
Genomic Characteristics and Comparative Genomics Analysis of Two Chinese Corynespora cassiicola Strains Causing Corynespora Leaf Fall (CLF) Disease

Abstract
1. Introduction
2. Materials and Methods
2.1. Fungal Growth Conditions and DNA Preparation
2.2. Pathogenicity Tests
2.3. Detection and Sequencing of the Cassiicolin-Encoding Genes
2.4. Genome Sequencing and Assembly
2.5. Gene Prediction and Annotation
2.6. Orthologous Gene Family, Phylogenetic, and Whole-Genome Synteny Analyses
2.7. Gene Family Expansion Analysis
2.8. Positive Selection
2.9. Annotation of Specific Gene Categories
2.10. Secondary Metabolite Gene Cluster Analysis
3. Results
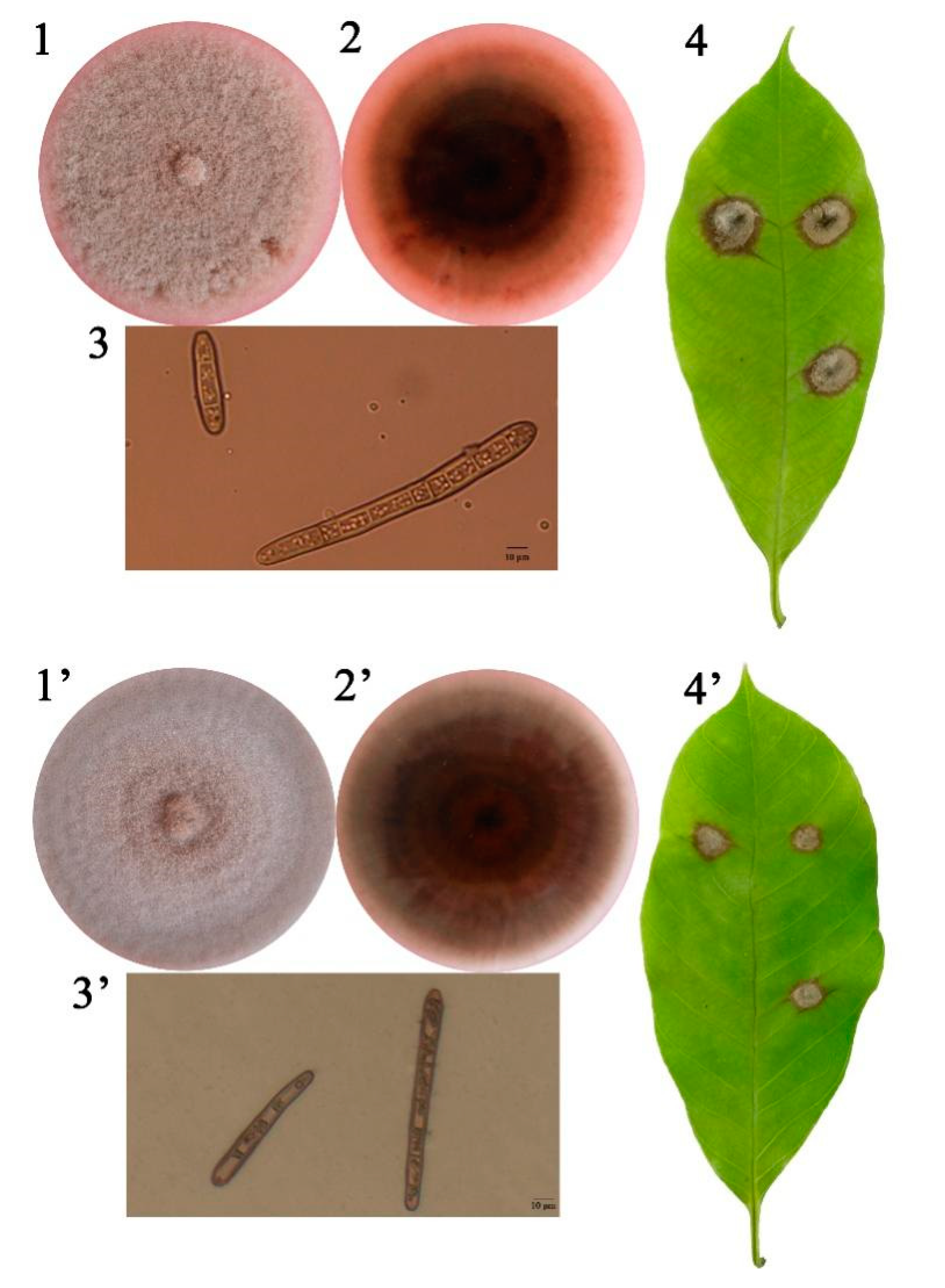
3.1. C. cassiicola Cas5 and Cas2 Isolates from China Differ in Virulence toward Rubber Tree Leaves
3.2. General Genome Features and Annotation
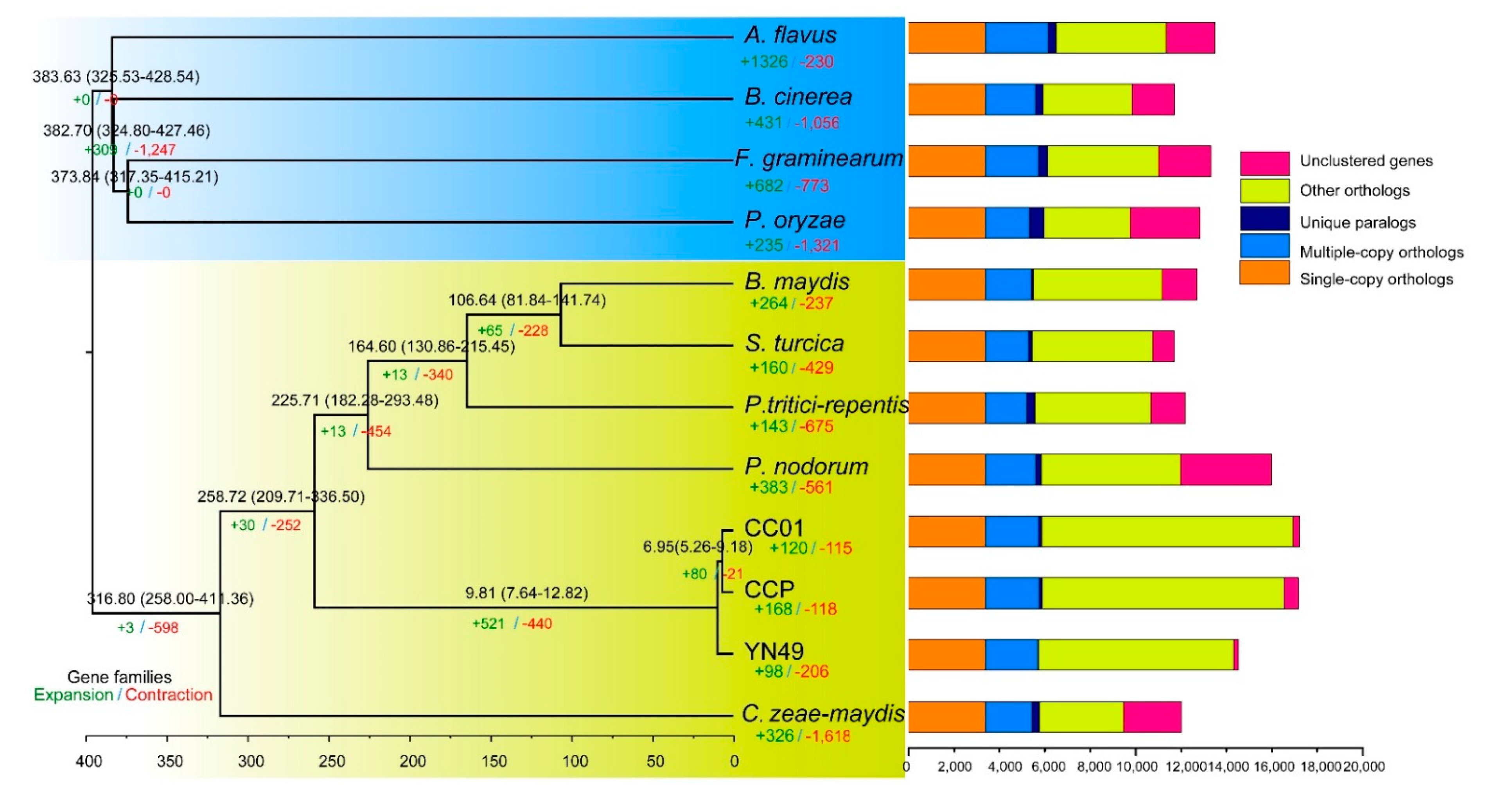
3.3. Analysis of Orthologues and Phylogenetic Relationships between C. cassiicola YN49 and CC01 and Other Fungi
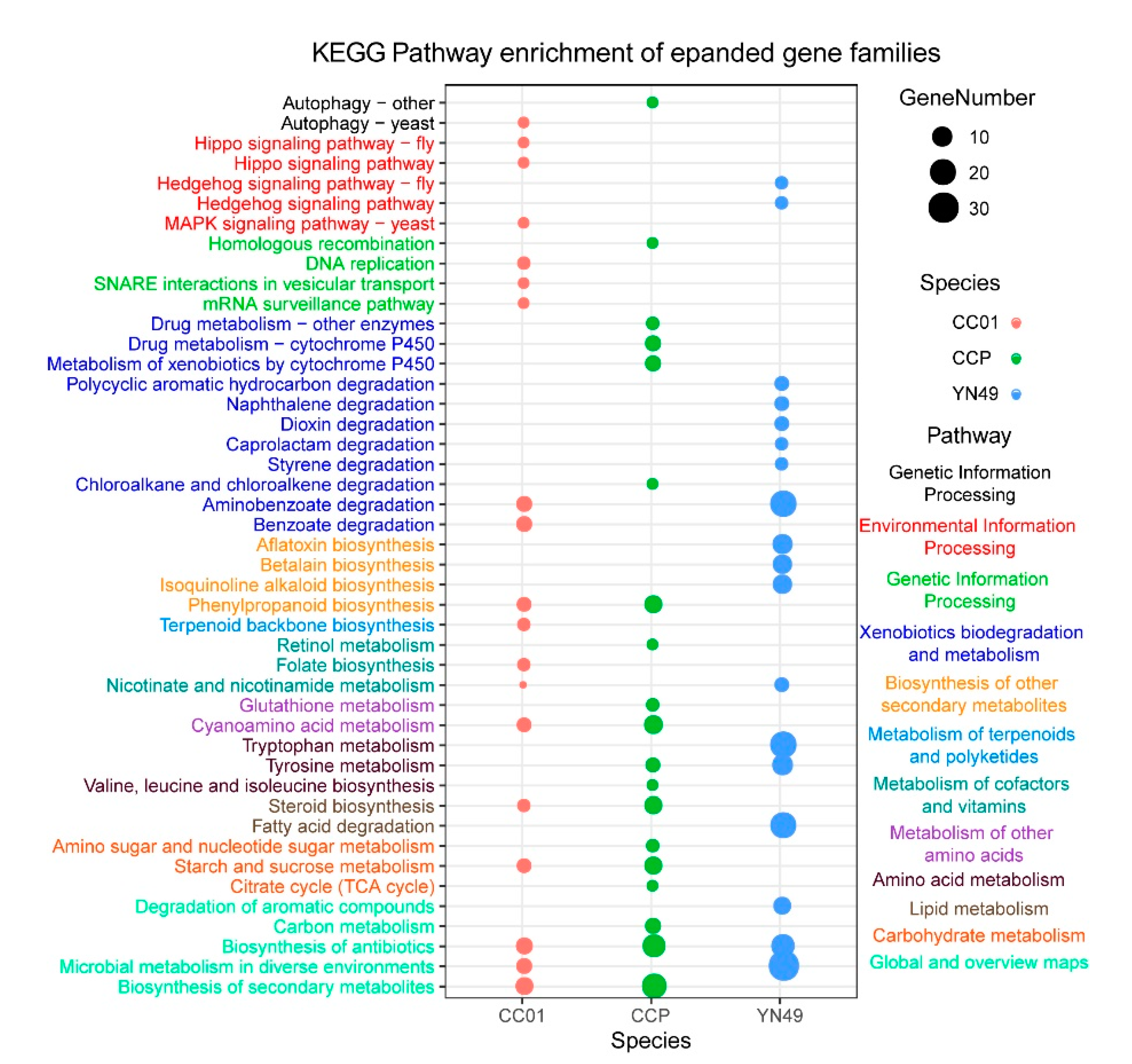
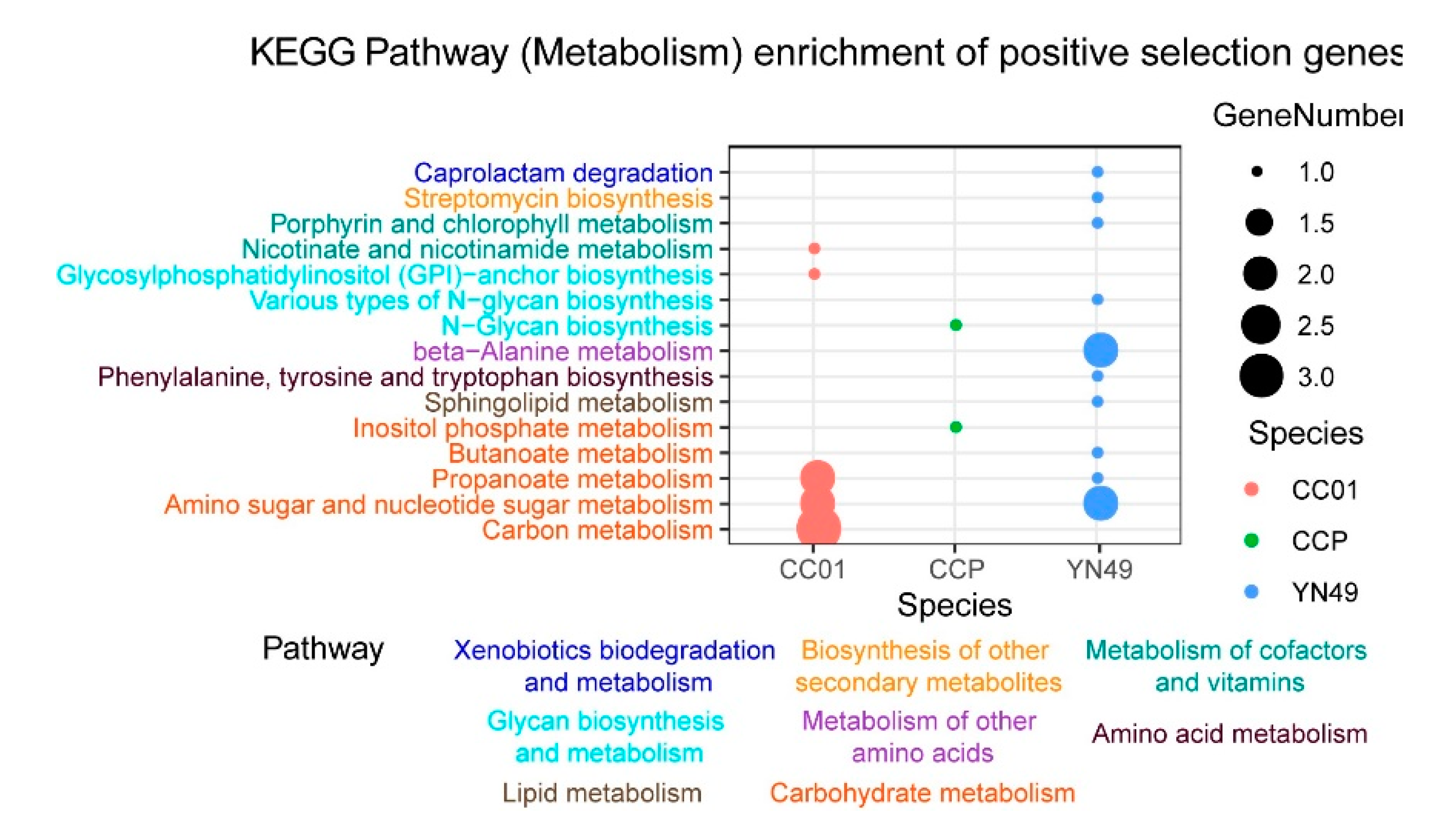
3.4. Gene Family Expansion and Contraction, and Positive Selection of Genes
3.5. Whole-Genome Synteny Comparisons between C. cassiicola YN49, CC01 and CCP
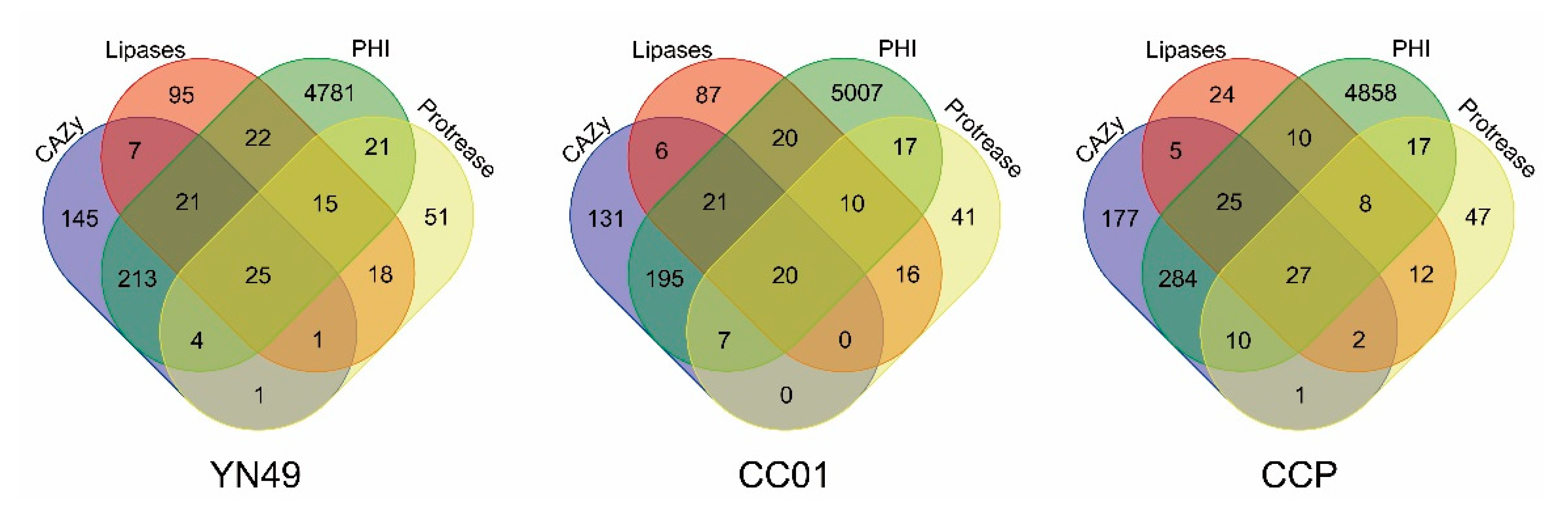
3.6. Secretome and Putative Pathogenicity Genes
3.7. Carbohydrate-Active Enzymes
3.8. Secondary Metabolite Gene Clusters
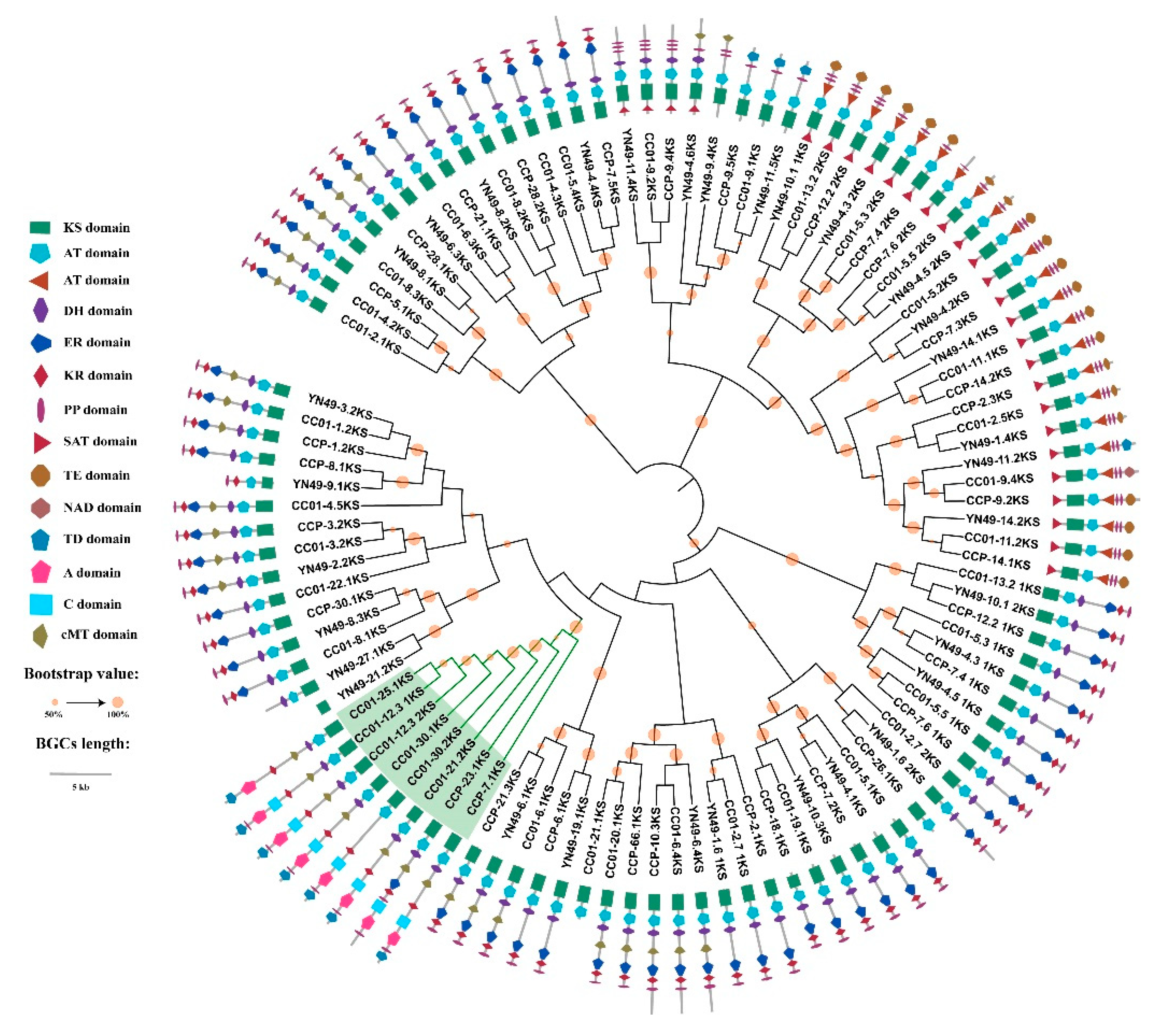
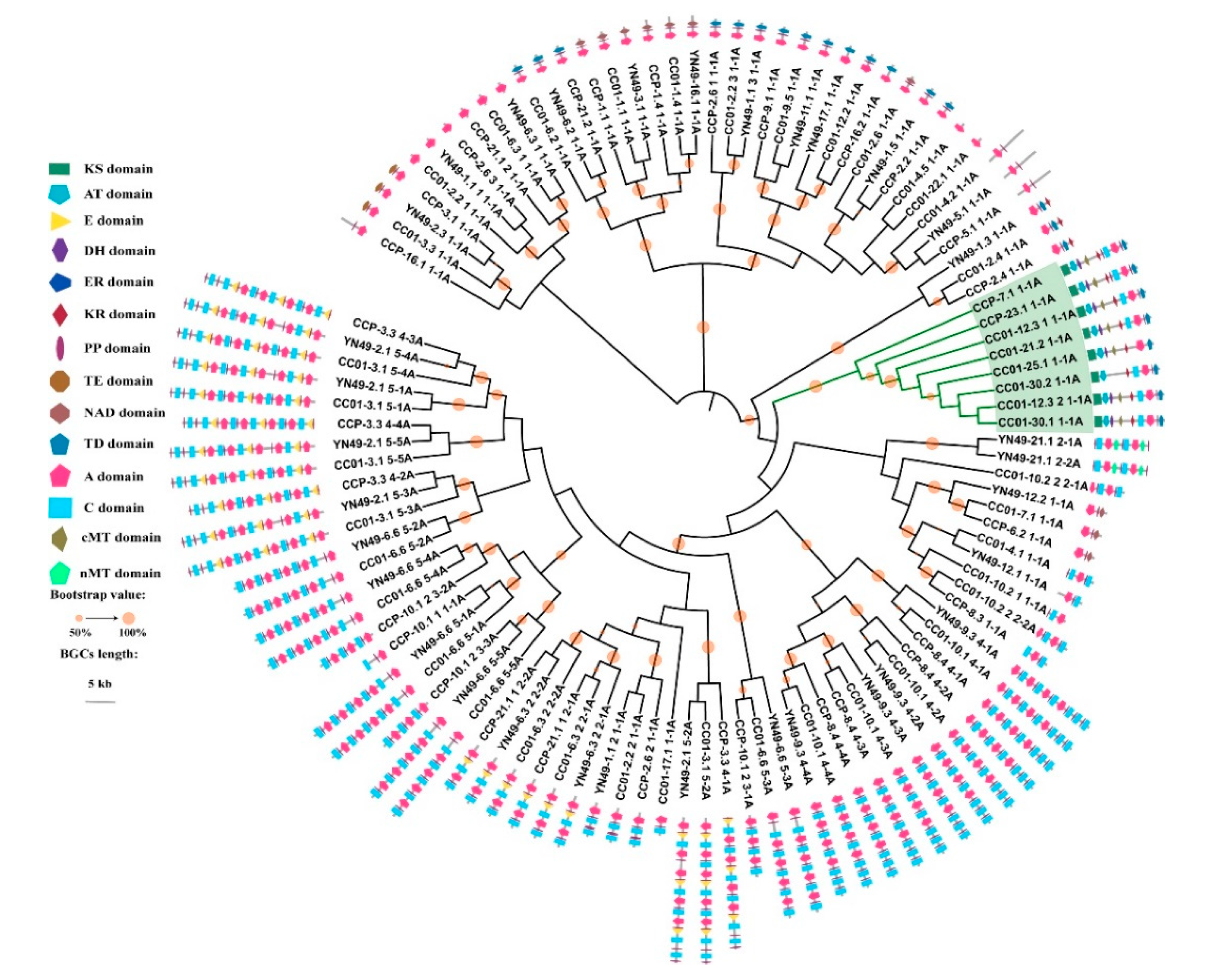
3.9. Phylogenomic Analysis of NRPS, PKS and PKS/NRPS Genes, and Domain Structure Analysis
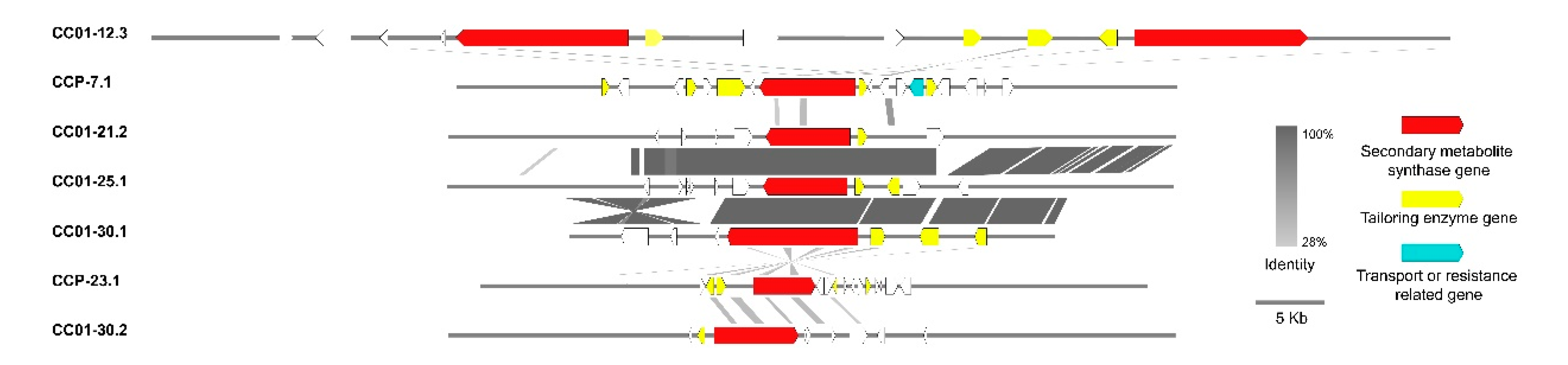
3.10. Synteny Analysis of PKS/NRPS Gene Clusters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schoch, C.; Crous, P.; Groenewald, J.; Boehm, E.; Burgess, T.; de Gruyter, J.; de Hoog, G.; Dixon, L.; Grube, M.; Gueidan, C.; et al. A class-wide phylogenetic assessment of Dothideomycetes. Stud. Mycol. 2009, 64, 1–15. [Google Scholar] [CrossRef]
- Yan, X.; Yu, C.; Fu, X.A.; Bao, F.; Du, D.; Wang, C.; Wang, N.; Wang, S.; Shi, Z.; Zhou, G.; et al. CARD 9 mutation linked to Corynespora cassiicola infection in a Chinese patient. Br. J. Dermatol. 2016, 174, 176–179. [Google Scholar] [CrossRef]
- Zhao, D.-L.; Shao, C.-L.; Gan, L.-S.; Wang, M.; Wang, C.-Y. Chromone Derivatives from a Sponge-Derived Strain of the Fungus Corynespora cassiicola. J. Nat. Prod. 2015, 78, 286–293. [Google Scholar] [CrossRef]
- Carris, L.M.; Glawe, D.A.; Gray, L.E. Isolation of the Soybean Pathogens Corynespora cassiicola and Phialophora gregata from Cysts of Heterodera glycines in Illinois. Mycologia 1986, 78, 503. [Google Scholar] [CrossRef]
- Deighton, F.C. XXXVIII. Preliminary list of fungi and diseases of plants in Sierra Leone. Bull. Misc. Inf. 1936, 7, 397–424. [Google Scholar]
- Liyanage, A.S.; Jayasinghe, C.; Liyanage, N.I.S.; Jayaratne, A.H.R. Corynespora leaf spot disease of rubber (Hevea brasiliensis) a new record. J. Rubber Res. Inst. Sri Lanka 1986, 65, 47–50. [Google Scholar]
- Dixon, L.J.; Schlub, R.L.; Pernezny, K.; Datnoff, L.E. Host Specialization and Phylogenetic Diversity of Corynespora cassiicola. Phytopathology 2009, 99, 1015–1027. [Google Scholar] [CrossRef]
- Tran, D.M.; Clément-Demange, A.; Déon, M.; Garcia, D.; Le Guen, V.; Clement-Vidal, A.; Soumahoro, M.; Masson, A.; Label, P.; Le, M.T.; et al. Genetic determinism of sensitivity to Corynespora cassiicola Exudates in rubber tree (Hevea brasiliensis). PLoS ONE 2016, 11, e0162807. [Google Scholar] [CrossRef]
- Jinji, P.; Xin, Z.; Yangxian, Q.; Yixian, X.; Huiqiang, Z.; He, Z. First record of Corynespora leaf fall disease of Hevea rubber tree in China. Australas. Plant Dis. Notes 2007, 2, 35–36. [Google Scholar] [CrossRef]
- Li, Z.; Fox, J.M. Mapping rubber tree growth in mainland Southeast Asia using time-series MODIS 250 m NDVI and statistical data. Appl. Geogr. 2012, 32, 420–443. [Google Scholar] [CrossRef]
- Chen, B.; Xiao, X.; Wu, Z.; Yun, T.; Kou, W.; Ye, H.; Lin, Q.; Doughty, R.; Dong, J.; Ma, J.; et al. Identifying establishment year and pre-conversion land cover of rubber plantations on Hainan Island, China using landsat data during 1987–2015. Remote Sens. 2018, 10, 1240. [Google Scholar] [CrossRef]
- Yang, X.; Blagodatsky, S.; Liu, F.; Beckschäfer, P.; Xu, J.; Cadisch, G. Rubber tree allometry, biomass partitioning and carbon stocks in mountainous landscapes of sub-tropical China. For. Ecol. Manag. 2017, 404, 84–99. [Google Scholar] [CrossRef]
- Déon, M.; Bourré, Y.; Gimenez, S.; Berger, A.; Bieysse, D.; de Lamotte, F.; Poncet, J.; Roussel, V.; Bonnot, F.; Oliver, G.; et al. Characterization of a cassiicolin-encoding gene from Corynespora cassiicola, pathogen of rubber tree (Hevea brasiliensis). Plant Sci. 2012, 185–186, 227–237. [Google Scholar] [CrossRef]
- Déon, M.; Fumanal, B.; Gimenez, S.; Bieysse, D.; Oliveira, R.R.; Shuib, S.S.; Breton, F.; Elumalai, S.; Vida, J.B.; Seguin, M.; et al. Diversity of the cassiicolin gene in Corynespora cassiicola and relation with the pathogenicity in Hevea brasiliensis. Fungal Biol. 2014, 118, 32–47. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Xie, X.; Shi, Y.; Chai, A.; Wang, Q.; Li, B. Variation of cassiicolin genes among Chinese isolates of Corynespora cassiicola. J. Microbiol. 2018, 56, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Lopez, D.; Ribeiro, S.; Label, P.; Fumanal, B.; Venisse, J.-S.; Kohler, A.; De Oliveira, R.R.; LaButti, K.; Lipzen, A.; Lail, K.; et al. Genome-wide analysis of corynespora cassiicola leaf fall disease putative effectors. Front. Microbiol. 2018, 9, 276. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, D.J. Growth of the plant cell wall. Nat. Rev. Mol. Cell Biol. 2005, 6, 850–861. [Google Scholar] [CrossRef] [PubMed]
- Xinchun, Z.; Xiangmin, L. Ultrastructural studies on the interaction of rubber tree with oidium heveae. Chin. J. Trop. Crop. 2010, 31, 2250–2254. [Google Scholar]
- Kubicek, C.P.; Starr, T.L.; Glass, N.L. Plant cell wall–degrading enzymes and their secretion in plant-pathogenic fungi. Annu. Rev. Phytopathol. 2014, 52, 427–451. [Google Scholar] [CrossRef] [PubMed]
- Zhi-Heng, L.; Ye, Q.I.; Xin-Yang, H.; Hong, Y.; Yue, H.; Rui, Z. Conditions and activity analysis of cell wall degrading enzymes produced from Corynespora cassiicola of brown spot of cucumber. China Veg. 2011, 8, 76–80. [Google Scholar]
- Breton, F.; Sanier, C.; D’Auzac, J. Role of cassiicolin, a host-selective toxin, in pathogenicity of Corynespora cassiicola, causal agent of a leaf fall disease of Hevea. Gastroenterology 2000, 120, A173–A174. [Google Scholar]
- Vining, L.C. Functions of secondary metabolites. Annu. Rev. Microbiol. 1990, 44, 395–427. [Google Scholar] [CrossRef]
- Pusztahelyi, T.; Holb, I.J. Pã³CsiI. Secondary metabolites in fungus-plant interactions. Front. Plant Sci. 2015, 6, 573. [Google Scholar] [CrossRef]
- Keller, N.P. Translating biosynthetic gene clusters into fungal armor and weaponry. Nat. Chem. Biol. 2015, 11, 671–677. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptivek-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Birney, E. Using GeneWise in the drosophila annotation experiment. Genome Res. 2000, 10, 547–548. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- De Bie, T.; Cristianini, N.; DeMuth, J.P.; Hahn, M.W. CAFE: A computational tool for the study of gene family evolution. Bioinformatics 2006, 22, 1269–1271. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Bedell, J.A.; Korf, I.; Gish, W. MaskerAid: A performance enhancement to RepeatMasker. Bioinformatics 2000, 16, 1040–1041. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef] [PubMed]
- Lombard, V.; Ramulu, H.G.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M. The Lipase Engineering Database: A navigation and analysis tool for protein families. Nucleic Acids Res. 2003, 31, 319–321. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Hahn, M.W.; De Bie, T.; Stajich, J.; Nguyen, C.; Cristianini, N. Estimating the tempo and mode of gene family evolution from comparative genomic data. Genome Res. 2005, 15, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Powell, A.J.; Conant, G.C.; E Brown, D.; Carbone, I.; A Dean, R. Altered patterns of gene duplication and differential gene gain and loss in fungal pathogens. BMC Genom. 2008, 9, 147. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Nandineni, M.R. Genome sequencing and comparative genomics reveal a repertoire of putative pathogenicity genes in chilli anthracnose fungus Colletotrichum truncatum. PLoS ONE 2017, 12, e0183567. [Google Scholar]
- Urban, M.; Pant, R.; Raghunath, A.; Irvine, A.G.; Pedro, H.; Hammond-Kosack, K.E. The Pathogen-Host Interactions database (PHI-base): Additions and future developments. Nucleic Acids Res. 2015, 43, D645–D655. [Google Scholar] [CrossRef]
- Blümke, A.; Falter, C.; Herrfurth, C.; Sode, B.; Bode, R.; Schäfer, W.; Feussner, I.; Voigt, C.A. Secreted fungal effector lipase releases free fatty acids to inhibit innate immunity-related callose formation during wheat head infection. Plant Physiol. 2014, 165, 346–358. [Google Scholar] [CrossRef]
- Chang, H.-X.; Yendrek, C.R.; Caetano-Anolles, G.; Hartman, G.L. Erratum to: Genomic characterization of plant cell wall degrading enzymes and in silico analysis of xylanases and polygalacturonases of Fusarium virguliforme. BMC Microbiol. 2017, 17, 110. [Google Scholar] [CrossRef]
- Yin, Z.; Liu, H.; Li, Z.; Ke, X.; Dou, D.; Gao, X.; Song, N.; Dai, Q.; Wu, Y.; Xu, J.; et al. Genome sequence of Valsa canker pathogens uncovers a potential adaptation of colonization of woody bark. New Phytol. 2015, 208, 1202–1216. [Google Scholar] [CrossRef]
- Robinson, S.L.; Christenson, J.K.; Wackett, L.P. Biosynthesis and chemical diversity of β-lactone natural products. Nat. Prod. Rep. 2018, 36, 458–475. [Google Scholar] [CrossRef]
- Tang, X.-X.; Yan, X.; Fu, W.-H.; Yi, L.-Q.; Tang, B.-W.; Yu, L.-B.; Fang, M.-J.; Wu, Z.; Qiu, Y.-K. New β-Lactone with tea pathogenic fungus inhibitory effect from marine-derived fungus MCCC3A00957. J. Agric. Food Chem. 2019, 67, 2877–2885. [Google Scholar] [CrossRef]
- Bushley, K.E.; Turgeon, B.G. Phylogenomics reveals subfamilies of fungal nonribosomal peptide synthetases and their evolutionary relationships. BMC Evol. Biol. 2010, 10, 26. [Google Scholar] [CrossRef]
- Kroken, S.; Glass, N.L.; Taylor, J.W.; Yoder, O.C.; Turgeon, B.G. Phylogenomic analysis of type I polyketide synthase genes in pathogenic and saprobic ascomycetes. Proc. Natl. Acad. Sci. USA 2003, 100, 15670–15675. [Google Scholar] [CrossRef]
- Lind, A.; Wisecaver, J.H.; Lameiras, C.; Wiemann, P.; Palmer, J.M.; Keller, N.P.; Rodrigues, F.; Goldman, G.H.; Rokas, A. Drivers of genetic diversity in secondary metabolic gene clusters within a fungal species. PLoS Biol. 2017, 15, e2003583. [Google Scholar] [CrossRef]
- SuryanarayananT, T.S.; Murali, T.S.; Thirunavukkarasu, N.; Rajulu, M.B.G.; Venkatesan, G.; Sukumar, R. Endophytic fungal communities in woody perennials of three tropical forest types of the Western Ghats, southern India. Biodivers. Conserv. 2011, 20, 913–928. [Google Scholar] [CrossRef]
- Gond, S.K.; Verma, V.C.; Kumar, A.; Kumar, V.; Kharwar, R.N. Study of endophytic fungal community from different parts of Aegle marmelos Correae (Rutaceae) from Varanasi (India). World J. Microbiol. Biotechnol. 2007, 23, 1371–1375. [Google Scholar] [CrossRef]
- Cai, L.; Ji, K.-F.; Hyde, K.D. Variation between freshwater and terrestrial fungal communities on decaying bamboo culms. Antonie van Leeuwenhoek 2006, 89, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.-X.; Zhang, X.; Pu, J.-J.; Liu, X.-M.; Lu, Y.; Zhang, H.; Zhang, H.-Q.; Lv, Y.-C.; Xie, Y.-X. Morphological and molecular analysis of genetic variability within isolates of Corynespora cassiicola from different hosts. Eur. J. Plant Pathol. 2011, 130, 83–95. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, X.; Li, K.; Wang, C.; Cai, L.; Zhuang, W.; Xiang, M.; Liu, X. Introgression and gene family contraction drive the evolution of lifestyle and host shifts of hypocrealean fungi. Mycology 2018, 9, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Aguileta, G.; Lengellé, J.; Chiapello, H.; Giraud, T.; Viaud, M.; Fournier, E.; Rodolphe, F.; Marthey, S.; Ducasse, A.; Gendrault, A.; et al. Genes under positive selection in a model plant pathogenic fungus, Botrytis. Infect. Genet. Evol. 2012, 12, 987–996. [Google Scholar] [CrossRef]
- Deng, X.; Wang, J.; Li, Y.; Wu, S.; Yang, S.; Chao, J.; Chen, Y.; Zhang, S.; Shi, M.; Tian, W. Comparative transcriptome analysis reveals phytohormone signalings, heat shock module and ROS scavenger mediate the cold-tolerance of rubber tree. Sci. Rep. 2018, 8, 4931. [Google Scholar] [CrossRef]
- Cheng, H.; Chen, X.; Fang, J.; An, Z.; Hu, Y.; Huang, H. Comparative transcriptome analysis reveals an early gene expression profile that contributes to cold resistance in Hevea brasiliensis (the Para rubber tree). Tree Physiol. 2018, 38, 1409–1423. [Google Scholar] [CrossRef]
- González-Fernández, R.; Prats, E.; Jorrín-Novo, J.V. Proteomics of Plant Pathogenic Fungi. J. Biomed. Biotechnol. 2010, 2010, 1–36. [Google Scholar] [CrossRef]
- Miller, M.E.; Zhang, Y.; Omidvar, V.; Sperschneider, J.; Schwessinger, B.; Raley, C.; Palmer, J.M.; Garnica, D.; Upadhyaya, N.; Rathjen, J.; et al. De Novo Assembly and Phasing of Dikaryotic Genomes from Two Isolates of Puccinia coronata f. sp. avenae, the Causal Agent of Oat Crown Rust. mBio 2018, 9. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, H.; Wang, C.; Xu, J.-R. Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genom. 2013, 14, 274. [Google Scholar] [CrossRef]
- Quoc, N.B.; Chau, N.N.B. The Role of Cell Wall Degrading Enzymes in Pathogenesis of Magnaporthe oryzae. Curr. Protein Pept. Sci. 2017, 18, 1019–1034. [Google Scholar] [CrossRef]
- Lara-Márquez, A.; Oyama, K.; Zavala-Páramo, M.G.; Villa-Rivera, M.G.; Conejo-Saucedo, U.; Cano-Camacho, H. Evolutionary analysis of pectin lyases of the genus colletotrichum. J. Mol. Evol. 2017, 85, 120–136. [Google Scholar] [CrossRef] [PubMed]
- Gan, P.; Ikeda, K.; Irieda, H.; Narusaka, M.; O’Connell, R.J.; Narusaka, Y.; Takano, Y.; Kubo, Y.; Shirasu, K. Comparative genomic and transcriptomic analyses reveal the hemibiotrophic stage shift of Colletotrichum fungi. New Phytol. 2013, 197, 1236–1249. [Google Scholar] [CrossRef]
- Liu, X.; Li, B.; Cai, J.; Zheng, X.; Feng, Y.; Huang, G. Colletotrichum Species Causing Anthracnose of Rubber Trees in China. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- Fernando, T.; Jayasinghe, C.; Wijesundera, R. Cell wall degrading enzyme secretion by Colletotrichum acutatum the causative fungus of secondary leaf fall of Hevea brasiliensis. Mycol. Res. 2001, 105, 195–201. [Google Scholar] [CrossRef]
- Barthe, P.; Pujade-Renaud, V.; Breton, F.; Gargani, D.; Thai, R.; Roumestand, C.; de Lamotte, F. Structural Analysis of Cassiicolin, a Host-selective Protein Toxin from Corynespora cassiicola. J. Mol. Biol. 2007, 367, 89–101. [Google Scholar] [CrossRef]
- Aragón, W.; Reina-Pinto, J.J.; Serrano, M. The intimate talk between plants and microorganisms at the leaf surface. J. Exp. Bot. 2017, 68, 5339–5350. [Google Scholar] [CrossRef]
- Feng, J.; Wang, F.; Liu, G.; Greenshields, D.; Shen, W.; Kaminskyj, S.; Hughes, G.R.; Peng, Y.; Selvaraj, G.; Zou, J.; et al. Analysis of a Blumeria graminis-Secreted Lipase Reveals the Importance of Host Epicuticular Wax Components for Fungal Adhesion and Development. Mol. Plant Microbe Interact. 2009, 22, 1601–1610. [Google Scholar] [CrossRef]
- Berestetskiy, A.O. A review of fungal phytotoxins: From basic studies to practical use. Appl. Biochem. Microbiol. 2008, 44, 453–465. [Google Scholar] [CrossRef]
- Ribeiro, S.; Tran, D.M.; Déon, M.; Clément-Demange, A.; Garcia, D.; Soumahoro, M.; Masson, A.; Pujade-Renaud, V. Gene deletion of Corynespora cassiicola cassiicolin Cas1 suppresses virulence in the rubber tree. Fungal Genet. Biol. 2019, 129, 101–114. [Google Scholar] [CrossRef]
- Xianbao, L.; Boxun, L.I.; Shan, C.; Guixiu, H. Diversity and pathogenicity of the cassiicolin gene in corynespora cassiicola of rubber tree in China. Chin. J. Trop. Crops 2016, 37, 1969–1973. [Google Scholar]
- Gao, S.; Zeng, R.; Xu, L.; Song, Z.; Gao, P.; Dai, F. Genome sequence and spore germination-associated transcriptome analysis of Corynespora cassiicola from cucumber. BMC Microbiol. 2020, 20, 1–20. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | YN49 | CC01 | HGCC | CCP | UM591 |
|---|---|---|---|---|---|
| Total genome size (Mb) | 45.1 | 47.1 | 42.7 | 44.8 | 41.4 |
| Genome coverage | 115× | 228× | 184× | 123× | 118× |
| G + C content (%) | 51.09 | 50.96 | 51.78 | 51.89 | 52.47 |
| Number of contigs | 32 | 33 | 1032 | 644 | 1941 |
| Contig N50 (bp) | 2,523,863 | 2,573,660 | / | 211,447 | 44,332 |
| Protein-coding genes | 14,504 | 17,219 | 14,631 | 17,167 | 14,744 |
| Substrates | Family | YN49 | CC01 | CCP |
|---|---|---|---|---|
| Cellulose | AA9 | 25 | 28 | 27 |
| GH1 | 2 | 1 | 3 | |
| GH12 | 2 | 2 | 2 | |
| GH3 | 13 | 12 | 13 | |
| GH45 | 1 | 2 | 2 | |
| GH5 | 31 | 27 | 37 | |
| GH6 | 3 | 2 | 2 | |
| GH7 | 5 | 5 | 5 | |
| Hemicellulose | CE1 | 8 | 8 | 9 |
| GH10 | 3 | 4 | 4 | |
| GH11 | 4 | 4 | 4 | |
| GH115 | 2 | 2 | 2 | |
| GH27 | 2 | 3 | 4 | |
| GH29 | 1 | 0 | 1 | |
| GH30 | 2 | 2 | 2 | |
| GH35 | 3 | 3 | 4 | |
| GH36 | 1 | 1 | 1 | |
| GH39 | 1 | 1 | 1 | |
| GH43 | 25 | 21 | 24 | |
| GH51 | 0 | 0 | 1 | |
| GH53 | 1 | 1 | 1 | |
| GH67 | 1 | 1 | 1 | |
| GH93 | 3 | 3 | 2 | |
| Pectin | CE12 | 4 | 2 | 5 |
| CE8 | 4 | 3 | 4 | |
| GH105 | 5 | 4 | 6 | |
| GH28 | 7 | 9 | 10 | |
| GH78 | 2 | 5 | 4 | |
| PL1 | 11 | 12 | 17 | |
| PL3 | 11 | 8 | 12 | |
| PL4 | 4 | 2 | 4 | |
| PL9 | 2 | 2 | 2 | |
| Cutin | CE5 | 5 | 4 | 7 |
| Total number | 194 | 184 | 223 |
| SM Cluster Types | YN49 | CC01 | CCP |
|---|---|---|---|
| NRPS | 16 | 15 | 15 |
| PKS (Type I) | 26 | 25 | 26 |
| PKS (Type III) | 1 | 1 | 1 |
| PKS/NRPS | 1 | 9 | 4 |
| PKS/Indole | 1 | 0 | 0 |
| NRPS/Indole | 0 | 1 | 0 |
| Indole | 1 | 1 | 1 |
| Terpene | 10 | 10 | 9 |
| Beta-lactone | 1 | 0 | 1 |
| Total | 57 | 62 | 57 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, B.; Yang, Y.; Cai, J.; Liu, X.; Shi, T.; Li, C.; Chen, Y.; Xu, P.; Huang, G. Genomic Characteristics and Comparative Genomics Analysis of Two Chinese Corynespora cassiicola Strains Causing Corynespora Leaf Fall (CLF) Disease. J. Fungi 2021, 7, 485. https://doi.org/10.3390/jof7060485
Li B, Yang Y, Cai J, Liu X, Shi T, Li C, Chen Y, Xu P, Huang G. Genomic Characteristics and Comparative Genomics Analysis of Two Chinese Corynespora cassiicola Strains Causing Corynespora Leaf Fall (CLF) Disease. Journal of Fungi. 2021; 7(6):485. https://doi.org/10.3390/jof7060485
Chicago/Turabian StyleLi, Boxun, Yang Yang, Jimiao Cai, Xianbao Liu, Tao Shi, Chaoping Li, Yipeng Chen, Pan Xu, and Guixiu Huang. 2021. "Genomic Characteristics and Comparative Genomics Analysis of Two Chinese Corynespora cassiicola Strains Causing Corynespora Leaf Fall (CLF) Disease" Journal of Fungi 7, no. 6: 485. https://doi.org/10.3390/jof7060485
APA StyleLi, B., Yang, Y., Cai, J., Liu, X., Shi, T., Li, C., Chen, Y., Xu, P., & Huang, G. (2021). Genomic Characteristics and Comparative Genomics Analysis of Two Chinese Corynespora cassiicola Strains Causing Corynespora Leaf Fall (CLF) Disease. Journal of Fungi, 7(6), 485. https://doi.org/10.3390/jof7060485