
Molecular Evolution of Lysine Biosynthesis in Agaricomycetes



 and
and 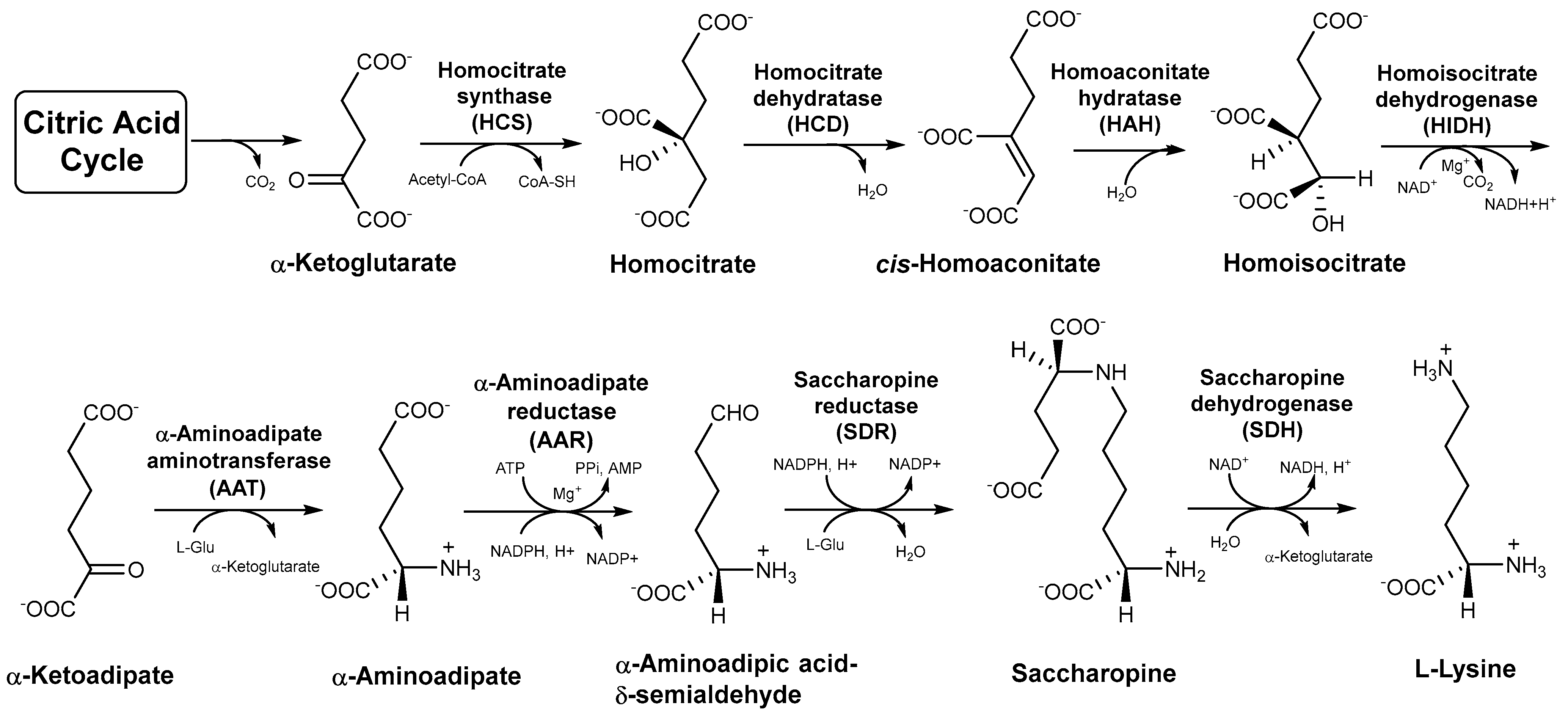{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Differential Expression Analysis of Genes by RNA-seq
2.2. Collection and Assembly of Sequences
2.3. Phylogenetic Analysis of Species and Enzymes Involved in the AAA Pathway
2.4. Conservative Analysis of the Primary and Secondary Structures of HCS, AAR, and SDH
2.5. 3-D Structure Predictions of HCS, AAR, and SDH
2.6. Conservative Motif Analysis of Binding Sites in 3-D Structures of HCS, AAR, and SDH
3. Results
3.1. RNA-seq Analysis of the Genes of the Eight Enzymes Involved in the AAA Pathway
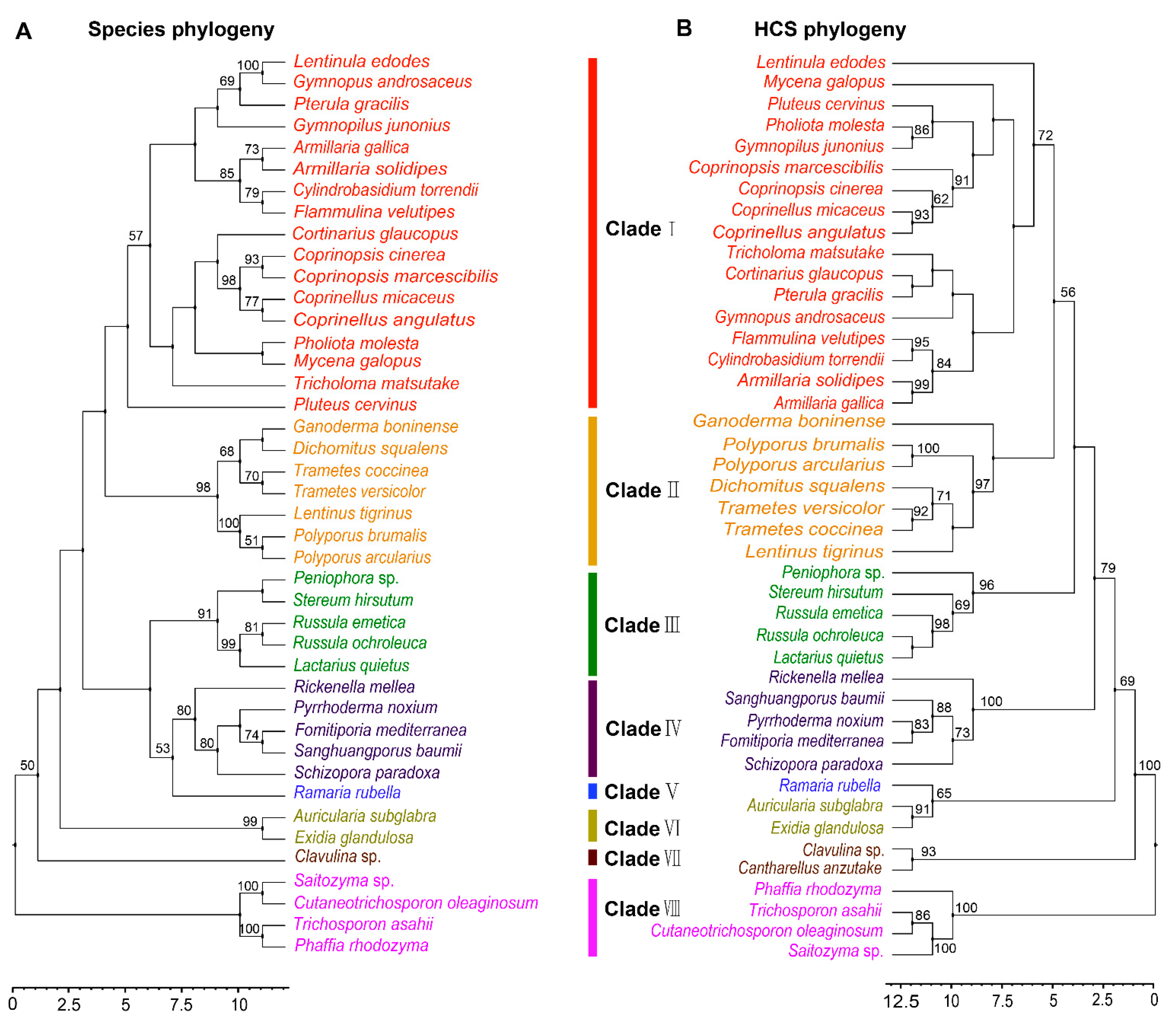
3.2. Phylogenetic Analysis of HCS in Agaricomycetes
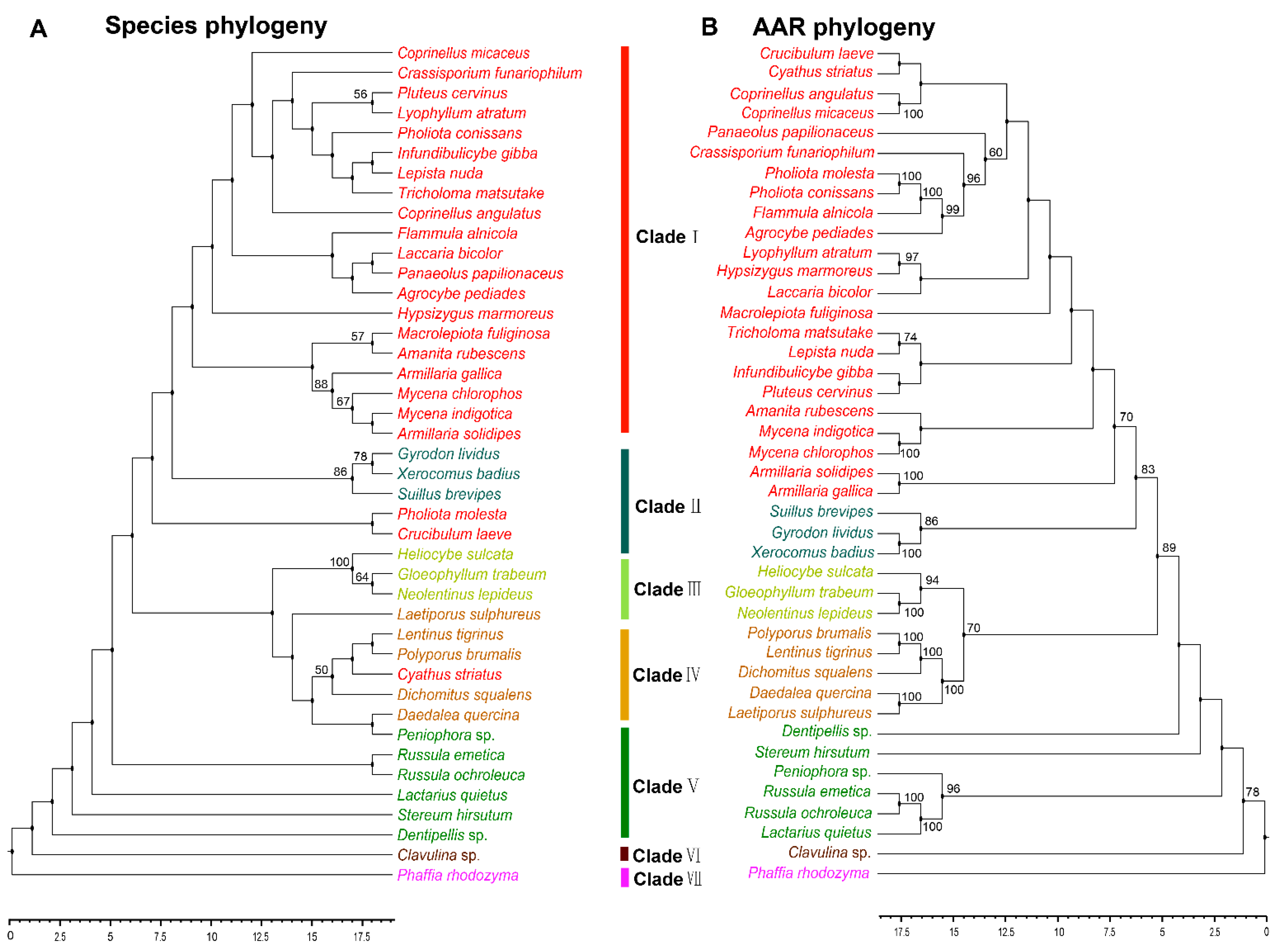
3.3. Phylogenetic Analysis of AAR in Agaricomycetes
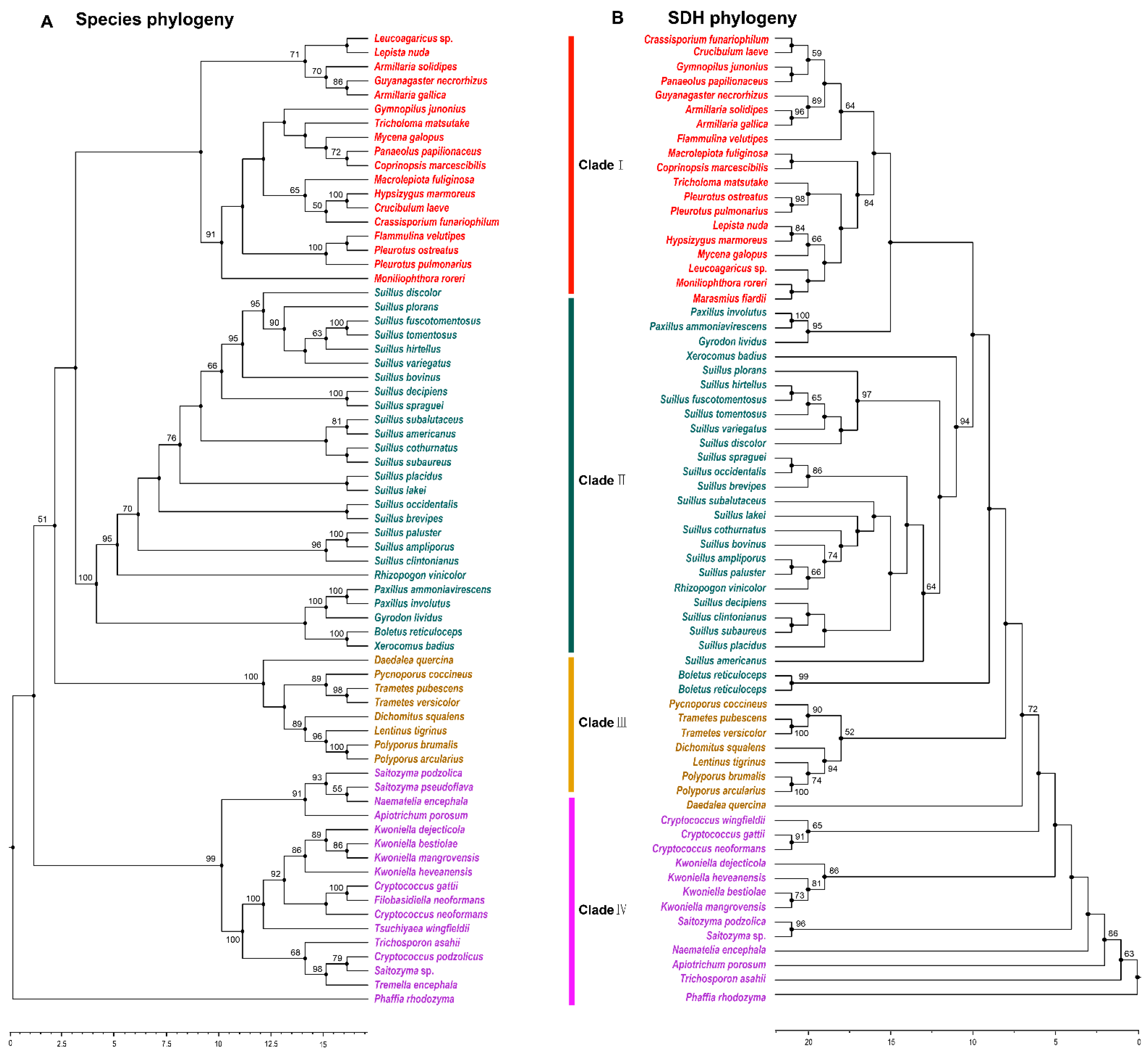
3.4. Phylogenetic Analysis of SDH in Agaricomycetes
3.5. Phylogenetic Analysis of Other Enzymes in the AAA Pathway
3.6. Conservation of Domains in the Primary Structures of HCS, AAR, and SDH
3.7. High Conservation of the Secondary Structures of HCS, AAR, and SDH
3.8. 3-D Structures of HCS, AAR, and SDH and the High Conservation of Binding Sites
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, F.; Wang, W.; Chen, B.Z.; Xie, B.G. Homocitrate synthase expression and lysine content in fruiting body of different developmental stages in Flammulina velutipes. Curr. Microbiol. 2015, 70, 821–828. [Google Scholar] [CrossRef]
- Xu, H.; Andi, B.; Qian, J.; West, A.H.; Cook, P.F. The α-aminoadipate pathway for lysine biosynthesis in fungi. Cell Biochem. Biophys. 2006, 46, 43–64. [Google Scholar] [CrossRef]
- Yin, J.; Li, Y.Y.; Han, H.; Zheng, J.; Wang, L.J.; Ren, W.K.; Chen, S.; Wu, F.; Fang, R.J.; Huang, X.G.; et al. Effects of lysine deficiency and Lys-Lys dipeptide on cellular apoptosis and amino acids metabolism. Mol. Nutr. Food Res. 2017, 61, 1600754. [Google Scholar] [CrossRef]
- Astrom, S.; Vonderdecken, A. Lysine deficiency reduces transcription activity and concentration of chromatin proteins reversibly in rat-liver. Acta Physiol. Scand. 1983, 117, 519–525. [Google Scholar] [CrossRef]
- Beluhan, S.; Ranogajec, A. Chemical composition and non-volatile components of Croatian wild edible mushrooms. Food Chem. 2011, 124, 1076–1082. [Google Scholar] [CrossRef]
- Manninen, H.; Rotola-Pukkila, M.; Aisala, H.; Hopia, A.; Laaksonen, T. Free amino acids and 5 ‘-nucleotides in Finnish forest mushrooms. Food Chem. 2018, 247, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Sunyaev, S.; Bork, P.; Dandekar, T. Metabolites: A helping hand for pathway evolution? Trends Biochem. Sci. 2003, 28, 336–341. [Google Scholar] [CrossRef]
- Cunchillos, C.; Lecointre, G. Ordering events of biochemical evolution. Biochimie 2007, 89, 555–573. [Google Scholar] [CrossRef]
- Fondi, M.; Brilli, M.; Emiliani, G.; Paffetti, D.; Fani, R. The primordial metabolism: An ancestral interconnection between leucine, arginine, and lysine biosynthesis. BMC Evol. Biol. 2007, 7 (Suppl. 2), S3. [Google Scholar] [CrossRef] [PubMed]
- Zabriskie, T.M.; Jackson, M.D. Lysine biosynthesis and metabolism in fungi. Nat. Prod. Rep. 2000, 17, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Amich, J.; Bignell, E. Amino acid biosynthetic routes as drug targets for pulmonary fungal pathogens: What is known and why do we need to know more? Curr. Opin. Microbiol. 2016, 32, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Schobel, F.; Jacobsen, I.D.; Brock, M. Evaluation of lysine biosynthesis as an antifungal drug target: Biochemical characterization of Aspergillus fumigatus homocitrate synthase and virulence studies. Eukaryot. Cell 2010, 9, 878–893. [Google Scholar] [CrossRef] [PubMed]
- Velasco, A.M.; Leguina, J.I.; Lazcano, A. Molecular evolution of the lysine biosynthetic pathways. J. Mol. Evol. 2002, 55, 445–459. [Google Scholar] [CrossRef]
- Miyauchi, S.; Kiss, E.; Kuo, A.; Drula, E.; Kohler, A.; Sanchez-Garcia, M.; Morin, E.; Andreopoulos, B.; Barry, K.W.; Bonito, G.; et al. Large-scale genome sequencing of mycorrhizal fungi provides insights into the early evolution of symbiotic traits. Nat. Commun. 2020, 11, 5125. [Google Scholar] [CrossRef]
- Liu, J.; Wang, J.; Zhang, D.; Shang, X.; Tan, Q. Analysis of genes related to lysine biosynthesis based on whole genome of Flammulina velutipes. Microbiol. China 2016, 43, 2225–2233. [Google Scholar] [CrossRef]
- Krizsan, K.; Almasi, E.; Merenyi, Z.; Sahu, N.; Viragh, M.; Koszo, T.; Mondo, S.; Kiss, B.; Balint, B.; Kues, U.; et al. Transcriptomic atlas of mushroom development reveals conserved genes behind complex multicellularity in fungi. Proc. Natl. Acad. Sci. USA 2019, 116, 7409–7418. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Aerts, A.; Ahren, D.; Brun, A.; Danchin, E.G.J.; Duchaussoy, F.; Gibon, J.; Kohler, A.; Lindquist, E.; Pereda, V.; et al. The genome of Laccaria bicolor provides insights into mycorrhizal symbiosis. Nature 2008, 452, 88–92. [Google Scholar] [CrossRef]
- He, M.-Q.; Zhao, R.-L.; Hyde, K.D.; Begerow, D.; Kemler, M.; Yurkov, A.; McKenzie, E.H.C.; Raspé, O.; Kakishima, M.; Sánchez-Ramírez, S.; et al. Notes, outline and divergence times of Basidiomycota. Fungal Divers. 2019, 99, 105–367. [Google Scholar] [CrossRef]
- Li, G.J.; Zhao, R.L.; Zhang, C.L.; Lin, F.C. A preliminary DNA barcode selection for the genus Russula (Russulales, Basidiomycota). Mycology 2019, 10, 61–74. [Google Scholar] [CrossRef]
- Geourjon, C.; Deleage, G. SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput. Appl. Biosci. 1995, 11, 681–684. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef]
- Zhang, C.; Freddolino, P.L.; Zhang, Y. COFACTOR: Improved protein function prediction by combining structure, sequence and protein-protein interaction information. Nucleic Acids Res. 2017, 45, W291–W299. [Google Scholar] [CrossRef]
- Bailey, T.L.; Williams, N.; Misleh, C.; Li, W.W. MEME: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006, 34, W369–W373. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Zhao, R.; Zhang, H.; Wei, P.; Qi, L.; Chen, G.; Yin, W.B.; Li, W. Rapid and accurate screening of lysine-producing edible mushrooms via the homocitrate synthase gene as a universal molecular marker. ACS Omega 2021, 6, 26910–26918. [Google Scholar] [CrossRef] [PubMed]
- Kalb, D.; Lackner, G.; Hoffmeister, D. Functional and phylogenetic divergence of fungal adenylate-forming reductases. Appl. Environ. Microbiol. 2014, 80, 6175–6183. [Google Scholar] [CrossRef] [PubMed]
- Nishida, H.; Nishiyama, M. What is characteristic of fungal lysine synthesis through the alpha-aminoadipate pathway? J. Mol. Evol. 2000, 51, 299–302. [Google Scholar] [CrossRef]
- Liu, J.; Li, Q.; Jiang, P.; Xu, Z.; Zhang, D.; Zhang, L.; Zhang, M.; Yu, H.; Song, C.; Tan, Q.; et al. Overexpression of the saccharopine dehydrogenase gene improves lysine biosynthesis in Flammulina velutipes. J. Basic Microbiol. 2019, 59, 890–900. [Google Scholar] [CrossRef]
- Stajich, J.E.; Wilke, S.K.; Ahren, D.; Au, C.H.; Birren, B.W.; Borodovsky, M.; Burns, C.; Canback, B.; Casselton, L.A.; Cheng, C.K.; et al. Insights into evolution of multicellular fungi from the assembled chromosomes of the mushroom Coprinopsis cinerea (Coprinus cinereus). Proc. Natl. Acad. Sci. USA 2010, 107, 11889–11894. [Google Scholar] [CrossRef]
- Sipos, G.; Prasanna, A.N.; Walter, M.C.; O’Connor, E.; Balint, B.; Krizsan, K.; Kiss, B.; Hess, J.; Varga, T.; Slot, J.; et al. Genome expansion and lineage-specific genetic innovations in the forest pathogenic fungi Armillaria. Nat. Ecol. Evol. 2017, 1, 1931–1941. [Google Scholar] [CrossRef]
- Floudas, D.; Held, B.W.; Riley, R.; Nagy, L.G.; Koehler, G.; Ransdell, A.S.; Younus, H.; Chow, J.; Chiniquy, J.; Lipzen, A.; et al. Evolution of novel wood decay mechanisms in Agaricales revealed by the genome sequences of Fistulina hepatica and Cylindrobasidium torrendii. Fungal Genet. Biol. 2015, 76, 78–92. [Google Scholar] [CrossRef]
- Bulfer, S.L.; Scott, E.M.; Couture, J.F.; Pillus, L.; Trievel, R.C. Crystal structure and functional analysis of homocitrate synthase, an essential enzyme in lysine biosynthesis. J. Biol. Chem. 2009, 284, 35769–35780. [Google Scholar] [CrossRef]
- Jogl, G.; Tong, L. Crystal structure of yeast acetyl-coenzyme A synthetase in complex with AMP. Biochemistry 2004, 43, 1425–1431. [Google Scholar] [CrossRef]
- Drake, E.J.; Miller, B.R.; Shi, C.; Tarrasch, J.T.; Sundlov, J.A.; Allen, C.L.; Skiniotis, G.; Aldrich, C.C.; Gulick, A.M. Structures of two distinct conformations of holo-non-ribosomal peptide synthetases. Nature 2016, 529, 235–238. [Google Scholar] [CrossRef]
- Andi, B.; Xu, H.; Cook, P.F.; West, A.H. Crystal structures of ligand-bound saccharopine dehydrogenase from Saccharomyces cerevisiae. Biochemistry 2007, 46, 12512–12521. [Google Scholar] [CrossRef]
- Burk, D.L.; Hwang, J.; Kwok, E.; Marrone, L.; Goodfellow, V.; Dmitrienko, G.I.; Berghuis, A.M. Structural studies of the final enzyme in the alpha-aminoadipate pathway-saccharopine dehydrogenase from Saccharomyces cerevisiae. J. Mol. Biol. 2007, 373, 745–754. [Google Scholar] [CrossRef]
- Kumar, V.P.; Thomas, L.M.; Bobyk, K.D.; Andi, B.; Cook, P.F.; West, A.H. Evidence in support of lysine 77 and histidine 96 as acid-base catalytic residues in saccharopine dehydrogenase from Saccharomyces cerevisiae. Biochemistry 2012, 51, 857–866. [Google Scholar] [CrossRef][Green Version]
- Krittanawong, C.; Isath, A.; Hahn, J.; Wang, Z.; Fogg, S.E.; Bandyopadhyay, D.; Jneid, H.; Virani, S.S.; Tang, W.H.W. Mushroom consumption and cardiovascular health: A systematic review. Am. J. Med. 2021, 134, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Storts, D.R.; Bhattacharjee, J.K. Properties of revertants of Lys2 and Lys5 mutants as well as alpha-aminoadipate-semialdehyde dehydrogenase from Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 1989, 161, 182–186. [Google Scholar] [CrossRef]
- Torruella, G.; Suga, H.; Riutort, M.; Pereto, J.; Ruiz-Trillo, I. The evolutionary history of lysine biosynthesis pathways within eukaryotes. J. Mol. Evol. 2009, 69, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Garnica, S.; Riess, K.; Schön, M.E.; Riess, K.; Schön, M.E. Divergence times and phylogenetic patterns of Sebacinales, a highly diverse and widespread fungal lineage. PLoS ONE 2016, 11, e0149531. [Google Scholar] [CrossRef] [PubMed]
- Irvin, S.D.; Bhattacharjee, J.K. A unique fungal lysine biosynthesis enzyme shares a common ancestor with tricarboxylic acid cycle and leucine biosynthetic enzymes found in diverse organisms. J. Mol. Evol. 1998, 46, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Ehmann, D.E.; Gehring, A.M.; Walsh, C.T. Lysine biosynthesis in Saccharomyces cerevisiae: Mechanism of alpha-aminoadipate reductase (Lys2) involves posttranslational phosphopantetheinylation by Lys5. Biochemistry 1999, 38, 6171–6177. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Z.; He, M.; Zhao, R.; Qi, L.; Chen, G.; Yin, W.-B.; Li, W. Molecular Evolution of Lysine Biosynthesis in Agaricomycetes. J. Fungi 2022, 8, 37. https://doi.org/10.3390/jof8010037
Song Z, He M, Zhao R, Qi L, Chen G, Yin W-B, Li W. Molecular Evolution of Lysine Biosynthesis in Agaricomycetes. Journal of Fungi. 2022; 8(1):37. https://doi.org/10.3390/jof8010037
Chicago/Turabian StyleSong, Zili, Maoqiang He, Ruilin Zhao, Landa Qi, Guocan Chen, Wen-Bing Yin, and Wei Li. 2022. "Molecular Evolution of Lysine Biosynthesis in Agaricomycetes" Journal of Fungi 8, no. 1: 37. https://doi.org/10.3390/jof8010037
APA StyleSong, Z., He, M., Zhao, R., Qi, L., Chen, G., Yin, W.-B., & Li, W. (2022). Molecular Evolution of Lysine Biosynthesis in Agaricomycetes. Journal of Fungi, 8(1), 37. https://doi.org/10.3390/jof8010037