, N.B.; Wee, C.-Y.; Abdullah, M.P. Antioxidant-mediated response of a susceptible papaya cultivar to a compatible strain of Erwinia mallotivora. Physiol. Mol. Plant Pathol. 2017, 98, 37–45. [Google Scholar] [CrossRef]
- Noor Shahida, Y.; Awang, Y.; Sijam, K.; Noriha, M.A.; Satar, M.G.M. Biochemical changes and leaf photosynthesis of Erwinia mallotivora infected papaya (Carica papaya) aeedlings. Am. J. Plant Physiol. 2016, 11, 12–22. [Google Scholar] [CrossRef]
- Pathania, N.; Justo, V.; Magdalita, P.; de la Cueva, F.; Herradura, L.; Waje, A.; Lobres, A.; Cueto, A.; Dillon, N.; Vawdrey, L.; et al. Integrated Disease Management Strategies for the Productive, Profitable and Sustainable Production of High Quality Papaya Fruit in the Southern Philippines and Australia—Final Report. Available online: https://www.aciar.gov.au/publication/technical-publications/integrated-disease-management-strategies-productive-profitable-and-sustainable (accessed on 20 July 2021).
- Mohd Said, N.A.; Abu Bakar, N.; Lau, H.Y. Label-free detection of Erwinia mallotivora DNA for papaya dieback disease using electrochemical impedance spectroscopy approach. ASM Sci. J. 2020, 4, 1–8. [Google Scholar]
- Ramachandran, K.; Manaf, U.A.; Zakaria, L. Molecular characterization and pathogenicity of Erwinia spp. associated with pineapple [Ananas comosus (L.) Merr.] and papaya (Carica papaya L.). J. Plant Prot. Res. 2015, 55, 396–404. [Google Scholar] [CrossRef]
- Bunawan, H.; Baharum, S.N. Papaya dieback in Malaysia: A step towards a new insight of disease resistance. Iran J. Biotechnol. 2015, 13, 1–2. [Google Scholar] [CrossRef][Green Version]
- Mohd Khairil, J.; Muhammad Munzir, M. Experiences in managing bacterial dieback disease of papaya in Malaysia. Acta Hortic. 2014, 1022, 125–132. [Google Scholar] [CrossRef]
- Suharjo, R.; Oktaviana, H.A.; Aeny, T.N.; Ginting, C.; Wardhana, R.A.; Nugroho, A.; Ratdiana, R. Erwinia mallotivora is the causal agent of papaya bacterial crown rot disease in Lampung Timur, Indonesia. Plant Prot. Sci. 2021, 57, 122–133. [Google Scholar] [CrossRef]
- Hanagasaki, T.; Yamashiro, M.; Gima, K.; Takushi, T.; Kawano, S. Characterization of Erwinia sp. causing black rot of papaya (Carica papaya) first recorded in Okinawa Main Island, Japan. Plant Pathol. 2021, 70, 932–942. [Google Scholar] [CrossRef]
- Wee, C.Y.; Muhammad Hanam, H.; Mohd Waznul Adly, M.Z.; Khairun, H.N. Expression of defense-related genes in papaya seedling infected with Erwinia mallotivora using real-time PCR. J. Trop. Agric. Fd. Sci. 2014, 42, 73–82. [Google Scholar]
- Hamid, M.H.; Rozano, L.; Chien Yeong, W.; Abdullah, J.O.; Saidi, N.B. Analysis of MAP kinase MPK4/MEKK1/MKK genes of Carica papaya L. comparative to other plant homologues. Bioinformation 2017, 13, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Juri, N.M.; Samsuddin, A.F.; Abdul-Murad, A.M.; Tamizi, A.A.; Shaharuddin, N.A.; Abu-Bakar, N. Discovery of pathogenesis related and effector genes of Erwinia mallotivora in Carica papaya (Eksotika I) seedlings via transcriptomic analysis. Int. J. Agric. Biol. 2020, 23, 1021–1032. [Google Scholar] [CrossRef]
- Juri, N.M.; Samsuddin, A.F.; Abdul-Murad, A.M.; Tamizi, A.A.; Hassan, M.A.; Abu-Bakar, N. In silico analysis and functional characterization of FHUB, a component of Erwinia mallotivora ferric hydroxamate uptake system. J. Teknol. 2020, 82, 83–90. [Google Scholar] [CrossRef]
- Abu-Bakar, N.; Juri, N.M.; Abu-Bakar, R.A.H.; Sohaime, M.Z.; Badrun, R.; Sarip, J.; Hassan, M.A.; Ahmad, K. Recombinant protein foliar application activates systemic acquired resistance and increases tolerance against papaya dieback disease. Asian J. Agric. Rural Dev. 2021, 11, 1–9. [Google Scholar] [CrossRef]
- Abu Bakar, N.S.; Saidi, N.B.; Rozano, L.; Abdullah, M.P.; Supian, S. In-silico characterization and expression analysis of NB-ARC genes in response to Erwinia mallotivora in Carica papaya. Sains Malays. 2021, 50, 2591–2602. [Google Scholar] [CrossRef]
- Tamizi, A.-A.; Abu-Bakar, N.; Samsuddin, A.-F.; Rozano, L.; Ahmad-Redzuan, R.; Abdul-Murad, A.-M. Characterisation and mutagenesis study of an alternative sigma factor gene (hrpL) from Erwinia mallotivora reveal its central role in papaya dieback disease. Biology 2020, 9, 323. [Google Scholar] [CrossRef] [PubMed]
- Sekeli, R.; Hamid, M.H.; Razak, R.A.; Wee, C.Y.; Ong-Abdullah, J. Malaysian Carica papaya L. var. Eksotika: Current research strategies fronting challenges. Front. Plant Sci. 2018, 9, 1380. [Google Scholar] [CrossRef]
- Sekeli, R.; Nazaruddin, N.H.; Tamizi, A.A.; Mat Amin, N.; Wee, C.Y.; Sarip, J.; Abdullah, N.; Saidi, N.I.; Abdul Razak, R.; Zulkifli, Z. Enhancing Eksotika papaya resistance to dieback disease through quorum quenching. J. Trop. Plant Physiol. 2019, 11, 1–9. [Google Scholar]
- Mohd-Azhar, H.; Sarip, J.; Ghazali, N.F.; Mohd Razikin, M.Z.; Mariatulqabtiah, A.R. Tolerance level of grafted papaya plants against papaya dieback disease. Malays. Appl. Biol. 2021, 50, 95–103. [Google Scholar]
- Mohd Azhar, H.; Johari, S.; Nur Sulastri, J.; Razali, M.; Muhammad Zulfa, M.R.; Noor Faimah, G.; Mariatulqabtiah, A.R. Field performance of selected papaya hybrids for tolerance to dieback disease. J. Trop. Agric. Fd. Sci. 2020, 48, 25–33. [Google Scholar]
- Sarip, J.; Radzuan, S.M.; Ghazali, M.F.; Norasiah, R. Viorica: A Papaya Variety Highly Tolerant to Dieback Disease. In Proceedings of the International Congress of the Malaysian Society for Microbiology, Penang, Malaysia, 7–10 December 2015. [Google Scholar]
- Mat Amin, N.; Nor Rahim, M.Y.; Ahmad Redzuan, R.; Tamizi, A.A.; Bunawan, H. Acyl homoserine lactonase genes from Bacillus species isolated from tomato rhizosphere soil in Malaysia. Res. J. Appl. Sci. 2016, 11, 656–659. [Google Scholar]
- Mat Amin, N.; Nor Rahim, M.Y.; Ahmad Redzuan, R.; Tamizi, A.A.; Bunawan, H. Quorum quenching bacteria isolated from rice and tomato rhizosphere soil in Malaysia. J. Appl. Biol. Sci. 2016, 10, 61–63. [Google Scholar]
- Blog Rasmi MARDI. Available online: https://blogmardi.wordpress.com/2017/08/02/ (accessed on 6 December 2021).
- All Cosmos. Available online: https://allcosmos.com/services-view/dieback-buster-95-2dbottle/ (accessed on 6 December 2021).
- Rivarez, M.P.S.; Parac, E.P.; Dimasingkil, S.F.M.; Magdalita, P.M. Influence of native endophytic bacteria on the growth and bacterial crown rot tolerance of papaya (Carica papaya). Eur. J. Plant Pathol. 2021, 161, 593–606. [Google Scholar] [CrossRef]
- Mohd Taha, M.D.; Mohd Jaini, M.F.; Saidi, N.B.; Abdul Rahim, R.; Md Shah, U.K.; Mohd Hashim, A. Biological control of Erwinia mallotivora, the causal agent of papaya dieback disease by indigenous seed-borne endophytic lactic acid bacteria consortium. PLoS ONE 2019, 14, e0224431. [Google Scholar] [CrossRef]
- Mukhopadhyay, R.; Kumar, D. Trichoderma: A beneficial antifungal agent and insights into its mechanism of biocontrol potential. Egypt J. Biol. Pest Control 2020, 30, 133. [Google Scholar] [CrossRef]
- Harman, G.E. Overview of mechanisms and uses of Trichoderma spp. Phytopathology 2006, 96, 190–194. [Google Scholar] [CrossRef]
- Keswani, C.; Mishra, S.; Sarma, B.K.; Singh, S.P.; Singh, H.B. Unraveling the efficient applications of secondary metabolites of various Trichoderma spp. Appl. Microbiol. Biotechnol. 2014, 98, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Cornejo, H.A.; Macías-Rodríguez, L.; Del-Val, E.; Larsen, J. Ecological functions of Trichoderma spp. and their secondary metabolites in the rhizosphere: Interactions with plants. FEMS Microbiol. Ecol. 2016, 92, fiw036. [Google Scholar] [CrossRef] [PubMed]
- Bitas, V.; Kim, H.S.; Bennett, J.W.; Kang, S. Sniffing on microbes: Diverse roles of microbial volatile organic compounds in plant health. Mol. Plant Microbe Interact. 2013, 26, 835–843. [Google Scholar] [CrossRef]
- Guo, Y.; Ghirardo, A.; Weber, B.; Schnitzler, J.P.; Benz, J.P.; Rosenkranz, M. Trichoderma species differ in their volatile profiles and in antagonism toward ectomycorrhiza Laccaria bicolor. Front. Microbiol. 2019, 10, 891. [Google Scholar] [CrossRef]
- Khan, R.A.A.; Najeeb, S.; Hussain, S.; Xie, B.; Li, Y. Bioactive secondary metabolites from Trichoderma spp. against phytopathogenic fungi. Microorganisms 2020, 8, 817. [Google Scholar] [CrossRef] [PubMed]
- Rush, T.A.; Shrestha, H.K.; Gopalakrishnan Meena, M.; Spangler, M.K.; Ellis, J.C.; Labbé, J.L.; Abraham, P.E. Bioprospecting Trichoderma: A systematic roadmap to screen genomes and natural products for biocontrol applications. Front. Fungal Biol. 2021, 2, 716511. [Google Scholar] [CrossRef]
- Vargas Gil, S.; Pastor, S.; Marcha, G.J. Quantitative isolation of biocontrol agents Trichoderma spp., Gliocladium spp. and actinomycetes from soil with culture media. Microbiol. Res. 2009, 164, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Yuan-Ying, S.; Lei, C. An optimized protocol of single spore isolation for fungi. Cryptogam. Mycol. 2013, 34, 349–356. [Google Scholar] [CrossRef]
- Magaldi, S.; Mata-Essayag, S.; Hartung de Capriles, C.; Perez, C.; Colella, M.T.; Olaizola, C.; Ontiveros, Y. Well diffusion for antifungal susceptibility testing. Int. J. Infect. Dis. 2004, 8, 39–45. [Google Scholar] [CrossRef]
- Valgas, C.; de Souza, S.M.; Smânia, E.F.A.; Smânia, A., Jr. Screening methods to determine antibacterial activity of natural products. Braz. J. Microbiol. 2007, 38, 369–380. [Google Scholar] [CrossRef]
- Kopchinskiy, A.; Komoń, M.; Kubicek, C.P.; Druzhinina, I.S. TrichoBLAST: A multilocus database for Trichoderma and Hypocrea identifications. Mycol. Res. 2005, 109, 658–660. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2021, 9, 357–359. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness. Methods Mol. Biol. 2019, 1962, 227–245. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.M.; Stajich, J. Funannotate v1.8.1: Eukaryotic Genome Annotation. Available online: https://zenodo.org/record/4054262#.YhxHjOpBxPY (accessed on 20 August 2021).
- Käll, L.; Krogh, A.; Sonnhammer, E.L.L. A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 2004, 338, 1027–1036. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef]
- Burge, S.W.; Daub, J.; Eberhardt, R.; Tate, J.; Barquist, L.; Nawrocki, E.P.; Eddy, S.R.; Gardner, P.P.; Bateman, A. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 2013, 41, D226–D232. [Google Scholar] [CrossRef]
- Arias-Carrasco, R.; Vásquez-Morán, Y.; Nakaya, H.I.; Maracaja-Coutinho, V. StructRNAfinder: An automated pipeline and web server for RNA families prediction. BMC Bioinform. 2018, 19, 55. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Moxon, S.; Marshall, M.; Khanna, A.; Eddy, S.R.; Bateman, A. Rfam: Annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005, 33, D121–D124. [Google Scholar] [CrossRef] [PubMed]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Mathelier, A. Intervene: A tool for intersection and visualization of multiple gene or genomic region sets. BMC Bioinform. 2017, 18, 287. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H.; Zhang, G.; Gu, Y.Q.; Coleman-Derr, D.; Xia, Q.; et al. OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019, 47, W52–W58. [Google Scholar] [CrossRef]
- Cai, F.; Druzhinina, I.S. In honor of John Bissett: Authoritative guidelines on molecular identification of Trichoderma. Fungal Divers. 2021, 107, 1–69. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Bolchacova, E.; Voigt, K.; Crous, P.W.; et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef]
- Druzhinina, I.; Kubicek, C.P. Species concepts and biodiversity in Trichoderma and Hypocrea: From aggregate species to species clusters? J. Zhejiang Univ. Sci. B 2005, 6, 100–112. [Google Scholar] [CrossRef]
- Liu, Y.J.; Whelen, S.; Hall, B.D. Phylogenetic relationships among ascomycetes: Evidence from an RNA polymerse II subunit. Mol. Biol. Evol. 1999, 16, 1799–1808. [Google Scholar] [CrossRef] [PubMed]
- Kubicek, C.P.; Steindorff, A.S.; Chenthamara, K.; Manganiello, G.; Henrissat, B.; Zhang, J.; Cai, F.; Kopchinskiy, A.G.; Kubicek, E.M.; Kuo, A.; et al. Evolution and comparative genomics of the most common Trichoderma species. BMC Genomics 2019, 20, 485. [Google Scholar] [CrossRef] [PubMed]
- Druzhinina, I.S.; Kopchinskiy, A.G.; Kubicek, E.M.; Kubicek, C.P. A complete annotation of the chromosomes of the cellulase producer Trichoderma reesei provides insights in gene clusters, their expression and reveals genes required for fitness. Biotechnol. Biofuels 2016, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- El Komy, M.H.; Saleh, A.A.; Eranthodi, A.; Molan, Y.Y. Characterization of novel Trichoderma asperellum isolates to select effective biocontrol agents against tomato Fusarium wilt. Plant Pathol. J. 2015, 31, 50–60. [Google Scholar] [CrossRef]
- Liu, L.; Tang, M.-C.; Tang, Y. Fungal Highly Reducing Polyketide Synthases Biosynthesize Salicylaldehydes That Are Precursors to Epoxycyclohexenol Natural Products. J. Am. Chem. Soc. 2019, 141, 19538–19541. [Google Scholar] [CrossRef]
- Munoz, F.M.; Demmler, G.J.; Travis, W.R.; Ogden, A.K.; Rossmann, S.N.; Rinaldi, M.G. Trichoderma longibrachiatum infection in a pediatric patient with aplastic anemia. J. Clin. Microbiol. 1997, 35, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Sautour, M.; Chrétien, M.L.; Valot, S.; Lafon, I.; Basmaciyana, L.; Legouge, C.; Verrier, T.; Gonssaud, B.; Abou-Hanna, H.; Dalle, F.; et al. First case of proven invasive pulmonary infection due to Trichoderma longibrachiatum in a neutropenic patient with acute leukemia. J. Mycol. Med. 2018, 28, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Waghunde, R.R.; Shelake, R.M.; Sabalpara, A.N. Trichoderma: A significant fungus for agriculture and environment. Afr. J. Agric. Res. 2016, 11, 1952–1965. [Google Scholar] [CrossRef]
- Zin, N.A.; Badaluddin, N.A. Biological functions of Trichoderma spp. for agriculture applications. Ann. Agric. Sci. 2020, 65, 168–178. [Google Scholar] [CrossRef]
- Thapa, S.; Rai, N.; Limbu, A.K.; Joshi, A. Impact of Trichoderma sp. in agriculture: A mini-review. J. Biol. Today’s World 2020, 9, 227. [Google Scholar]
- Singh, A.; Sarma, B.K.; Singh, H.B.; Upadhyay, R.S. Trichoderma: A silent worker of plant rhizosphere. In Biotechnology and Biology of Trichoderma, 1st ed.; Gupta, V.K., Schmoll, M., Herrera-Estrella, A., Upadhyay, R.S., Druzhinina, I., Tuohy, M.G., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 533–542. [Google Scholar]
- Stocco, M.; Mónaco, C.; Cordo, C. A comparison of preservation methods for Trichoderma harzianum cultures. Rev. Iberoam. Micol. 2010, 27, 213. [Google Scholar] [CrossRef]
- Sood, M.; Kapoor, D.; Kumar, V.; Sheteiwy, M.S.; Ramakrishnan, M.; Landi, M.; Araniti, F.; Sharma, A. Trichoderma: The “secrets” of a multitalented biocontrol agent. Plants 2020, 9, 762. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov. E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Z.; Akdemir, A.; Tremmel, G.; Imoto, S.; Miyano, S.; Shibuya, T.; Yamaguchi, R. Nanopore basecalling from a perspective of instance segmentation. BMC Bioinform. 2020, 21, 136. [Google Scholar] [CrossRef]
- Li, M.F.; Li, G.H.; Zhang, K.Q. Non-volatile metabolites from Trichoderma spp. Metabolites 2019, 9, 58. [Google Scholar] [CrossRef]
- Parker, S.R.; Cutler, H.G.; Schreiner, P.R. Koninginin C: A biologically active natural product from Trichoderma koningii. Biosci. Biotechnol. Biochem. 1995, 59, 1126–1127. [Google Scholar] [CrossRef][Green Version]
- Liu, K.; Yang, Y.B.; Chen, J.L.; Miao, C.P.; Wang, Q.; Zhou, H.; Chen, Y.W.; Li, Y.Q.; Ding, Z.T.; Zhao, L.X. Koninginins N-Q, polyketides from the endophytic fungus Trichoderma koningiopsis harbored in Panax notoginseng. Nat. Prod. Bioprospect. 2016, 6, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Shenouda, M.L.; Cox, R.J. Molecular methods unravel the biosynthetic potential of Trichoderma species. RSC Adv. 2021, 11, 3622–3635. [Google Scholar] [CrossRef]
- Berkaew, P.; Soonthornchareonnon, N.; Salasawadee, K.; Chanthaket, R.; Isaka, M. Aurocitrin and Related Polyketide Metabolites from the Wood-Decay Fungus Hypocrea sp. BCC 14122. J. Nat. Prod. 2008, 71, 902–904. [Google Scholar] [CrossRef] [PubMed]
- Hyde, K.D.; Xu, J.; Rapior, S.; Jeewon, R.; Lumyong, S.; Niego, A.G.T.; Abeywickrama, P.D.; Aluthmuhandiram, J.V.S.; Brahamanage, R.S.; Brooks, S.; et al. The amazing potential of fungi: 50 ways we can exploit fungi industrially. Fungal Divers. 2019, 97, 1–136. [Google Scholar] [CrossRef]
- Kjærbølling, I.; Mortensen, U.H.; Vesth, T.; Andersen, M.R. Strategies to establish the link between biosynthetic gene clusters and secondary metabolites. Fungal Genet. Biol. 2019, 130, 107–121. [Google Scholar] [CrossRef]
- Alberti, F.; Foster, G.D.; Bailey, A.M. Natural products from filamentous fungi and production by heterologous expression. Appl. Microbiol. Biotechnol. 2017, 101, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Harvey, C.J.B.; Tang, M.; Schlecht, U.; Horecka, J.; Fischer, C.R.; Lin, H.C.; Li, J.; Naughton, B.; Cherry, J.; Miranda, M.; et al. HEx: A heterologous expression platform for the discovery of fungal natural products. Sci. Adv. 2018, 4, eaar5459. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Soil Sample ID | Location Site, State | Coordinates |
|---|---|---|
| Mardi-2018-TK | Tanjong Karang, Selangor | 3°26′27.8″ N 101°08′50.1″ E |
| Mardi-2018-R | Rembau, Negeri Sembilan | 2°34′26.0″ N 102°05′48.5″ E |
| Mardi-2018-P | Raub, Pahang | 3°49′19.3″ N 101°50′38.4″ E |
| Mardi-2018-C | Mardi Serdang, Selangor | 2°59′24.9″ N 101°41′49.5″ E |
| Mardi-2018-BK | Baling, Kedah | 5°40′05.6″ N 100°49′47.5″ E |
| Treatment | Diameter of Inhibition Zone (mm) | Fungal Genus (Based on Morphology) | Origin of the Fungal Isolate | ||||
|---|---|---|---|---|---|---|---|
| Replicate | Mean | Standard Deviation | |||||
| 1 | 2 | 3 | |||||
| − control | 0 | 0 | 0 | 0 | 0 | - | - |
| + control | 17 | 17 | 17 | 17.0 | 0 | - | - |
| Isolate UKM-M-UW RA5 | 42 | 39 | 42 | 41.0 | 1.73 | Trichoderma | Rembau, Negeri Sembilan |
| Isolate UKM-M-UW RA6 | 33 | 26 | 34 | 31.0 | 4.36 | Trichoderma | Rembau, Negeri Sembilan |
| Isolate UKM-M-UW RA3a | 34 | 28 | 27 | 29.7 | 3.79 | Trichoderma | Rembau, Negeri Sembilan |
| Isolate UKM-M-UW RA1 | 20 | 23 | 26 | 23.0 | 3.0 | Trichoderma | Rembau, Negeri Sembilan |
| Isolate UKM-M-UW C2 | 15 | 22 | 23 | 20.0 | 4.36 | Trichoderma | Serdang, Selangor |
| Isolate | Locus | Closest Match Organism | NCBI Accession Number | Coverage (%) | Identity (%) |
|---|---|---|---|---|---|
| UKM-M-UW RA3a | ITS | Trichoderma sp. strain ZMQRS9 | MT446202.1 | 100 | 99.83 |
| Tef1 | Trichoderma koningiopsis strain LESF360 | KT278986.1 | 100 | 99.65 | |
| Rpb2 | Trichoderma koningiopsis isolate Tkois1 | MT081443.1 | 100 | 99.77 | |
| UKM-M-UW RA5 | ITS | Trichoderma koningiopsis strain 18ASMA001 | MT520621.1 | 100 | 100 |
| Tef1 | Trichoderma koningiopsis strain VSL155 | MT058870.1 | 100 | 99.33 | |
| Rpb2 | Trichoderma koningiopsis isolate Tkois1 | MT081443.1 | 100 | 99.60 | |
| UKM-M-UW RA6 | ITS | Trichoderma koningiopsis strain 18ASMA001 | MT520621.1 | 100 | 100 |
| Tef1 | Trichoderma koningiopsis strain LESF360 | KT278986.1 | 100 | 100 | |
| Rpb2 | Trichoderma koningiopsis isolate Tkois1 | MT081443.1 | 100 | 100 |
| Parameter | RA3a | RA5 | RA6 |
|---|---|---|---|
| Number of contigs | 14 | 11 | 13 |
| Total contigs length | 36,531,570 | 36,477,170 | 36,470,223 |
| Mean contig size | 2,609,397.86 | 3,316,106.36 | 2,805,401.77 |
| Contig size first quartile | 1,043,387 | 3,650,583 | 981,951 |
| Median contig size | 2,049,512 | 3,895,316 | 3,855,011 |
| Contig size third quartile | 5,555,030 | 6,876,866 | 5,268,312 |
| Longest contig | 6,903,293 | 6,995,056 | 6,877,006 |
| Shortest contig | 6075 | 6406 | 5219 |
| Contigs > 500 nt | 14 (100%) | 11 (100%) | 13 (100%) |
| Contigs > 1K nt | 14 (100%) | 11 (100%) | 13 (100%) |
| Contigs > 10K nt | 13 (92.86%) | 10 (90.91%) | 12 (92.31%) |
| Contigs > 100K nt | 11 (78.57%) | 8 (72.73%) | 10 (76.92%) |
| Contigs > 1M nt | 10 (71.43%) | 7 (63.64%) | 8 (61.54) |
| N50 | 5,555,030 | 5,554,967 | 3,979,290 |
| L50 | 3 | 3 | 4 |
| N80 | 2,447,863 | 3,862,469 | 3,855,011 |
| L80 | 6 | 6 | 6 |
| BUSCO Scaffold Stat | RA3a | RA5 | RA6 |
|---|---|---|---|
| Percentage BUSCO | 97.7% | 97.7% | 97.8% |
| Complete BUSCO’s | 4392 | 4391 | 4394 |
| Complete and single copy BUSCO’s | 4378 | 4379 | 4381 |
| Complete and duplicate BUSCO’s | 14 | 12 | 13 |
| Fragmented BUSCO’s | 20 | 20 | 20 |
| Missing BUSCO’s | 82 | 83 | 80 |
| Total BUSCO groups searched | 4494 | ||
| BUSCO Scaffold Stat | RA3a | RA5 | RA6 |
|---|---|---|---|
| Percentage BUSCO | 92.3% | 88.0% | 87.4% |
| Complete BUSCO’s | 4146 | 3955 | 3927 |
| Complete and single copy BUSCO’s | 4137 | 3946 | 3922 |
| Complete and duplicate BUSCO’s | 9 | 9 | 5 |
| Fragmented BUSCO’s | 108 | 219 | 245 |
| Missing BUSCO’s | 240 | 320 | 322 |
| Total BUSCO groups searched | 4494 | ||
| Fungal Strain | Total Clusters | NRPS-Like | PKS | Terpene | Hybrid NRPS/PKS | Hybrid PKS/Terpene |
|---|---|---|---|---|---|---|
| UKM-M-UW RA3a | 43 | 17 | 14 | 7 | 4 | 1 |
| UKM-M-UW RA5 | 40 | 16 | 11 | 8 | 4 | 1 |
| UKM-M-UW RA6 | 41 | 17 | 12 | 7 | 4 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tamizi, A.-A.; Mat-Amin, N.; Weaver, J.A.; Olumakaiye, R.T.; Akbar, M.A.; Jin, S.; Bunawan, H.; Alberti, F. Genome Sequencing and Analysis of Trichoderma (Hypocreaceae) Isolates Exhibiting Antagonistic Activity against the Papaya Dieback Pathogen, Erwinia mallotivora. J. Fungi 2022, 8, 246. https://doi.org/10.3390/jof8030246
Tamizi A-A, Mat-Amin N, Weaver JA, Olumakaiye RT, Akbar MA, Jin S, Bunawan H, Alberti F. Genome Sequencing and Analysis of Trichoderma (Hypocreaceae) Isolates Exhibiting Antagonistic Activity against the Papaya Dieback Pathogen, Erwinia mallotivora. Journal of Fungi. 2022; 8(3):246. https://doi.org/10.3390/jof8030246
Chicago/Turabian StyleTamizi, Amin-Asyraf, Noriha Mat-Amin, Jack A. Weaver, Richard T. Olumakaiye, Muhamad Afiq Akbar, Sophie Jin, Hamidun Bunawan, and Fabrizio Alberti. 2022. "Genome Sequencing and Analysis of Trichoderma (Hypocreaceae) Isolates Exhibiting Antagonistic Activity against the Papaya Dieback Pathogen, Erwinia mallotivora" Journal of Fungi 8, no. 3: 246. https://doi.org/10.3390/jof8030246
APA StyleTamizi, A.-A., Mat-Amin, N., Weaver, J. A., Olumakaiye, R. T., Akbar, M. A., Jin, S., Bunawan, H., & Alberti, F. (2022). Genome Sequencing and Analysis of Trichoderma (Hypocreaceae) Isolates Exhibiting Antagonistic Activity against the Papaya Dieback Pathogen, Erwinia mallotivora. Journal of Fungi, 8(3), 246. https://doi.org/10.3390/jof8030246