
Transcriptome Analysis Identifies a Gene Cluster for the Biosynthesis of Biruloquinone, a Rare Phenanthraquinone, in a Lichen-Forming Fungus Cladonia macilenta


Abstract
1. Introduction
2. Materials and Methods
2.1. Genome Annotation and Biosynthetic Gene Cluster Identification
2.2. Phylogenetic Analysis
2.3. RNA-Seq Experiment
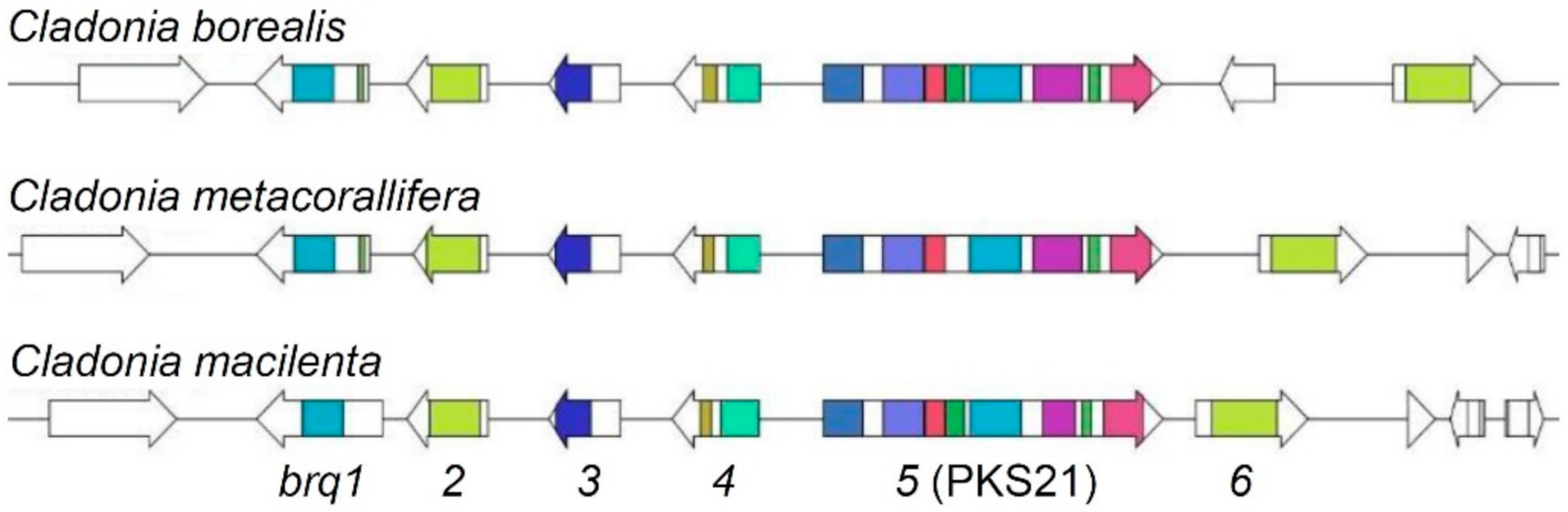
2.4. Identification of Syntenic Gene Clusters
2.5. Data Availability
3. Results
3.1. The Biosynthesis of Biruloquinone
3.2. Phylogenetic Placement of C. macilenta PKS
3.3. Transcriptome Analysis of C. macilenta PKS
3.4. Identification of Biruloquinone Biosynthesis Gene Clusters
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Calcott, M.J.; Ackerley, D.F.; Knight, A.; Keyzers, R.A.; Owen, J.G. Secondary metabolism in the lichen symbiosis. Chem. Soc. Rev. 2018, 47, 1730–1760. [Google Scholar] [CrossRef] [PubMed]
- Stocker-Wörgötter, E. Metabolic diversity of lichen-forming ascomycetous Fungi: Culturing, polyketide and shikimate metabolite production, and PKS genes. Nat. Prod. Rep. 2008, 25, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Stocker-Wörgötter, E.; Cordeiro, L.M.C.; Iacomini, M. Accumulation of potential pharmaceutically relevant lichen metabolites in lichens and cultured lichen symbionts. In Studies in Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2013; Volume 39, pp. 337–380. [Google Scholar]
- Culberson, C.F. Supplement to “Chemical and Botanical Guide to Lichen Products”. Bryologist 1970, 73, 177. [Google Scholar] [CrossRef]
- Stenroos, S.; Ahti, T. Phylogeny of the genus Cladonia s.lat. (Cladoniaceae, Ascomycetes) inferred from molecular, morphological, and chemical data. Cladistics 2002, 18, 237–278. [Google Scholar] [CrossRef]
- Luo, H.; Li, C.; Kim, J.C.; Liu, Y.; Jung, J.S.; Koh, Y.J.; Hur, J.-S. Biruloquinone, an acetylcholinesterase inhibitor produced by lichen-forming Fungus Cladonia macilenta. J. Microbiol. Biotechnol. 2013, 23, 161–166. [Google Scholar] [CrossRef]
- Krivoshchekova, O.E.; Stepanenko, L.S.; Mishchenko, N.P.; Denisenko, V.A.; Maksimov, O.B. A study of aromatic metabolites of lichens of the family Parmeliaceae. II. Pigments. Chem. Nat. Compd. 1983, 19, 270–274. [Google Scholar] [CrossRef]
- Arnone, A.; Nasini, G.; de Pava, O.V. A reinvestigation of the structure of biruloquinone, a 9,10-phenanthrenequinone isolated from Mycosphaerella rubella. Phytochemistry 1991, 30, 2729–2731. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Hara, K.; Kawakami, H.; Komine, M. Lichen substances and their biological activities. In Recent Advances in Lichenology; Upreti, D.K., Divakar, P.K., Shukla, V., Bajpai, R., Eds.; Springer India: New Delhi, India, 2015; pp. 181–199. [Google Scholar]
- Müller, K. Pharmaceutically relevant metabolites from lichens. Appl. Microbiol. Biotechnol. 2001, 56, 9–16. [Google Scholar] [CrossRef]
- Yu, J.; Chang, P.-K.; Ehrlich, K.C.; Cary, J.W.; Bhatnagar, D.; Cleveland, T.E.; Payne, G.A.; Linz, J.E.; Woloshuk, C.P.; Bennett, J.W. Clustered pathway genes in aflatoxin biosynthesis. Appl. Environ. Microbiol. 2004, 70, 1253–1262. [Google Scholar] [CrossRef]
- Hendrickson, L.; Davis, C.R.; Roach, C.; Nguyen, D.K.; Aldrich, T.; McAda, P.C.; Reeves, C.D. Lovastatin biosynthesis in Aspergillus terreus: Characterization of blocked mutants, enzyme activities and a multifunctional polyketide synthase gene. Chem. Biol. 1999, 6, 429–439. [Google Scholar] [CrossRef]
- Meehan, M.J.; Xie, X.; Zhao, X.; Xu, W.; Tang, Y.; Dorrestein, P.C. FT-ICR-MS characterization of intermediates in the biosynthesis of the α-methylbutyrate side chain of lovastatin by the 277 KDa polyketide synthase LovF. Biochemistry 2011, 50, 287–299. [Google Scholar] [CrossRef]
- Moriwaki, A.; Kihara, J.; Kobayashi, T.; Tokunaga, T.; Arase, S.; Honda, Y. Insertional mutagenesis and characterization of a polyketide synthase gene (PKS1) required for melanin biosynthesis in Bipolaris oryzae. FEMS Microbiol. Lett. 2004, 238, 1–8. [Google Scholar]
- Cheng, Q.; Kinney, K.A.; Whitman, C.P.; Szaniszlo, P.J. Characterization of two polyketide synthase genes in Exophiala lecanii-Corni, a melanized fungus with bioremediation potential. Bioorg. Chem. 2004, 32, 92–108. [Google Scholar] [CrossRef]
- Fulton, T.R.; Ibrahim, N.; Losada, M.C.; Grzegorski, D.; Tkacz, J.S. A melanin polyketide synthase (PKS) gene from Nodulisporium sp. that shows homology to the Pks1 gene of Colletotrichum lagenarium. Mol. Gen. Genet. 1999, 262, 714–720. [Google Scholar] [CrossRef]
- Zhang, A.; Lu, P.; Dahl-Roshak, A.M.; Paress, P.S.; Kennedy, S.; Tkacz, J.S.; An, Z. Efficient disruption of a polyketide synthase gene (Pks1) required for melanin synthesis through Agrobacterium-mediated transformation of Glarea lozoyensis. Mol. Gen. Genom. 2003, 268, 645–655. [Google Scholar] [CrossRef]
- Kroken, S.; Glass, N.L.; Taylor, J.W.; Yoder, O.C.; Turgeon, B.G. Phylogenomic analysis of type I polyketide synthase genes in pathogenic and saprobic ascomycetes. Proc. Natl. Acad. Sci. USA 2003, 100, 15670–15675. [Google Scholar] [CrossRef]
- Jeong, M.-H.; Kim, J.A.; Yu, N.H.; Jung, J.S.; Hong, S.G.; Cheong, Y.H.; Hur, J.-S. Isolation and characterization of a non-reducing polyketide synthase gene in Cladonia macilenta. Mycoscience 2015, 56, 49–57. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, Z.; Shao, C.-L.; Wang, C.-Y. Analysis of the sequences, structures, and functions of product-releasing enzyme domains in fungal polyketide synthases. Front. Microbiol. 2017, 8, 1685. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-Y.; Choi, J.; Kim, J.A.; Jeong, M.-H.; Kim, S.; Lee, Y.-H.; Hur, J.-S. Draft genome sequence of Cladonia macilenta KoLRI003786, a lichen-forming fungus producing biruloquinone. Genome Announc. 2013, 1, e00695-13. [Google Scholar] [CrossRef]
- Humann, J.L.; Lee, T.; Ficklin, S.; Main, D. Structural and functional annotation of eukaryotic genomes with GenSAS. Methods Mol. Biol. 2019, 1962, 29–51. [Google Scholar]
- Stanke, M.; Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 2003, 19, ii215–ii225. [Google Scholar] [CrossRef] [PubMed]
- Ter-Hovhannisyan, V.; Lomsadze, A.; Chernoff, Y.O.; Borodovsky, M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008, 18, 1979–1990. [Google Scholar] [CrossRef] [PubMed]
- Richmond, T. Identification of complete gene structures in genomic DNA. Genome Biol. 2000, 1, reports222. [Google Scholar] [CrossRef]
- Delcher, A.L.; Harmon, D.; Kasif, S.; White, O.; Salzberg, S.L. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999, 27, 4636–4641. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. AntiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef]
- Ziemert, N.; Podell, S.; Penn, K.; Badger, J.H.; Allen, E.; Jensen, P.R. The natural product domain seeker NaPDoS: A phylogeny based bioinformatic tool to classify secondary metabolite gene diversity. PLoS ONE 2012, 7, e34064. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML Version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Navarro-Muñoz, J.C.; Selem-Mojica, N.; Mullowney, M.W.; Kautsar, S.A.; Tryon, J.H.; Parkinson, E.I.; De Los Santos, E.L.C.; Yeong, M.; Cruz-Morales, P.; Abubucker, S.; et al. A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol. 2020, 16, 60–68. [Google Scholar] [CrossRef]
- Newman, A.G.; Vagstad, A.L.; Belecki, K.; Scheerer, J.R.; Townsend, C.A. Analysis of the cercosporin polyketide synthase CTB1 reveals a new fungal thioesterase function. Chem. Commun. 2012, 48, 11772–11774. [Google Scholar] [CrossRef]
- Ahuja, M.; Chiang, Y.-M.; Chang, S.-L.; Praseuth, M.B.; Entwistle, R.; Sanchez, J.F.; Lo, H.-C.; Yeh, H.-H.; Oakley, B.R.; Wang, C.C.C. Illuminating the diversity of aromatic polyketide synthases in Aspergillus nidulans. J. Am. Chem. Soc. 2012, 134, 8212–8221. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, Z.; Shao, C.-L.; Wang, J.-L.; Bai, H.; Wang, C.-Y. Bioinformatical analysis of the sequences, structures and functions of fungal polyketide synthase product template domains. Sci. Rep. 2015, 5, 10463. [Google Scholar] [CrossRef]
- Calchera, A.; Dal Grande, F.; Bode, H.B.; Schmitt, I. Biosynthetic gene content of the ‘perfume lichens’ Evernia prunastri and Pseudevernia furfuracea. Molecules 2019, 24, 203. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, R.L.; Abdel-Hameed, M.; Sorensen, J.L. Lichen biosynthetic gene clusters. Part I. Genome sequencing reveals a rich biosynthetic potential. J. Nat. Prod. 2018, 81, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Geng, C.; Yuan, X.; Hua, M.; Tian, F.; Li, C. Identification of a putative polyketide synthase gene involved in usnic acid biosynthesis in the lichen Nephromopsis pallescens. PLoS ONE 2018, 13, e0199110. [Google Scholar] [CrossRef] [PubMed]
- Armaleo, D.; Sun, X.; Culberson, C. Insights from the first putative biosynthetic gene cluster for a lichen depside and depsidone. Mycologia 2011, 103, 741–754. [Google Scholar] [CrossRef]
- Elshobary, M.E.; Becker, M.G.; Kalichuk, J.L.; Chan, A.C.; Belmonte, M.F.; Piercey-Normore, M.D. Tissue-specific localization of polyketide synthase and other associated genes in the lichen, Cladonia rangiferina, using laser microdissection. Phytochemistry 2018, 156, 142–150. [Google Scholar] [CrossRef]
- Yin, W.; Keller, N.P. Transcriptional regulatory elements in fungal secondary metabolism. J. Microbiol. 2011, 49, 329–339. [Google Scholar] [CrossRef]
- Kim, W.; Park, J.-J.; Gang, D.R.; Peever, T.L.; Chen, W. A novel type pathway-specific regulator and dynamic genome environments of a solanapyrone biosynthesis gene cluster in the fungus Ascochyta rabiei. Eukaryot. Cell 2015, 14, 1102–1113. [Google Scholar] [CrossRef]
- Molina, M.C.; Crespo, A.; Vicente, C.; Elix, J.A. Differences in the composition of phenolics and fatty acids of cultured mycobiont and thallus of Physconia distorta. Plant Physiol. Biochem. 2003, 41, 175–180. [Google Scholar] [CrossRef]
- Brunauer, G.; Hager, A.; Grube, M.; Türk, R.; Stocker-Wörgötter, E. Alterations in secondary metabolism of aposymbiotically grown mycobionts of Xanthoria elegans and cultured resynthesis stages. Plant Physiol. Biochem. 2007, 45, 146–151. [Google Scholar] [CrossRef]
- Ejiri, H.; Sankawa, U.; Shibata, S. Graciliformin and its acetates in Cladonia graciliformis. Phytochemistry 1975, 14, 277–279. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Matsubara, H.; Kinoshita, Y.; Kinoshita, K.; Koyama, K.; Takahashi, K.; Ahmadjiam, V.; Kurokawa, T.; Yoshimura, I. Naphthazarin derivatives from cultures of the lichen Cladonia cristatella. Phytochemistry 1996, 43, 1239–1242. [Google Scholar] [CrossRef]
- Dallery, J.; Adelin, É.; Le Goff, G.; Pigné, S.; Auger, A.; Ouazzani, J.; O’Connell, R.J. H3K4 trimethylation by CclA regulates pathogenicity and the production of three families of terpenoid secondary metabolites in Colletotrichum higginsianum. Mol. Plant Pathol. 2019, 20, 831–842. [Google Scholar] [CrossRef]
- Bachleitner, S.; Sørensen, J.L.; Gacek-Matthews, A.; Sulyok, M.; Studt, L.; Strauss, J. Evidence of a demethylase-independent role for the H3K4-specific histone demethylases in Aspergillus nidulans and Fusarium graminearum secondary metabolism. Front. Microbiol. 2019, 10, 1759. [Google Scholar] [CrossRef]
- Studt, L.; Janevska, S.; Arndt, B.; Boedi, S.; Sulyok, M.; Humpf, H.-U.; Tudzynski, B.; Strauss, J. Lack of the COMPASS component Ccl1 reduces H3K4 trimethylation levels and affects transcription of secondary metabolite genes in two plant–pathogenic Fusarium species. Front. Microbiol. 2017, 7, 2144. [Google Scholar] [CrossRef]
- Studt, L.; Rösler, S.M.; Burkhardt, I.; Arndt, B.; Freitag, M.; Humpf, H.-U.; Dickschat, J.S.; Tudzynski, B. Knock-down of the methyltransferase Kmt6 relieves H3K27me3 and results in induction of cryptic and otherwise silent secondary metabolite gene clusters in Fusarium fujikuroi: Relief of H3K27me3 induces silent SM gene clusters. Environ. Microbiol. 2016, 18, 4037–4054. [Google Scholar] [CrossRef]
- Gacek-Matthews, A.; Berger, H.; Sasaki, T.; Wittstein, K.; Gruber, C.; Lewis, Z.A.; Strauss, J. KdmB, a Jumonji histone H3 demethylase, regulates genome-wide H3K4 trimethylation and is required for normal induction of secondary metabolism in Aspergillus nidulans. PLoS Genet. 2016, 12, e1006222. [Google Scholar] [CrossRef]
- Gacek-Matthews, A.; Noble, L.M.; Gruber, C.; Berger, H.; Sulyok, M.; Marcos, A.T.; Strauss, J.; Andrianopoulos, A. KdmA, a histone H3 demethylase with bipartite function, differentially regulates primary and secondary metabolism in Aspergillus nidulans: Histone H3K9 demethylase function in growth. Mol. Microbiol. 2015, 96, 839–860. [Google Scholar] [CrossRef]
- Soukup, A.A.; Chiang, Y.-M.; Bok, J.W.; Reyes-Dominguez, Y.; Oakley, B.R.; Wang, C.C.C.; Strauss, J.; Keller, N.P. Overexpression of the Aspergillus nidulans histone 4 acetyltransferase EsaA increases activation of secondary metabolite production: H4K12 acetylation indicates SM production potential. Mol. Microbiol. 2012, 86, 314–330. [Google Scholar] [CrossRef]
- Reyes-Dominguez, Y.; Bok, J.W.; Berger, H.; Shwab, E.K.; Basheer, A.; Gallmetzer, A.; Scazzocchio, C.; Keller, N.; Strauss, J. Heterochromatic marks are associated with the repression of secondary metabolism clusters in Aspergillus nidulans: Heterochromatin regulation of secondary metabolism. Mol. Microbiol. 2010, 76, 1376–1386. [Google Scholar] [CrossRef]
- Bok, J.W.; Chiang, Y.-M.; Szewczyk, E.; Reyes-Dominguez, Y.; Davidson, A.D.; Sanchez, J.F.; Lo, H.-C.; Watanabe, K.; Strauss, J.; Oakley, B.R.; et al. Chromatin-level regulation of biosynthetic gene clusters. Nat. Chem. Biol. 2009, 5, 462–464. [Google Scholar] [CrossRef]
- Shwab, E.K.; Bok, J.W.; Tribus, M.; Galehr, J.; Graessle, S.; Keller, N.P. Histone deacetylase activity regulates chemical diversity in Aspergillus. Eukaryot. Cell 2007, 6, 1656–1664. [Google Scholar] [CrossRef] [PubMed]
- Stenroos, S.; Pino-Bodas, R.; Hyvönen, J.; Lumbsch, H.T.; Ahti, T. Phylogeny of the family Cladoniaceae (Lecanoromycetes, Ascomycota) based on sequences of multiple loci. Cladistics 2019, 35, 351–384. [Google Scholar] [CrossRef]
- Arai, Y.; Kinoshita, K.; Usuniwa, Y.; Yamamoto, Y.; Koyama, K.; Takahashi, K. Search for the substances of cultured lichen mycobiont—Focusing on Cladonia fruticlosa. Lichenology 2012, 10, 199. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ORF a (Cma) | Size (aa) | BLASTP Homolog (Accession) | % Identity (Coverage) | Conserved Domain | E-Value |
|---|---|---|---|---|---|
| 00864 | 325 | extradiol dioxygenase (XP_037155234) | 71 (100) | Memo-like protein (Pfam01875) | 6 × 10−77 |
| 00865 | 393 | hypothetical protein (CAF9912135) | 26 (31) | (not detected) | - |
| 00866 | 737 | hypothetical protein (CAF9931462) | 60 (96) | (not detected) | - |
| 00867 | 699 | GAL4-type transcription factor (PMD28964), brq1 | 56 (80) | fungal-specific transcription factor (Pfam04082) | 1 × 10−14 |
| 00868 | 452 | MFS transporter (OCK96312), brq2 | 75 (99) | Major Facilitator Superfamily (Pfam07690) | 1 × 10−21 |
| 00869 | 425 | O-methyltransferase (KAA6412585), brq3 | 74 (99) | O-methyltransferase (Pfam00891) | 4 × 10−14 |
| 00870 | 430 | FAD-dependent monooxygenase (KAA6409270), brq4 | 72 (99) | NAD(P)-binding Rossmann-like domain (Pfam13450) | 2 × 10−7 |
| 00871 | 1971 | polyketide synthase (QIX11496), brq5 | 94 (100) | ketoacyl synthase (Pfam00109) | 8 × 10−88 |
| 00872 | 590 | MFS transporter (KAA6410135), brq6 | 71 (96) | Major Facilitator Superfamily (Pfam07690) | 3 × 10−32 |
| 00873 | 167 | hypothetical protein (XP_018191177) | 30 (60) | (not detected) | - |
| 00874 | 191 | hypothetical protein (CAF9930055) | 86 (100) | CS domain (Pfam04969) | 5 × 10−19 |
| 00875 | 159 | hypothetical protein (XP_037155314) | 87 (98) | Polysaccharide biosynthesis (Pfam04669) | 7 × 10−38 |
| 00876 | 970 | ABC efflux pump (SLM35520) | 50 (99) | ABC transporter transmembrane region (Pfam00664) | 3 × 10−35 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, W.; Jeong, M.-H.; Yun, S.-H.; Hur, J.-S. Transcriptome Analysis Identifies a Gene Cluster for the Biosynthesis of Biruloquinone, a Rare Phenanthraquinone, in a Lichen-Forming Fungus Cladonia macilenta. J. Fungi 2021, 7, 398. https://doi.org/10.3390/jof7050398
Kim W, Jeong M-H, Yun S-H, Hur J-S. Transcriptome Analysis Identifies a Gene Cluster for the Biosynthesis of Biruloquinone, a Rare Phenanthraquinone, in a Lichen-Forming Fungus Cladonia macilenta. Journal of Fungi. 2021; 7(5):398. https://doi.org/10.3390/jof7050398
Chicago/Turabian StyleKim, Wonyong, Min-Hye Jeong, Sung-Hwan Yun, and Jae-Seoun Hur. 2021. "Transcriptome Analysis Identifies a Gene Cluster for the Biosynthesis of Biruloquinone, a Rare Phenanthraquinone, in a Lichen-Forming Fungus Cladonia macilenta" Journal of Fungi 7, no. 5: 398. https://doi.org/10.3390/jof7050398
APA StyleKim, W., Jeong, M.-H., Yun, S.-H., & Hur, J.-S. (2021). Transcriptome Analysis Identifies a Gene Cluster for the Biosynthesis of Biruloquinone, a Rare Phenanthraquinone, in a Lichen-Forming Fungus Cladonia macilenta. Journal of Fungi, 7(5), 398. https://doi.org/10.3390/jof7050398