
Hybrid Assembly Improves Genome Quality and Completeness of Trametes villosa CCMB561 and Reveals a Huge Potential for Lignocellulose Breakdown

 ,
,  ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Strain and Extraction of Genomic DNA
2.2. MinION Library Preparation and Sequencing
2.3. Illumina Library Preparation and Sequencing
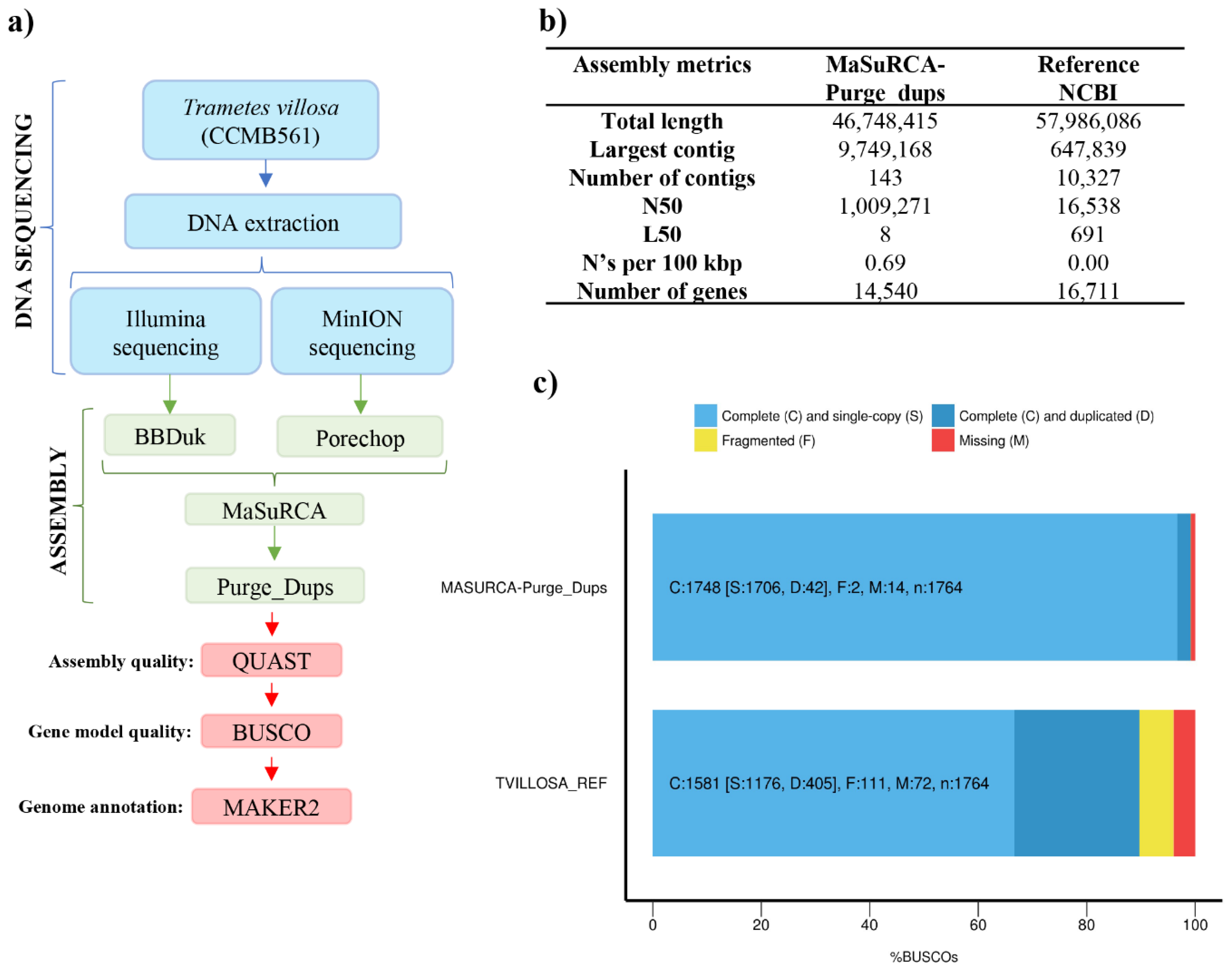
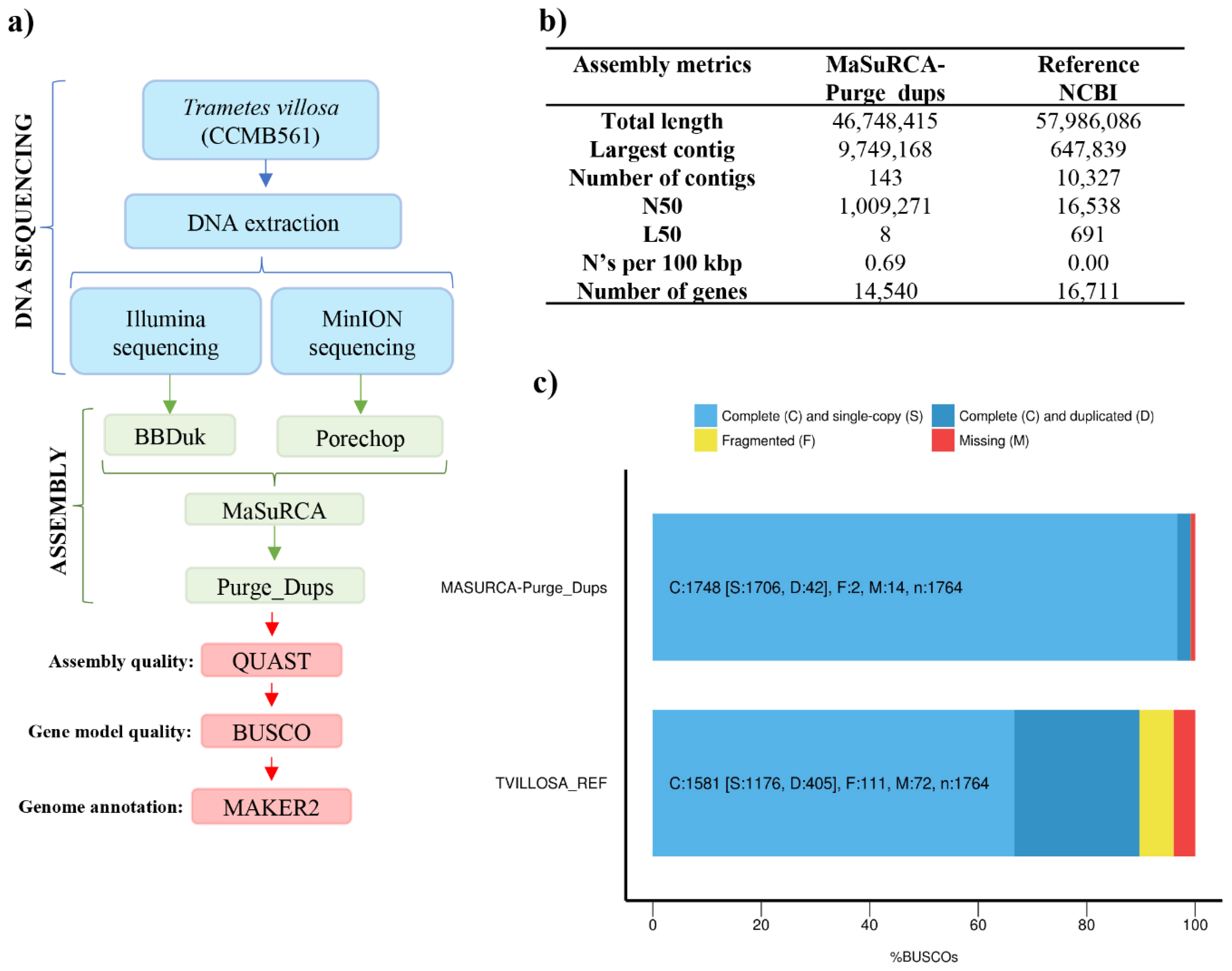
2.4. De Novo Genome Assembly and Assessment
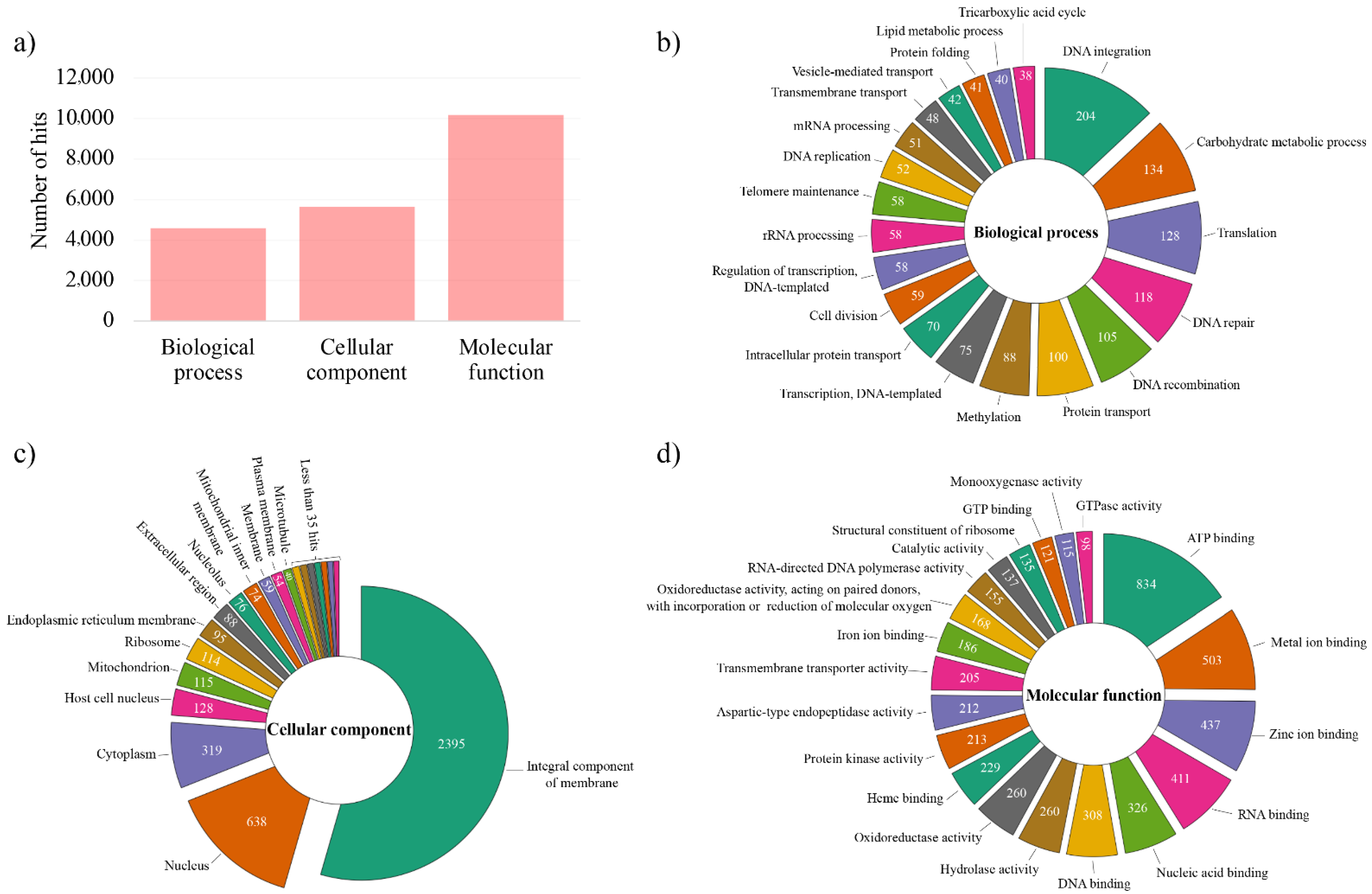
2.5. Genome Annotation and Gene Ontology Analyses
2.6. Repeat Annotation
2.7. Comparative Genomics and Phylogenomics
2.8. CAZy Annotation and Potential for Lignocellulose Degradation
3. Results and Discussion
3.1. Illumina and MinION Sequencing
3.2. Genome Assembly and Assessment
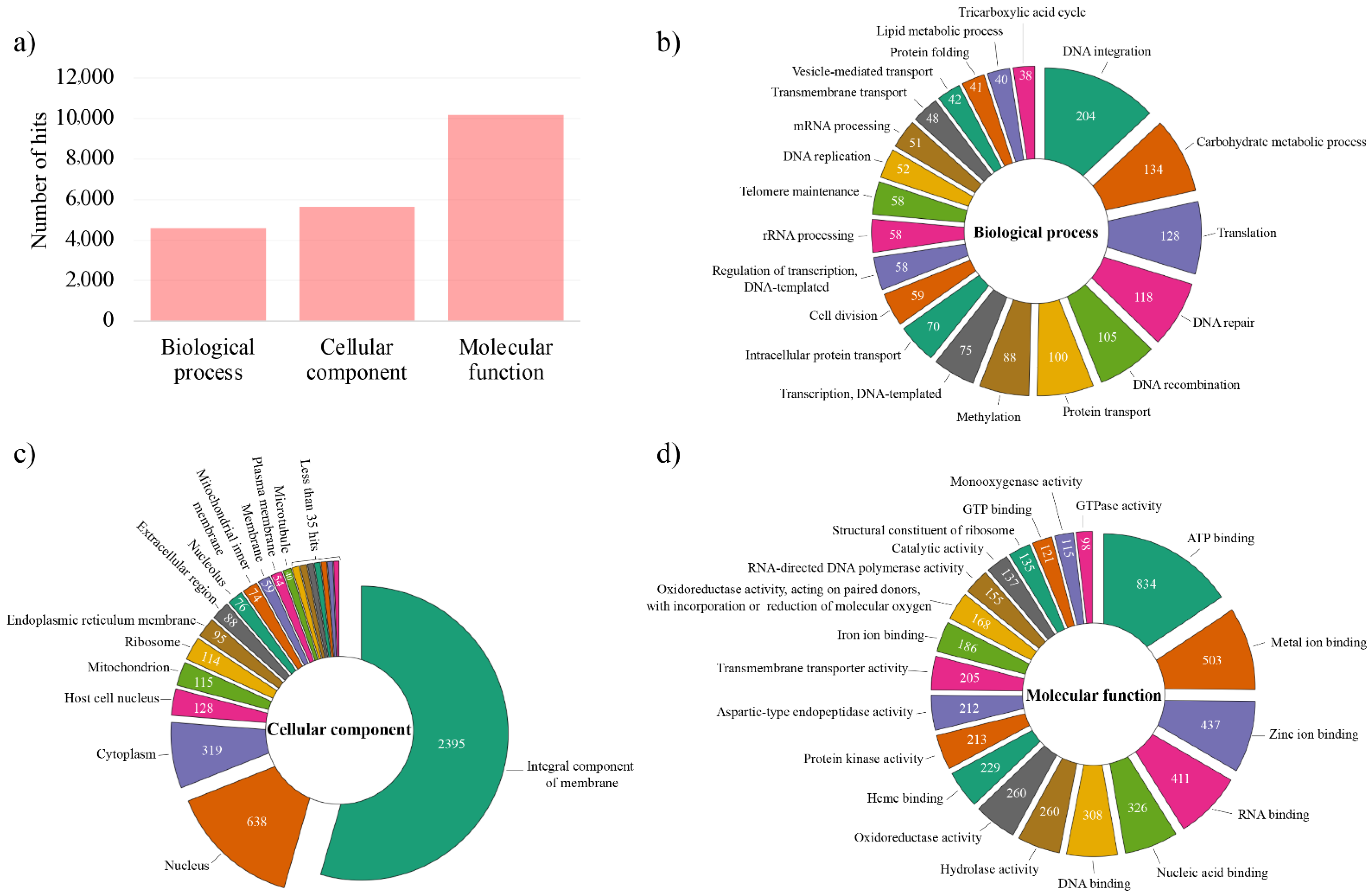
3.3. Genome Annotation and Gene Ontology (GO) Analysis
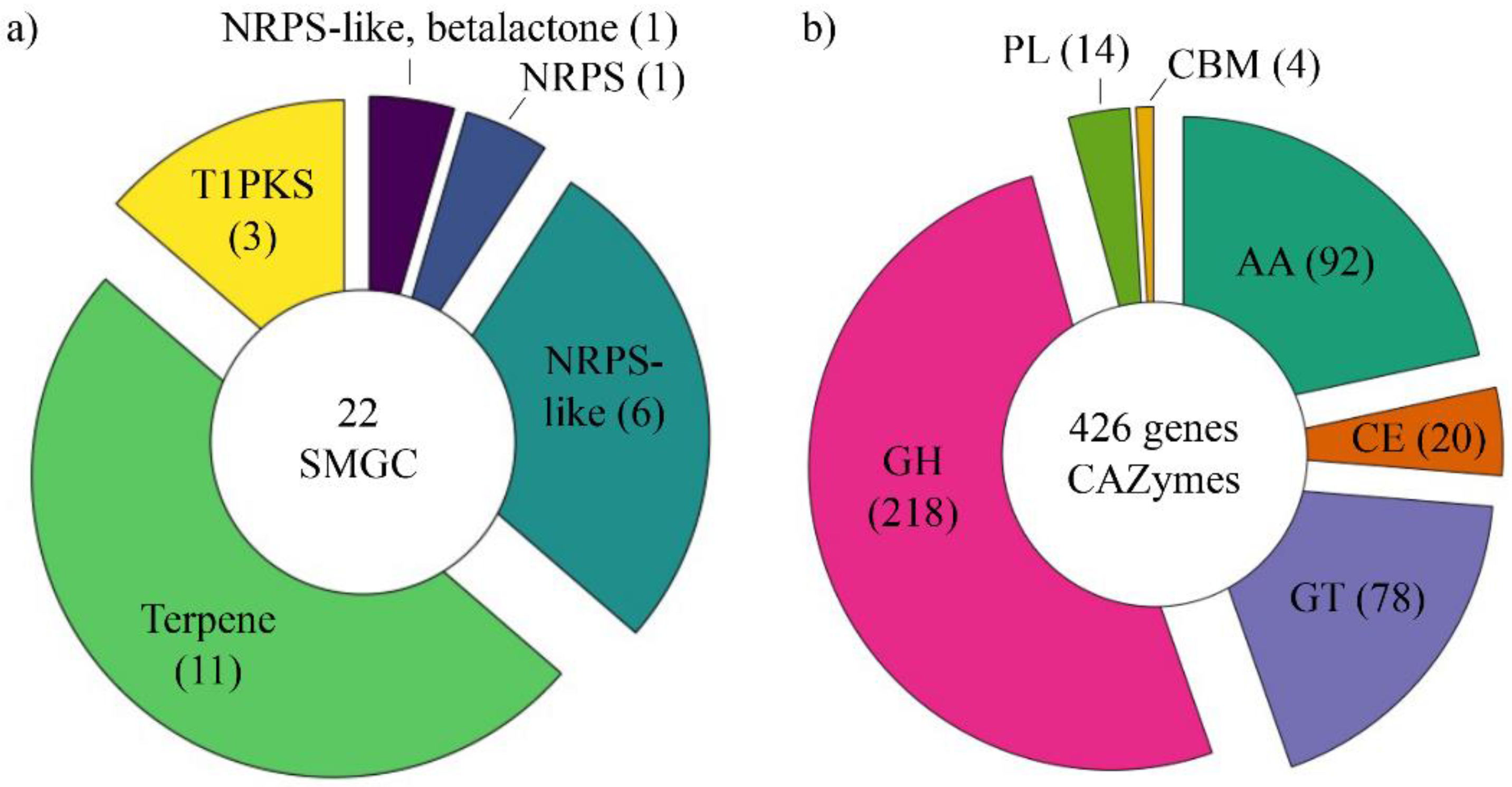
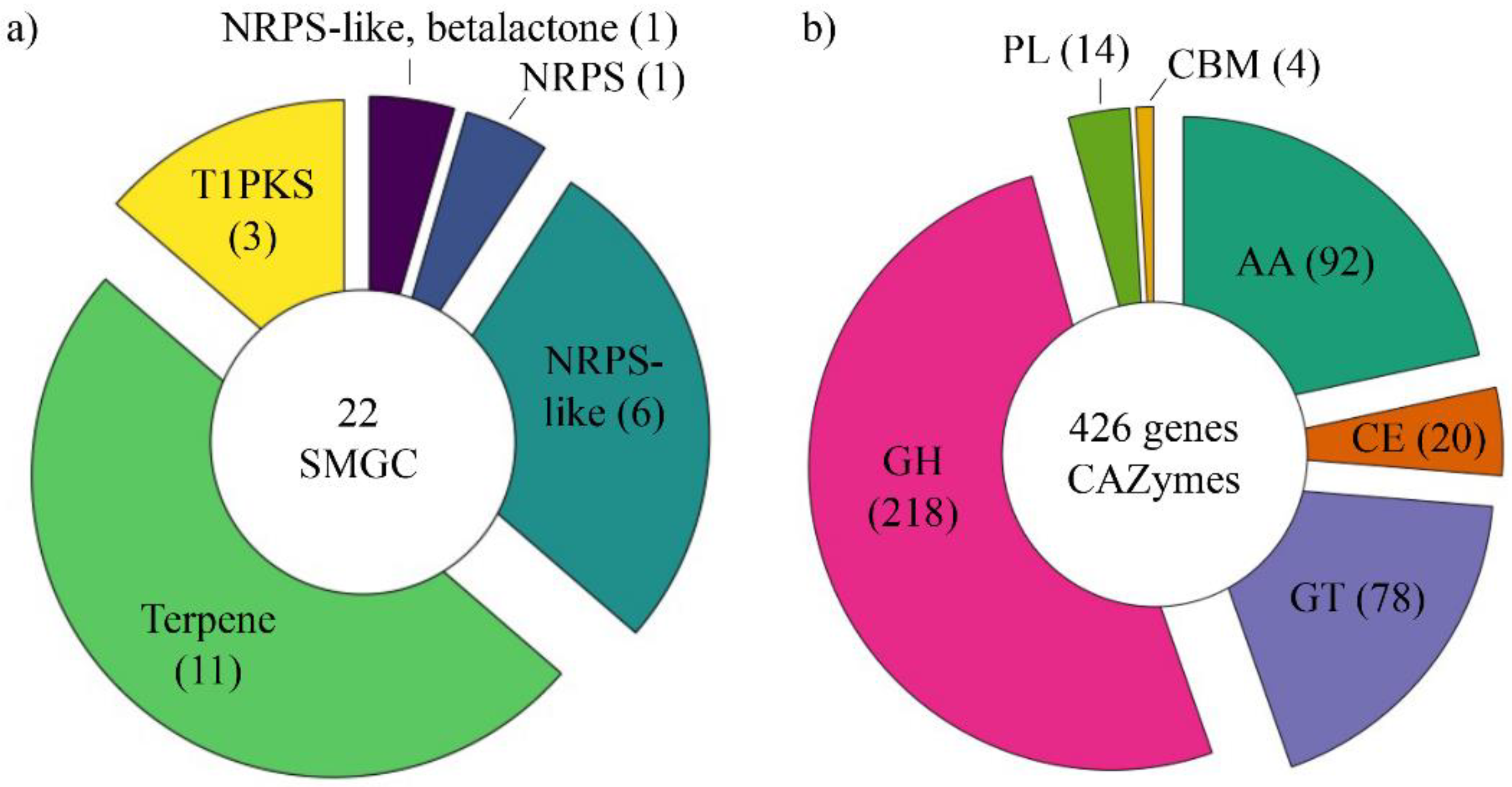
3.4. Annotation of Secondary Metabolite Gene Clusters (SMGCs) and CAZymes of Trametes Villosa CCMB561
3.4.1. Secondary Metabolite Gene Clusters (SMGCs)
3.4.2. CAZome Annotation
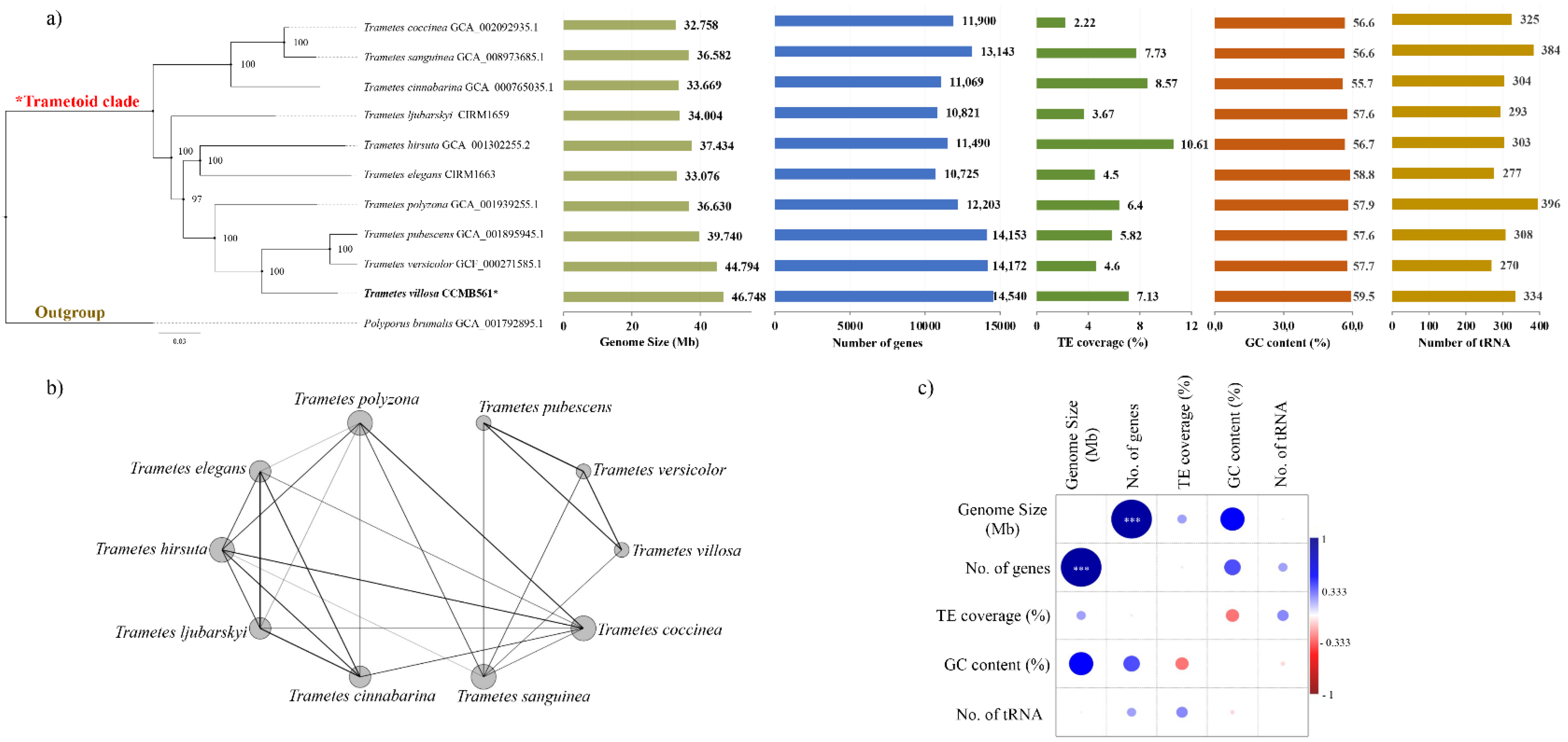
3.5. Comparative Genomics and Phylogenomics of the Genus Trametes
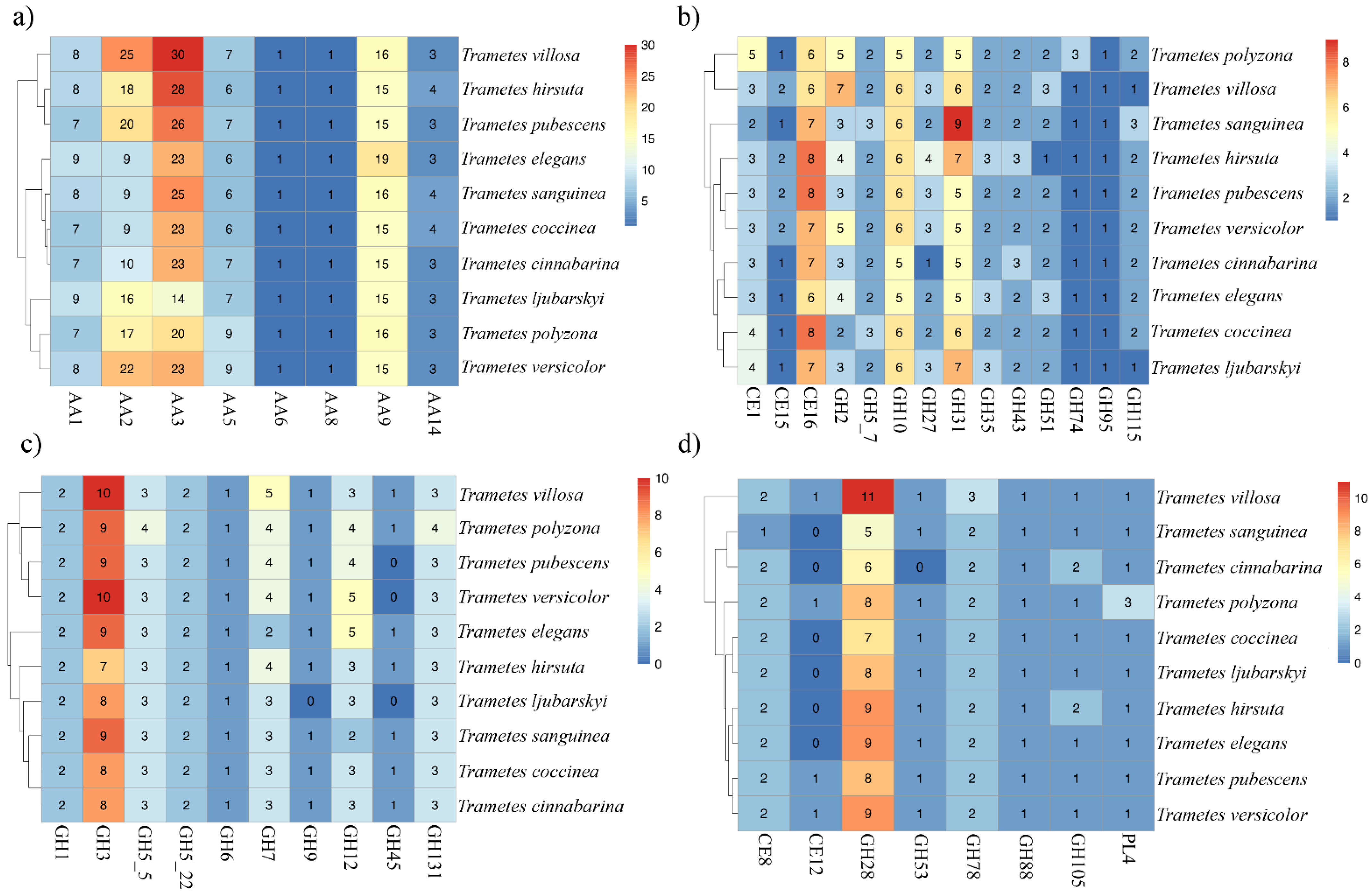
3.6. Potential for Lignocellulose Breakdown by Trametes spp.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Srivastava, N.; Rawat, R.; Singh Oberoi, H.; Ramteke, P.W. A Review on Fuel Ethanol Production from Lignocellulosic Biomass. Int. J. Green Energy 2015, 12, 949–960. [Google Scholar] [CrossRef]
- Isikgor, F.H.; Becer, C.R. Lignocellulosic biomass: A sustainable platform for the production of bio-based chemicals and polymers. Polym. Chem. 2015, 6, 4497–4559. [Google Scholar] [CrossRef] [Green Version]
- Houfani, A.A.; Anders, N.; Spiess, A.C.; Baldrian, P.; Benallaoua, S. Insights from enzymatic degradation of cellulose and hemicellulose to fermentable sugars—A review. Biomass Bioenergy 2020, 134, 105481. [Google Scholar] [CrossRef]
- Hernández-Beltrán, J.U.; Hernández-De Lira, I.O.; Cruz-Santos, M.M.; Saucedo-Luevanos, A.; Hernández-Terán, F.; Balagurusamy, N. Insight into Pretreatment Methods of Lignocellulosic Biomass to Increase Biogas Yield: Current State, Challenges, and Opportunities. Appl. Sci. 2019, 9, 3721. [Google Scholar] [CrossRef] [Green Version]
- Bilal, M.; Nawaz, M.Z.; Iqbal, H.M.N.; Hou, J.; Mahboob, S.; Al-Ghanim, K.A.; Cheng, H. Engineering Ligninolytic Consortium for Bioconversion of Lignocelluloses to Ethanol and Chemicals. Protein Pept. Lett. 2018, 25, 108–119. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, Y.; Zhang, Y.; Yang, X.; Yang, E.; Xu, H.; Yang, Q.; Chagan, I.; Cui, X.; Chen, W.; et al. Lignin degradation potential and draft genome sequence of Trametes trogii S0301. Biotechnol. Biofuels 2019, 12, 256. [Google Scholar] [CrossRef]
- Floudas, D.; Binder, M.; Riley, R.; Barry, K.; Blanchette, R.A.; Henrissat, B.; Martínez, A.T.; Otillar, R.; Spatafora, J.W.; Yadav, J.S.; et al. The Paleozoic Origin of Enzymatic Lignin Decomposition Reconstructed from 31 Fungal Genomes. Science 2012, 336, 1715–1719. [Google Scholar] [CrossRef] [Green Version]
- Andriani, A.; Maharani, A.; Yanto, D.H.Y.; Pratiwi, H.; Astuti, D.; Nuryana, I.; Agustriana, E.; Anita, S.H.; Juanssilfero, A.B.; Perwitasari, U.; et al. Sequential production of ligninolytic, xylanolytic, and cellulolytic enzymes by Trametes hirsuta AA-017 under different biomass of Indonesian sorghum accessions-induced cultures. Bioresour. Technol. Rep. 2020, 12, 100562. [Google Scholar] [CrossRef]
- Vasina, D.V.; Moiseenko, K.V.; Fedorova, T.V.; Tyazhelova, T.V. Lignin-degrading peroxidases in white-rot fungus Trametes hirsuta 072. Absolute expression quantification of full multigene family. PLoS ONE 2017, 12, e0173813. [Google Scholar] [CrossRef]
- Mäkinen, M.; Kuuskeri, J.; Laine, P.; Smolander, O.-P.; Kovalchuk, A.; Zeng, Z.; Asiegbu, F.O.; Paulin, L.; Auvinen, P.; Lundell, T. Genome description of Phlebia radiata 79 with comparative genomics analysis on lignocellulose decomposition machinery of phlebioid fungi. BMC Genom. 2019, 20, 430. [Google Scholar] [CrossRef] [Green Version]
- Atilano-Camino, M.M.; Álvarez-Valencia, L.H.; García-González, A.; García-Reyes, R.B. Improving laccase production from Trametes versicolor using lignocellulosic residues as cosubstrates and evaluation of enzymes for blue wastewater biodegradation. J. Environ. Manag. 2020, 275, 111231. [Google Scholar] [CrossRef] [PubMed]
- Paës, G.; Navarro, D.; Benoit, Y.; Blanquet, S.; Chabbert, B.; Chaussepied, B.; Coutinho, P.M.; Durand, S.; Grigoriev, I.V.; Haon, M.; et al. Tracking of enzymatic biomass deconstruction by fungal secretomes highlights markers of lignocellulose recalcitrance. Biotechnol. Biofuels 2019, 12, 76. [Google Scholar] [CrossRef] [PubMed]
- Espósito, E.; Azevedo, J.L. De Fungos—Uma Introdução à Biologia, Bioquímica e Biotecnologia, 2nd ed.; EDUCS, Editora da Universidade de Caxias do Sul: Caxias do Sul, Brasil, 2004; pp. 9–510. [Google Scholar]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Chandra, R. Ligninolytic enzymes and its mechanisms for degradation of lignocellulosic waste in environment. Heliyon 2020, 6, e03170. [Google Scholar] [CrossRef] [PubMed]
- Mendonça Maciel, M.J.; Castro e Silva, A.; Telles Ribeiro, H.C. Industrial and biotechnological applications of ligninolytic enzymes of the basidiomycota: A review. Electron. J. Biotechnol. 2010, 13. [Google Scholar] [CrossRef]
- Sista Kameshwar, A.K.; Qin, W. Comparative study of genome-wide plant biomass-degrading CAZymes in white rot, brown rot and soft rot fungi. Mycology 2018, 9, 93–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel, G. Fungal and Bacterial Biodegradation: White Rots, Brown Rots, Soft Rots, and Bacteria. In: Deterioration and protection of sustainable biomaterials. Am. Chem. Soc. 2014, 23–58. [Google Scholar] [CrossRef]
- Giweta, M. Role of litter production and its decomposition, and factors affecting the processes in a tropical forest ecosystem: A review. J. Ecol. Environ. 2020, 44, 11. [Google Scholar] [CrossRef]
- Hage, H.; Miyauchi, S.; Virágh, M.; Drula, E.; Min, B.; Chaduli, D.; Navarro, D.; Favel, A.; Norest, M.; Lesage-Meessen, L.; et al. Gene family expansions and transcriptome signatures uncover fungal adaptations to wood decay. Environ. Microbiol. 2021, 23, 5716–5732. [Google Scholar] [CrossRef]
- Couturier, M.; Navarro, D.; Chevret, D.; Henrissat, B.; Piumi, F.; Ruiz-Dueñas, F.J.; Martinez, A.T.; Grigoriev, I.V.; Riley, R.; Lipzen, A.; et al. Enhanced degradation of softwood versus hardwood by the white-rot fungus Pycnoporus coccineus. Biotechnol. Biofuels 2015, 8, 216. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, A.R.; Tyazhelova, T.V.; Moiseenko, K.V.; Vasina, D.V.; Mosunova, O.V.; Fedorova, T.V.; Maloshenok, L.G.; Landesman, E.O.; Bruskin, S.A.; Psurtseva, N.V.; et al. Draft Genome Sequence of the Fungus Trametes hirsuta 072. Genome Announc. 2015, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, D.S.S.; Kato, R.B.; Miranda, F.M.; da Costa Pinheiro, K.; Fonseca, P.L.C.; Tomé, L.M.R.; Vaz, A.B.M.; Badotti, F.; Ramos, R.T.J.; Brenig, B.; et al. Draft genome sequence of Trametes villosa (Sw.) Kreisel CCMB561, a tropical white-rot Basidiomycota from the semiarid region of Brazil. Data Br. 2018, 18, 1581–1587. [Google Scholar] [CrossRef] [PubMed]
- Busk, P.K.; Lange, M.; Pilgaard, B.; Lange, L. Several Genes Encoding Enzymes with the Same Activity Are Necessary for Aerobic Fungal Degradation of Cellulose in Nature. PLoS ONE 2014, 9, e114138. [Google Scholar] [CrossRef]
- Granchi, Z.; Peng, M.; Chi-A-Woeng, T.; de Vries, R.P.; Hildén, K.; Mäkelä, M.R. Genome Sequence of the Basidiomycete White-Rot Fungus Trametes pubescens FBCC735. Genome Announc. 2017, 5. [Google Scholar] [CrossRef] [Green Version]
- Miyauchi, S.; Rancon, A.; Drula, E.; Hage, H.; Chaduli, D.; Favel, A.; Grisel, S.; Henrissat, B.; Herpoël-Gimbert, I.; Ruiz-Dueñas, F.J.; et al. Integrative visual omics of the white-rot fungus Polyporus brumalis exposes the biotechnological potential of its oxidative enzymes for delignifying raw plant biomass. Biotechnol. Biofuels 2018, 11, 201. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Jia, G.; Sun, H.; Sun, T.; Hou, D. Genome sequence of the fungus Pycnoporus sanguineus, which produces cinnabarinic acid and pH- and thermo- stable laccases. Gene 2020, 742, 144586. [Google Scholar] [CrossRef]
- Carneiro, R.T.; Lopes, M.A.; Silva, M.L.; Santos, V.D.; Souza, V.B.; Sousa, A.O.; Pirovani, C.P.; Koblitz, M.G.; Benevides, R.G.; Góes-Neto, A. Trametes villosa Lignin Peroxidase (TvLiP): Genetic and Molecular Characterization. J. Microbiol. Biotechnol. 2017, 27, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Coniglio, R.O.; Díaz, G.V.; Fonseca, M.I.; Castrillo, M.L.; Piccinni, F.E.; Villalba, L.L.; Campos, E.; Zapata, P.D. Enzymatic hydrolysis of barley straw for biofuel industry using a novel strain of Trametes villosa from Paranaense rainforest. Prep. Biochem. Biotechnol. 2020, 50, 753–762. [Google Scholar] [CrossRef]
- Yamanaka, R.; Soares, C.F.; Matheus, D.R.; Machado, K.M.G. Lignolytic enzymes produced by Trametes villosa ccb176 under different culture conditions. Braz. J. Microbiol. 2008, 39, 78–84. [Google Scholar] [CrossRef] [Green Version]
- Silva, M.L.; de Souza, V.B.; da Silva Santos, V.; Kamida, H.M.; de Vasconcellos-Neto, J.R.; Góes-Neto, A.; Koblitz, M.G. Production of Manganese Peroxidase by Trametes villosa on Unexpensive Substrate and Its Application in the Removal of Lignin from Agricultural Wastes. Adv. Biosci. Biotechnol. 2014, 5, 1067–1077. [Google Scholar] [CrossRef] [Green Version]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and Direct Sequencing of Fungal Ribosomal RNA Genes for Phylogenetics. In PCR Protocols; Elsevier: Amsterdam, The Netherlands, 1990; pp. 315–322. [Google Scholar]
- Tomé, L.M.R.; Badotti, F.; Assis, G.B.N.; Fonseca, P.L.C.; da Silva, G.A.; da Silveira, R.M.B.; Costa-Rezende, D.H.; dos Santos, E.R.D.; de Carvalho Azevedo, V.A.; Figueiredo, H.C.P.; et al. Proteomic fingerprinting for the fast and accurate identification of species in the Polyporoid and Hymenochaetoid fungi clades. J. Proteom. 2019, 203, 103390. [Google Scholar] [CrossRef] [PubMed]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranallo-Benavidez, T.R.; Jaron, K.S.; Schatz, M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020, 11, 1432. [Google Scholar] [CrossRef] [Green Version]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k -mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.H.-W.; Vogel, A.; Denton, A.K.; Istace, B.; Wormit, A.; van de Geest, H.; Bolger, M.E.; Alseekh, S.; Maß, J.; Pfaff, C.; et al. De Novo Assembly of a New Solanum pennellii Accession Using Nanopore Sequencing. Plant Cell 2017, 29, 2336–2348. [Google Scholar] [CrossRef] [Green Version]
- Zimin, A.V.; Marçais, G.; Puiu, D.; Roberts, M.; Salzberg, S.L.; Yorke, J.A. The MaSuRCA genome assembler. Bioinformatics 2013, 29, 2669–2677. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Guan, D.; McCarthy, S.A.; Wood, J.; Howe, K.; Wang, Y.; Durbin, R. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 2020, 36, 2896–2898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantarel, B.L.; Korf, I.; Robb, S.M.C.; Parra, G.; Ross, E.; Moore, B.; Holt, C.; Sanchez Alvarado, A.; Yandell, M. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2007, 18, 188–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holt, C.; Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 2011, 12, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, M.S.; Holt, C.; Moore, B.; Yandell, M. Genome Annotation and Curation Using MAKER and MAKER-P. Curr. Protoc. Bioinform. 2014, 48, 4.11.1–4.11.39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanke, M.; Steinkamp, R.; Waack, S.; Morgenstern, B. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res. 2004, 32, W309–W312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lomsadze, A. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 2005, 33, 6494–6506. [Google Scholar] [CrossRef]
- Tarailo-Graovac, M.; Chen, N. Using Repeat Masker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinform. 2009, 25, 4.10.1–4.10.14. [Google Scholar] [CrossRef] [PubMed]
- Geib, S.M.; Hall, B.; Derego, T.; Bremer, F.T.; Cannoles, K.; Sim, S.B. Genome Annotation Generator: A simple tool for generating and correcting WGS annotation tables for NCBI submission. Gigascience 2018, 7, giy018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araujo, F.A.; Barh, D.; Silva, A.; Guimarães, L.; Ramos, R.T.J. GO FEAT: A rapid web-based functional annotation tool for genomic and transcriptomic data. Sci. Rep. 2018, 8, 1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z. Automated De Novo Identification of Repeat Sequence Families in Sequenced Genomes. Genome Res. 2002, 12, 1269–1276. [Google Scholar] [CrossRef] [Green Version]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21, i351–i358. [Google Scholar] [CrossRef] [Green Version]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [Green Version]
- Dutreux, F.; Da Silva, C.; D’Agata, L.; Couloux, A.; Gay, E.J.; Istace, B.; Lapalu, N.; Lemainque, A.; Linglin, J.; Noel, B.; et al. De novo assembly and annotation of three Leptosphaeria genomes using Oxford Nanopore MinION sequencing. Sci. Data 2018, 5, 180235. [Google Scholar] [CrossRef] [PubMed]
- Minei, R.; Hoshina, R.; Ogura, A. De novo assembly of middle-sized genome using MinION and Illumina sequencers. BMC Genom. 2018, 19, 700. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, L.M.; Borelli, G.; Camargo, A.P.; de Assis, M.A.; de Ferraz, S.M.F.; Fiamenghi, M.B.; José, J.; Mofatto, L.S.; Nagamatsu, S.T.; Persinoti, G.F.; et al. Bioinformatics applied to biotechnology: A review towards bioenergy research. Biomass Bioenergy 2019, 123, 195–224. [Google Scholar] [CrossRef]
- Maggiori, C.; Raymond-Bouchard, I.; Brennan, L.; Touchette, D.; Whyte, L. MinION sequencing from sea ice cryoconites leads to de novo genome reconstruction from metagenomes. Sci. Rep. 2021, 11, 21041. [Google Scholar] [CrossRef]
- Miyauchi, S.; Navarro, D.; Grigoriev, I.V.; Lipzen, A.; Riley, R.; Chevret, D.; Grisel, S.; Berrin, J.-G.; Henrissat, B.; Rosso, M.-N. Visual Comparative Omics of Fungi for Plant Biomass Deconstruction. Front. Microbiol. 2016, 7, 1335. [Google Scholar] [CrossRef]
- Sun, X.; He, C.; Fang, Z.; Xiao, Y. Expression and characterization of NADPH-cytochrome P450 reductase from Trametes versicolor in Escherichia coli. Sheng Wu Gong Cheng Xue Bao 2018, 34, 1156–1168. [Google Scholar] [CrossRef]
- Chen, W.; Lee, M.-K.; Jefcoate, C.; Kim, S.-C.; Chen, F.; Yu, J.-H. Fungal Cytochrome P450 Monooxygenases: Their Distribution, Structure, Functions, Family Expansion, and Evolutionary Origin. Genome Biol. Evol. 2014, 6, 1620–1634. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Ruan, R.; Li, H. The completed genome sequence of the pathogenic ascomycete fungus Penicillium digitatum. Genomics 2021, 113, 439–446. [Google Scholar] [CrossRef]
- Osbourn, A. Secondary metabolic gene clusters: Evolutionary toolkits for chemical innovation. Trends Genet. 2010, 26, 449–457. [Google Scholar] [CrossRef]
- Xiao, H.; Zhong, J.-J. Production of Useful Terpenoids by Higher-Fungus Cell Factory and Synthetic Biology Approaches. Trends Biotechnol. 2016, 34, 242–255. [Google Scholar] [CrossRef]
- Brandenburger, E.; Gressler, M.; Leonhardt, R.; Lackner, G.; Habel, A.; Hertweck, C.; Brock, M.; Hoffmeister, D. A Highly Conserved Basidiomycete Peptide Synthetase Produces a Trimeric Hydroxamate Siderophore. Appl. Environ. Microbiol. 2017, 83, e01478-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miethke, M.; Marahiel, M.A. Siderophore-Based Iron Acquisition and Pathogen Control. Microbiol. Mol. Biol. Rev. 2007, 71, 413–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levasseur, A.; Drula, E.; Lombard, V.; Coutinho, P.M.; Henrissat, B. Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels 2013, 6, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidar, A.; Albuquerque, E.D.; Voshol, G.P.; Ram, A.F.J.; Vijgenboom, E.; Punt, P.J. Carbohydrate Binding Modules: Diversity of Domain Architecture in Amylases and Cellulases from Filamentous Microorganisms. Front. Bioeng. Biotechnol. 2020, 8, 871. [Google Scholar] [CrossRef]
- Van den Brink, J.; de Vries, R.P. Fungal enzyme sets for plant polysaccharide degradation. Appl. Microbiol. Biotechnol. 2011, 91, 1477–1492. [Google Scholar] [CrossRef] [Green Version]
- Justo, A.; Hibbett, D.S. Phylogenetic classification of Trametes (Basidiomycota, Polyporales) based on a five-marker dataset. Taxon 2011, 60, 1567–1583. [Google Scholar] [CrossRef]
- Mohanta, T.K.; Bae, H. The diversity of fungal genome. Biol. Proced. Online 2015, 17, 8. [Google Scholar] [CrossRef] [Green Version]
- Muszewska, A.; Steczkiewicz, K.; Stepniewska-Dziubinska, M.; Ginalski, K. Cut-and-Paste Transposons in Fungi with Diverse Lifestyles. Genome Biol. Evol. 2017, 9, 3463–3477. [Google Scholar] [CrossRef] [Green Version]
- Muszewska, A.; Steczkiewicz, K.; Stepniewska-Dziubinska, M.; Ginalski, K. Transposable elements contribute to fungal genes and impact fungal lifestyle. Sci. Rep. 2019, 9, 4307. [Google Scholar] [CrossRef]
- Castanera, R.; Borgognone, A.; Pisabarro, A.G.; Ramírez, L. Biology, dynamics, and applications of transposable elements in basidiomycete fungi. Appl. Microbiol. Biotechnol. 2017, 101, 1337–1350. [Google Scholar] [CrossRef]
- Raina, M.; Ibba, M. tRNAs as regulators of biological processes. Front. Genet. 2014, 5, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stajich, J.E. Fungal genomes and insights into the evolution of the kingdom. Microbiol. Spectr. 2017, 5, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.W.S. Structure and Action Mechanism of Ligninolytic Enzymes. Appl. Biochem. Biotechnol. 2009, 157, 174–209. [Google Scholar] [CrossRef] [PubMed]
- Dashtban, M.; Schraft, H.; Syed, T.A.; Qin, W. Fungal biodegradation and enzymatic modification of lignin. Int. J. Biochem. Mol. Biol. 2010, 1, 36–50. [Google Scholar] [PubMed]
- Couturier, M.; Ladevèze, S.; Sulzenbacher, G.; Ciano, L.; Fanuel, M.; Moreau, C.; Villares, A.; Cathala, B.; Chaspoul, F.; Frandsen, K.E.; et al. Lytic xylan oxidases from wood-decay fungi unlock biomass degradation. Nat. Chem. Biol. 2018, 14, 306–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Illumina | MinION | |
|---|---|---|
| Total reads number | 48,347,940 | 1,043,247 |
| Total reads bases (bp) | 5,798,237,268 | 4,189,223,607 |
| Coverage | 129× | 93× |
| Longest read (bp) | 151 | 21,613 |
| Mean reads length (bp) | 138 | 4476 |
| GC content (%) | 57.5 | 56 |
| Assembly Short Reads (Illumina) | Assembly Oxford Nanopore (MinION) | Hybrid Assembly (Illumina and Oxford Nanopore) | ||||||
|---|---|---|---|---|---|---|---|---|
| Assembly/ Software | MaSuRCa | CANU | CANU-smartdenovo | RACON | FLYE | SPADES | MaSuRCa | MaSuRCa-Purge_Dups |
| Number of contigs (≥0 bp) | 4026 | 1836 | 337 | 1836 | 882 | 12,829 | 264 | 143 |
| Number of contigs (≥500 bp) | 3930 | 1836 | 337 | 1836 | 881 | 1940 | 264 | 143 |
| Largest contig | 470,636 | 1,594,329 | 1,660,310 | 1,605,280 | 1,891,910 | 1,207,893 | 4,772,416 | 9,749,168 |
| Total length (≥500 bp) | 58,820,861 | 63,704,316 | 42,774,667 | 63,971,542 | 49,876,064 | 65,406,907 | 62,711,988 | 46,748,415 |
| GC (%) | 59.40 | 59.36 | 59.39 | 59.41 | 59.35 | 59.39 | 59.39 | 59.45 |
| N50 | 27,657 | 103,641 | 238,816 | 104,325 | 204,679 | 282,055 | 598,690 | 1,009,271 |
| L50 | 503 | 115 | 43 | 114 | 55 | 69 | 21 | 8 |
| # N’s per 100 kbp | 0.00 | 0.00 | 0.00 | 0.00 | 2.41 | 227.07 | 0.16 | 0.69 |
| Complete (%) | Single-Copy (%) | Duplicated (%) | Fragmented (%) | Missing (%) | |
|---|---|---|---|---|---|
| CANU | 80.7 | 65.2 | 15.5 | 6.7 | 12.6 |
| CANU-smartdenovo | 76.2 | 73.8 | 2.4 | 8.6 | 15.2 |
| FLYE | 90.2 | 85.2 | 5.0 | 3.9 | 5.9 |
| MaSuRCa (Hybrid) | 99.0 | 64.0 | 35.0 | 0.2 | 0.8 |
| MaSuRCa (Illumina) | 97.4 | 64..3 | 33.1 | 0.9 | 1.7 |
| MaSuRCa-Purge_Dups | 99.1 | 96.7 | 2.4 | 0.1 | 0.8 |
| RACON | 88.2 | 70.0 | 18.2 | 4.5 | 7.3 |
| SPADES | 99.1 | 41.6 | 57.5 | 0.2 | 0.7 |
| ID Fungo | Total No. TE | Total TE Coverage% | Retroelements | DNA Transposons | Helitron | Unclassified | |||
|---|---|---|---|---|---|---|---|---|---|
| SINEs | LINEs | LTR Elements | |||||||
| Ty1/Copia | Gypsy/DIRS1 | ||||||||
| Trametes cinnabarina | 252 | 8.57 | 0 | 144 | 337 | 450 | 390 | 0 | 3079 |
| Trametes coccinea | 104 | 2.22 | 0 | 0 | 119 | 137 | 0 | 0 | 1187 |
| Trametes elegans | 129 | 4.50 | 0 | 0 | 210 | 451 | 14 | 0 | 1841 |
| Trametes hirsuta | 191 | 10.61 | 0 | 73 | 264 | 387 | 65 | 42 | 1949 |
| Trametes ljubarskyi | 172 | 3.67 | 0 | 0 | 317 | 263 | 89 | 0 | 2219 |
| Trametes polyzona | 349 | 6.41 | 0 | 113 | 416 | 912 | 105 | 69 | 5043 |
| Trametes pubescens | 303 | 5.82 | 19 | 106 | 107 | 160 | 258 | 30 | 3385 |
| Trametes sanguinea | 191 | 7.73 | 0 | 41 | 184 | 328 | 32 | 0 | 1855 |
| Trametes versicolor | 234 | 4.6 | 0 | 50 | 38 | 144 | 11 | 94 | 4165 |
| Trametes villosa | 274 | 7.13 | 17 | 97 | 186 | 503 | 74 | 70 | 4437 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomé, L.M.R.; da Silva, F.F.; Fonseca, P.L.C.; Mendes-Pereira, T.; Azevedo, V.A.d.C.; Brenig, B.; Badotti, F.; Góes-Neto, A. Hybrid Assembly Improves Genome Quality and Completeness of Trametes villosa CCMB561 and Reveals a Huge Potential for Lignocellulose Breakdown. J. Fungi 2022, 8, 142. https://doi.org/10.3390/jof8020142
Tomé LMR, da Silva FF, Fonseca PLC, Mendes-Pereira T, Azevedo VAdC, Brenig B, Badotti F, Góes-Neto A. Hybrid Assembly Improves Genome Quality and Completeness of Trametes villosa CCMB561 and Reveals a Huge Potential for Lignocellulose Breakdown. Journal of Fungi. 2022; 8(2):142. https://doi.org/10.3390/jof8020142
Chicago/Turabian StyleTomé, Luiz Marcelo Ribeiro, Felipe Ferreira da Silva, Paula Luize Camargos Fonseca, Thairine Mendes-Pereira, Vasco Ariston de Carvalho Azevedo, Bertram Brenig, Fernanda Badotti, and Aristóteles Góes-Neto. 2022. "Hybrid Assembly Improves Genome Quality and Completeness of Trametes villosa CCMB561 and Reveals a Huge Potential for Lignocellulose Breakdown" Journal of Fungi 8, no. 2: 142. https://doi.org/10.3390/jof8020142
APA StyleTomé, L. M. R., da Silva, F. F., Fonseca, P. L. C., Mendes-Pereira, T., Azevedo, V. A. d. C., Brenig, B., Badotti, F., & Góes-Neto, A. (2022). Hybrid Assembly Improves Genome Quality and Completeness of Trametes villosa CCMB561 and Reveals a Huge Potential for Lignocellulose Breakdown. Journal of Fungi, 8(2), 142. https://doi.org/10.3390/jof8020142