
Microbiota Associated with Different Developmental Stages of the Dry Rot Fungus Serpula lacrymans



Abstract
1. Introduction
2. Materials and Methods
2.1. Biological Material
2.2. Isolation of Bacteria from S. lacrymans Fruiting Bodies, Mycelia and Rhizomorphs
2.3. Characterization of Isolated Bacteria
2.3.1. Molecular Identification and Phylogenetic Placement
2.3.2. Biopolymer Degradation Tests and Evaluation
2.3.3. Co-Culture of S. lacrymans with Isolated Bacteria
2.4. Fluorescence In Situ Hybridization (FISH)
2.4.1. Sampling and Fixation Conditions
2.4.2. Isolation of the Surface Community by Cuticle Tape Lift
2.4.3. FISH Probes and Fluorochromes
2.4.4. (In Tube) FISH
2.4.5. Multiplex FISH
2.4.6. FISH for Imprints of the Surface Community on Adhesive Tape
2.4.7. Nucleic Acid and Cell Wall Staining
3. Results
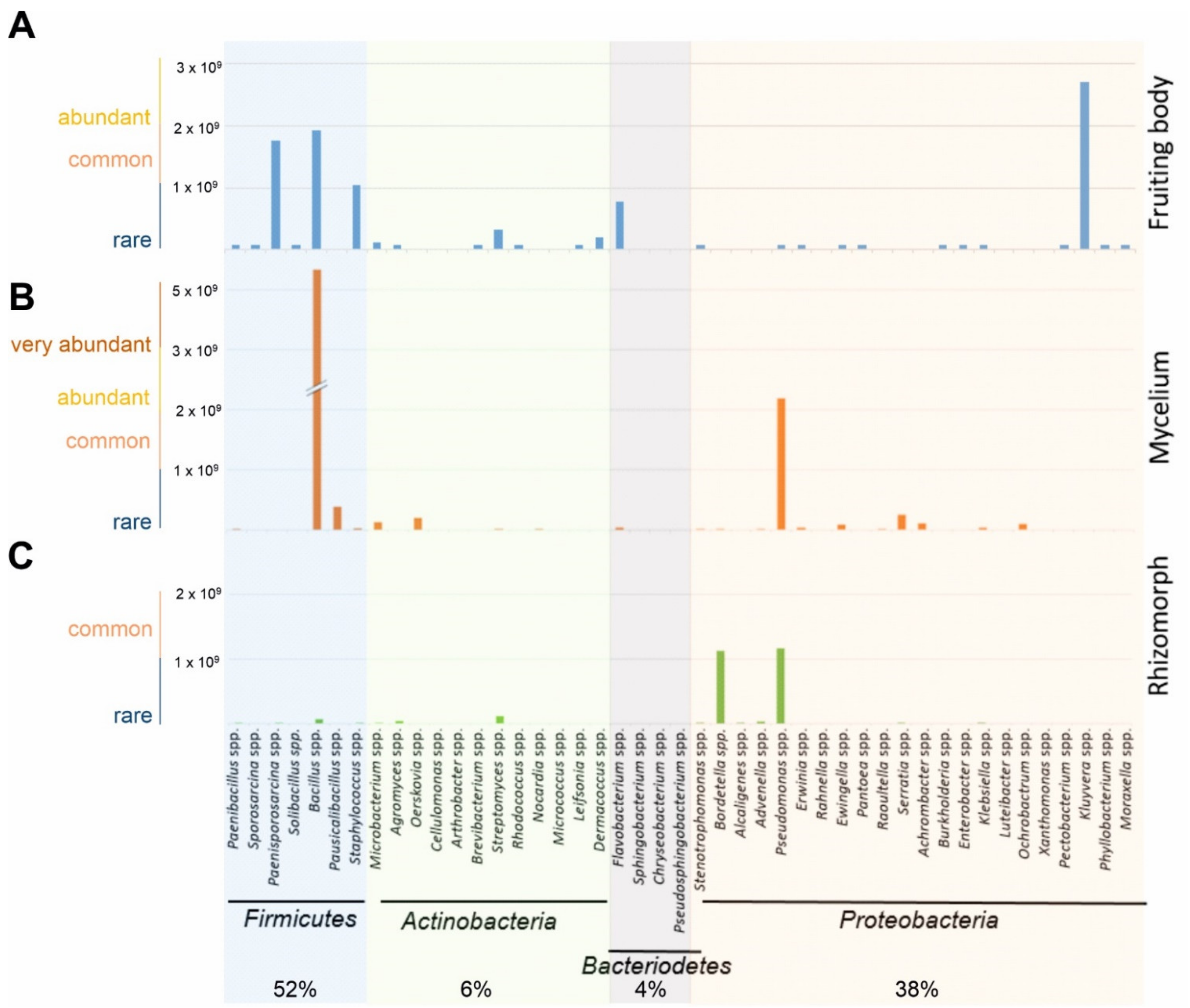
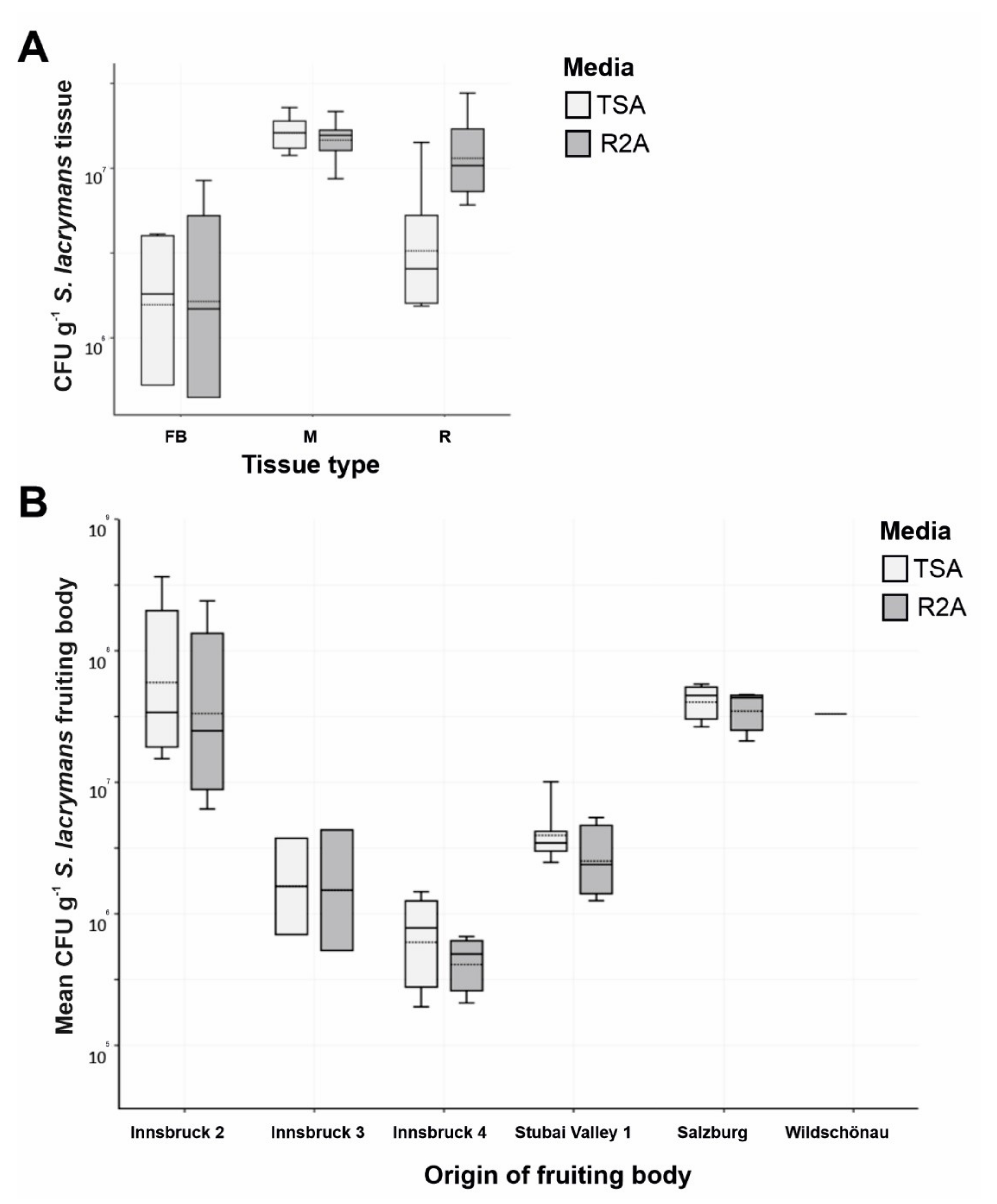
3.1. Microbial Diversity Associated with S. lacrymans Fruiting Bodies, Mycelia and Rhizomorphs
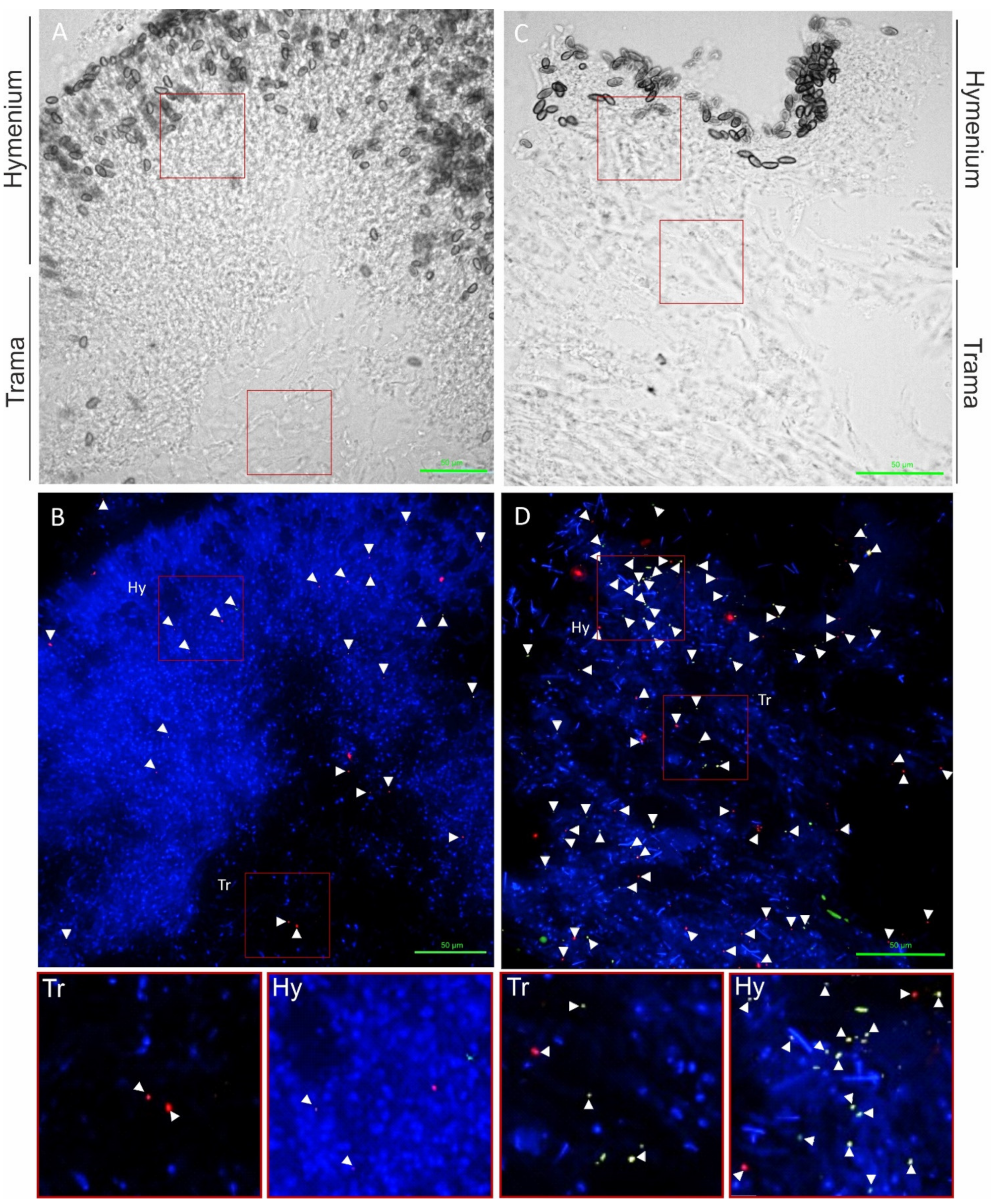
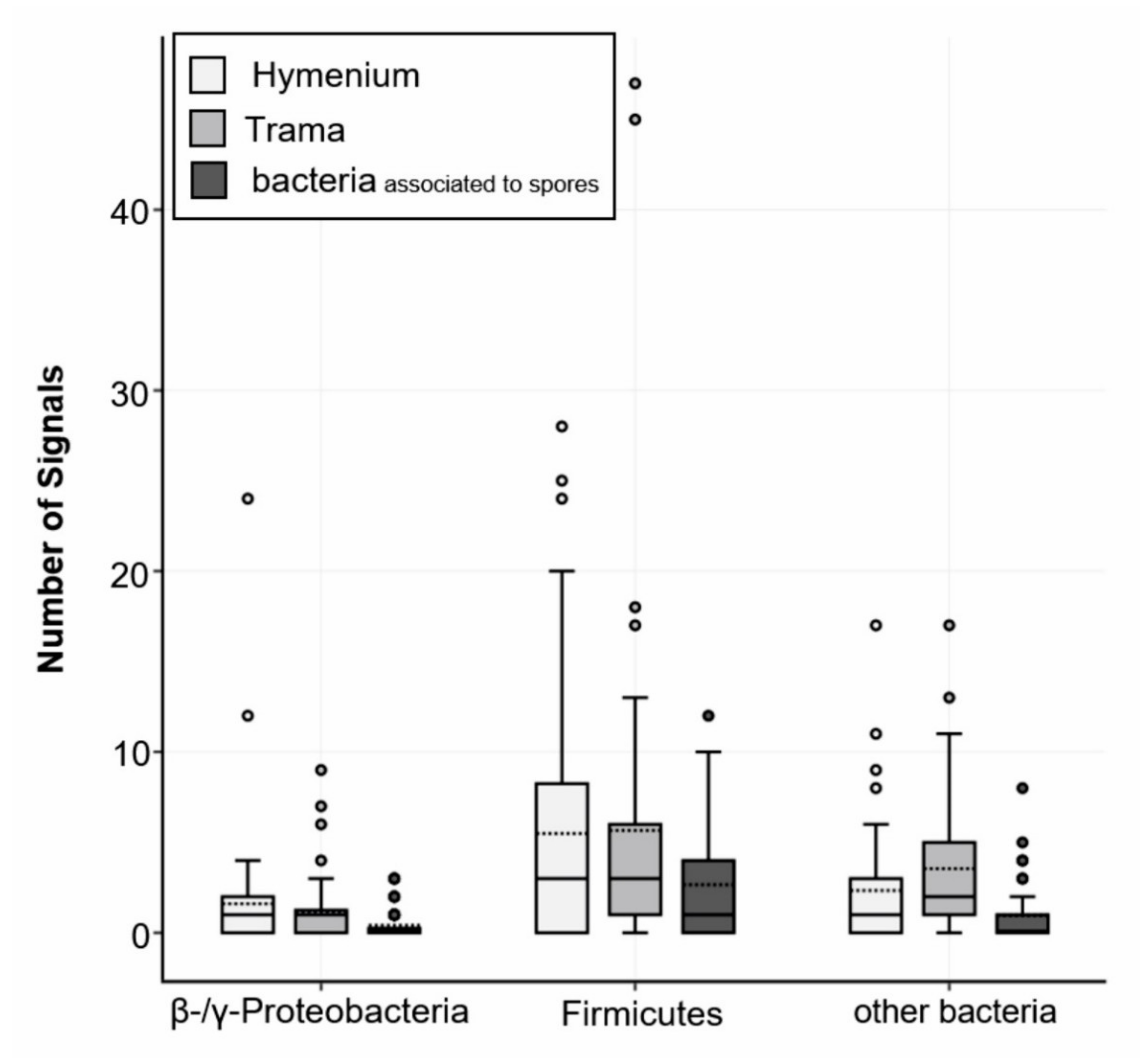
3.2. Detection and Localization of Bacterial Interaction Partners with Fluorescence In Situ Hybridization (FISH)
3.3. Enzymatic Activities of Isolated Bacteria
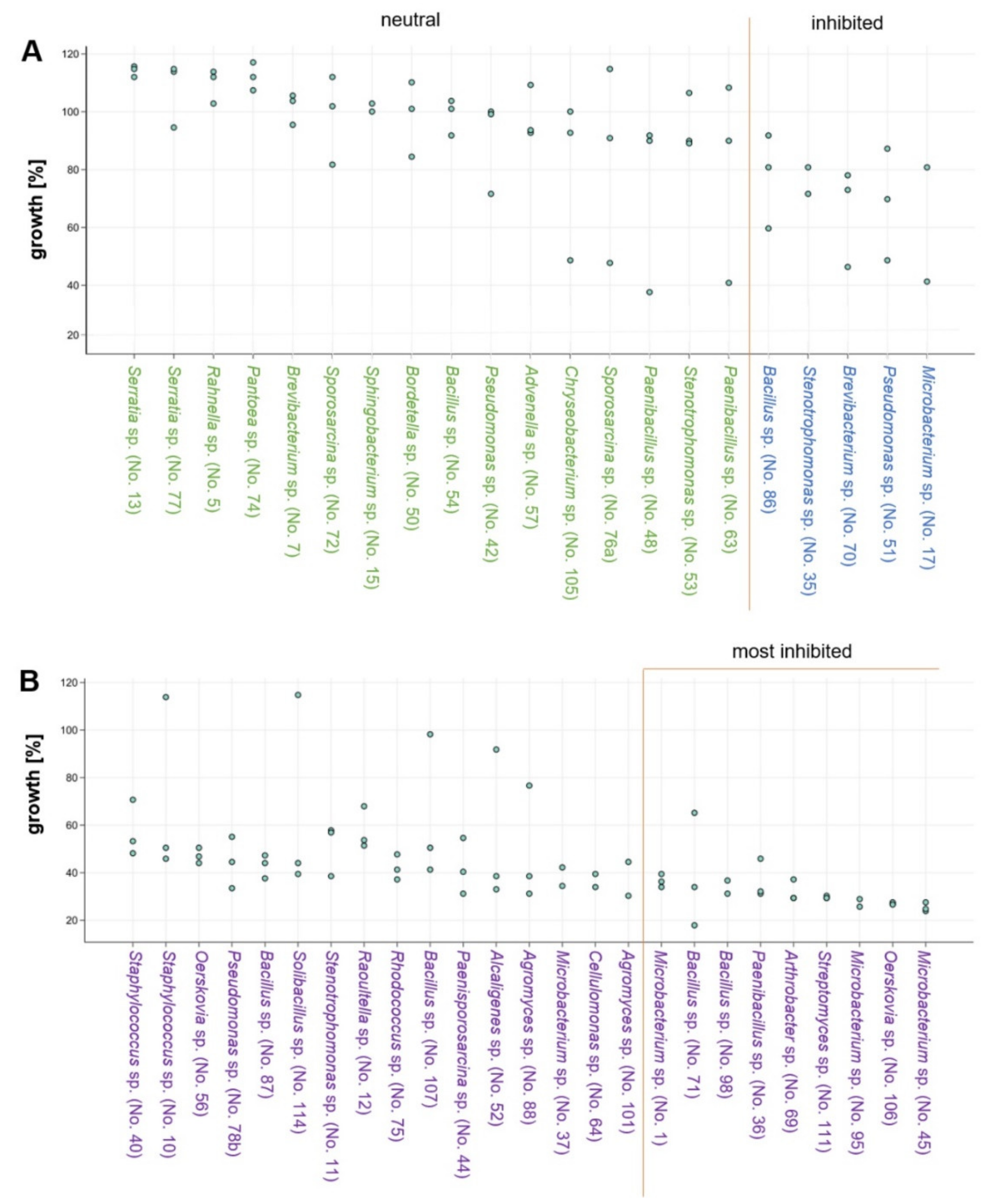
3.4. Effects of Co-Cultivation on Fungal and Bacterial Growth
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jennings, D.H. Serpula lacrymans: Fundamental Biology and Control Strategies; Bravery, A.F., Ed.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 1991. [Google Scholar]
- Boddy, L.; Frankland, J.C.; van West, P. Ecology of Saprotrophic Basidiomycetes; Academic Press: London, UK, 2007. [Google Scholar]
- Singh, J. Dry rot and other wood-destroying fungi: Their occurrence, biology, pathology and control. Indoor Built Environ. 1999, 8, 3–20. [Google Scholar] [CrossRef]
- Boutelje, J.B.; Bravery, A.F. Observations on the Bacterial Attack of Piles Supporting a Stockholm Building; Svenska Träforskningsinstitutet: Stockholm, Sweden, 1968. [Google Scholar]
- Clausen, C.A. Bacterial associations with decaying wood: A review. Int. Biodeterior. Biodegrad. 1996, 37, 101–107. [Google Scholar] [CrossRef]
- Boer, W.; Folman, L.B.; Summerbell, R.C.; Boddy, L. Living in a fungal world: Impact of fungi on soil bacterial niche development. FEMS Microbiol. Rev. 2005, 29, 795–811. [Google Scholar] [CrossRef]
- Rayner, A.D.M.; Boddy, L. Fungal Decomposition of Wood: Its Biology and Ecology; John Wiley & Sons Ltd.: Chichester, UK, 1988; 587p. [Google Scholar]
- Lynd, L.R.; Weimer, P.J.; van Zyl, W.H.; Pretorius, I.S. Microbial Cellulose Utilization: Fundamentals and Biotechnology. Microbiol. Mol. Biol. Rev. 2002, 66, 506. [Google Scholar] [CrossRef]
- Kumari, D.; Reddy, M.S.; Upadhyay, R.C. Diversity of cultivable bacteria associated with fruiting bodies of wild Himalayan Cantharellus spp. Ann. Microbiol. 2013, 63, 845–853. [Google Scholar] [CrossRef]
- Zhang, H.-B.; Yang, M.-X.; Tu, R. Unexpectedly high bacterial diversity in decaying wood of a conifer as revealed by a molecular method. Int. Biodeterior. Biodegrad. 2008, 62, 471–474. [Google Scholar] [CrossRef]
- Brown, M.E.; Chang, M.C. Exploring bacterial lignin degradation. Curr. Opin. Chem. Biol. 2014, 19, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Valášková, V.; de Boer, W.; Klein Gunnewiek, P.J.A.; Pospíšek, M.; Baldrian, P. Phylogenetic composition and properties of bacteria coexisting with the fungus Hypholoma fasciculare in decaying wood. ISME J. 2009, 3, 1218. [Google Scholar] [CrossRef] [PubMed]
- Noll, M.; Jirjis, R. Microbial communities in large-scale wood piles and their effects on wood quality and the environment. Appl. Microbiol. Biotechnol. 2012, 95, 551–563. [Google Scholar] [CrossRef]
- Pottier, D.; Andre, V.; Rioult, J.P.; Bourreau, A.; Duhamel, C.; Bouchart, V.K.; Richard, E.; Guibert, M.; Verite, P.; Garon, D. Airborne molds and mycotoxins in Serpula lacrymans-damaged homes. Atmos. Pollut. Res. 2014, 5, 325–334. [Google Scholar] [CrossRef]
- Beldman, G.; Searle-Van Leeuwen, M.F.; Rombouts, F.M.; Voragen, F.G.J. The cellulase of Trichoderma viride. Eur. J. Biochem. 1985, 146, 301–308. [Google Scholar] [CrossRef]
- Eastwood, D.C.; Floudas, D.; Binder, M.; Majcherczyk, A.; Schneider, P.; Aerts, A.; Asiegbu, F.O.; Baker, S.E.; Barry, K.; Bendiksby, M.; et al. The Plant Cell Wall-Decomposing Machinery Underlies the Functional Diversity of Forest Fungi. Science 2011, 333, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Arantes, V.; Goodell, B. Current Understanding of Brown-Rot Fungal Biodegradation Mechanisms: A Review. In Deterioration and Protection of Sustainable Biomaterials; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2014; Volume 1158, pp. 3–21. [Google Scholar]
- de Boer, W.; van der Wal, A. Interactions between Saprotrophic Basidiomycetes and Bacteria. In British Mycological Society Symposia Series; Academic Press: Cambridge, UK, 2008; Volume 28, pp. 143–153. [Google Scholar]
- Rinta-Kanto, J.M.; Sinkko, H.; Rajala, T.; Al-Soud, W.A.; Sørensen, S.J.; Tamminen, M.V.; Timonen, S. Natural decay process affects the abundance and community structure of Bacteria and Archaea in Picea abies logs. FEMS Microbiol. Ecol. 2016, 92. [Google Scholar] [CrossRef]
- Folman, L.; Gunnewiek, P.; Boddy, L.; de Boer, W. Impact of white-rot fungi on numbers and community composition of bacteria colonizing beech wood from forest soil. FEMS Microbiol. Ecol. 2008, 63, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Hervé, V.; Le Roux, X.; Uroz, S.; Gelhaye, E.; Frey-Klett, P. Diversity and structure of bacterial communities associated with Phanerochaete chrysosporium during wood decay. Environ. Microbiol. 2014, 16, 2238–2252. [Google Scholar] [CrossRef]
- Tornberg, K.; Bååth, E.; Olsson, S. Fungal growth and effects of different wood decomposing fungi on the indigenous bacterial community of polluted and unpolluted soils. Biol. Fertil. Soils 2003, 37, 190–197. [Google Scholar] [CrossRef]
- Warmink, J.A.; van Elsas, J.D. Selection of bacterial populations in the mycosphere of Laccaria proxima: Is type III secretion involved? ISME J. 2008, 2, 887–900. [Google Scholar] [CrossRef]
- Stopnisek, N.; Zühlke, D.; Carlier, A.; Barberán, A.; Fierer, N.; Becher, D.; Riedel, K.; Eberl, L.; Weisskopf, L. Molecular mechanisms underlying the close association between soil Burkholderia and fungi. ISME J. 2016, 10, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Greaves, H. The bacterial factor in wood decay. Wood Sci. Technol. 1971, 5, 6–16. [Google Scholar] [CrossRef]
- Weightman, A.J.; Boddy, L.; Johnston, S.R. Bacteria in decomposing wood and their interactions with wood-decay fungi. FEMS Microbiol. Ecol. 2016, 92. [Google Scholar] [CrossRef]
- Yurkov, A.; Krüger, D.; Begerow, D.; Arnold, N.; Tarkka, M.T. Basidiomycetous Yeasts from Boletales Fruiting Bodies and Their Interactions with the Mycoparasite Sepedonium chrysospermum and the Host Fungus Paxillus. Microb. Ecol. 2012, 63, 295–303. [Google Scholar] [CrossRef]
- Mieszkin, S.; Deveau, A.; Labbé, J.; Lastovetsky, O.A.; Millet, L.J.; Vajna, B.; Bonfante, P.; Krom, B.P.; Olsson, S.; van Elsas, J.D.; et al. Bacterial–fungal interactions: Ecology, mechanisms and challenges. FEMS Microbiol. Rev. 2018, 42, 335–352. [Google Scholar] [CrossRef]
- Cowling, E.; Merrill, W. Nitrogen in wood and its role in wood deterioration. Can. J. Bot. 2011, 44, 1539–1554. [Google Scholar] [CrossRef]
- Purahong, W.; Wubet, T.; Lentendu, G.; Schloter, M.; Pecyna, M.J.; Kapturska, D.; Hofrichter, M.; Krüger, D.; Buscot, F. Life in leaf litter: Novel insights into community dynamics of bacteria and fungi during litter decomposition. Mol. Ecol. 2016, 25, 4059–4074. [Google Scholar] [CrossRef]
- Bordenstein, S.R.; Theis, K.R. Host Biology in Light of the Microbiome: Ten Principles of Holobionts and Hologenomes. PLoS Biol. 2015, 13, e1002226. [Google Scholar] [CrossRef] [PubMed]
- Benucci, G.M.N.; Bonito, G.M. The Truffle Microbiome: Species and Geography Effects on Bacteria Associated with Fruiting Bodies of Hypogeous Pezizales. Microb. Ecol. 2016, 72, 4–8. [Google Scholar] [CrossRef]
- Cullings, K.; Stott, M.; Marinkovich, N.; DeSimone, J.; Bhardwaj, S. Phylum-level diversity of the microbiome of the extremophilic basidiomycete fungus Pisolithus arhizus (Scop.) Rauschert: An island of biodiversity in a thermal soil desert. MicrobiologyOpen 2020, 9, e1062. [Google Scholar] [CrossRef] [PubMed]
- Bahram, M.; Vanderpool, D.; Pent, M.; Hiltunen, M.; Ryberg, M. The genome and microbiome of a dikaryotic fungus (Inocybe terrigena, Inocybaceae) revealed by metagenomics. Environ. Microbiol. Rep. 2018, 10, 155–166. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, Q.; Li, J.; Lian, B. Bacterial diversity among the fruit bodies of ectomycorrhizal and saprophytic fungi and their corresponding hyphosphere soils. Sci. Rep. 2018, 8, 11672. [Google Scholar] [CrossRef] [PubMed]
- Tarkka, M.T.; Drigo, B.; Deveau, A. Mycorrhizal microbiomes. Mycorrhiza 2018, 28, 403–409. [Google Scholar] [CrossRef]
- Frey-Klett, P.; Burlinson, P.; Deveau, A.; Barret, M.; Tarkka, M.; Sarniguet, A. Bacterial-fungal interactions: Hyphens between agricultural, clinical, environmental, and food microbiologists. Microbiol. Mol. Biol. Rev. MMBR 2011, 75, 583–609. [Google Scholar] [CrossRef]
- Minerdi, D.; Moretti, M.; Gilardi, G.; Barberio, C.; Gullino, M.L.; Garibaldi, A. Bacterial ectosymbionts and virulence silencing in a Fusarium oxysporum strain. Environ. Microbiol. 2008, 10, 1725–1741. [Google Scholar] [CrossRef] [PubMed]
- Splivallo, R.; Deveau, A.; Valdez, N.; Kirchhoff, N.; Frey-Klett, P.; Karlovsky, P. Bacteria associated with truffle-fruiting bodies contribute to truffle aroma. Environ. Microbiol. 2015, 17, 2647–2660. [Google Scholar] [CrossRef] [PubMed]
- Vahdatzadeh, M.; Deveau, A.; Splivallo, R. The Role of the Microbiome of Truffles in Aroma Formation: A Meta-Analysis Approach. Appl. Environ. Microbiol. 2015, 81, 6946. [Google Scholar] [CrossRef] [PubMed]
- Benoit, I.; van den Esker, M.H.; Patyshakuliyeva, A.; Mattern, D.J.; Blei, F.; Zhou, M.; Dijksterhuis, J.; Brakhage, A.A.; Kuipers, O.P.; de Vries, R.P.; et al. Bacillus subtilis attachment to Aspergillus niger hyphae results in mutually altered metabolism. Environ. Microbiol. 2015, 17, 2099–2113. [Google Scholar] [CrossRef]
- Schulz-Bohm, K.; Tyc, O.; de Boer, W.; Peereboom, N.; Debets, F.; Zaagman, N.; Janssens, T.K.S.; Garbeva, P. Fungus-associated bacteriome in charge of their host behavior. Fungal Genet. Biol. 2017, 102, 38–48. [Google Scholar] [CrossRef]
- Tauber, J.P.; Matthäus, C.; Lenz, C.; Hoffmeister, D.; Popp, J. Analysis of basidiomycete pigments in situ by Raman spectroscopy. J. Biophotonics 2018, 11, e201700369. [Google Scholar] [CrossRef]
- Tauber, J.P.; Gallegos-Monterrosa, R.; Kovacs, A.T.; Shelest, E.; Hoffmeister, D. Dissimilar pigment regulation in Serpula lacrymans and Paxillus involutus during inter-kingdom interactions. Microbiology 2018, 164, 65–77. [Google Scholar] [CrossRef]
- Tauber, J.P.; Schroeckh, V.; Shelest, E.; Brakhage, A.A.; Hoffmeister, D. Bacteria induce pigment formation in the basidiomycete Serpula lacrymans. Environ. Microbiol. 2016, 18, 5218–5227. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Leschine, S. Degradation of polymers: Cellulose, xylan, pectin, starch. In Handbook on Clostridia; Durre, P., Ed.; Taylor & Francis Group: Boca Raton, FL, USA, 2005; pp. 101–132. [Google Scholar]
- Apun, K.; Jong, B.C.; Salleh, M.A. Screening and isolation of a cellulolytic and amylolytic Bacillus from sago pith waste. J. Gen. Appl. Microbiol. 2000, 46, 263–267. [Google Scholar] [CrossRef][Green Version]
- Bibi, N.; Ali, S.; Tabassum, R. Isolation and Identification of Novel IndigenousBacterial Strain as a Low Cost Pectinase Source. Braz. Arch. Biol. Technol. 2018, 61. [Google Scholar] [CrossRef]
- Yin, L.-J.; Huang, P.-S.; Lin, H.-H. Isolation of Cellulase-Producing Bacteria and Characterization of the Cellulase from the Isolated Bacterium Cellulomonas sp. YJ5. J. Agric. Food Chem. 2010, 58, 9833–9837. [Google Scholar] [CrossRef] [PubMed]
- Subajini, M.; Sandrasegarampillai, B.; Vasanthy, A. Screening and identification of a thermophilic and alkalophilic bacterium producing xylanase. Adv. Appl. Sci. Res. 2012, 3, 242–250. [Google Scholar]
- Sridevi, B.; Charya, M.A.S. Isolation, identification and screening of potential cellulase-free xylanase producing fungi. Afr. J. Biotechnol. 2013, 10, 4624–4630. [Google Scholar]
- Adesina, F.; Onilude, A. Isolation, identification and screening of xylanase and glucanase-producing microfungi from degrading wood in Nigeria. Afr. J. Agric. Res. 2013, 8, 4414–4421. [Google Scholar] [CrossRef]
- Freitag, M.; Morrell, J.J. Wood sandwich tests of potential biological control agents for basidiomycetous decay fungi. Mater. Org. 1990, 25, 63–70. [Google Scholar]
- Srinivasan, A.B.; Staines, H.J. Effect of Media Composition on the Antagonistic Properties of Trichoderma spp. against Wood Decay Fungi; International Research Group on Wood Preservation Documetn No. IRG/WP/1538-92; IRG Secretariat: Stockholm, Sweden, 1992. [Google Scholar]
- Remus-Emsermann, M.N.P.; Lücker, S.; Müller, D.B.; Potthoff, E.; Daims, H.; Vorholt, J.A. Spatial distribution analyses of natural phyllosphere-colonizing bacteria on Arabidopsis thaliana revealed by fluorescence in situ hybridization. Environ. Microbiol. 2014, 16, 2329–2340. [Google Scholar] [CrossRef]
- Amann, R.I.; Binder, B.J.; Olson, R.J.; Chisholm, S.W.; Devereux, R.; Stahl, D.A. Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl. Environ. Microbiol. 1990, 56, 1919–1925. [Google Scholar] [CrossRef]
- Daims, H.; Brühl, A.; Amann, R.; Schleifer, K.-H.; Wagner, M. The Domain-specific Probe EUB338 is Insufficient for the Detection of all Bacteria: Development and Evaluation of a more Comprehensive Probe Set. Syst. Appl. Microbiol. 1999, 22, 434–444. [Google Scholar] [CrossRef]
- Neef, A. Anwendung der In Situ Einzelzell-Identifizierung von Bakterien zur Populationsanalyse in komplexen mikrobiellen Biozönosen. Ph.D. Thesis, Technische Universität, Munich, Germany, 1997. [Google Scholar]
- Manz, W.; Amann, R.; Ludwig, W.; Wagner, M.; Schleifer, K.-H. Phylogenetic Oligodeoxynucleotide Probes for the Major Subclasses of Proteobacteria: Problems and Solutions. Syst. Appl. Microbiol. 1992, 15, 593–600. [Google Scholar] [CrossRef]
- Meier, H.; Amann, R.; Ludwig, W.; Schleifer, K.H. Specific Oligonucleotide Probes for in situ Detection of a Major Group of Gram-positive Bacteria with low DNA G+C Content. Syst. Appl. Microbiol. 1999, 22, 186–196. [Google Scholar] [CrossRef]
- Roller, C.; Wagner, M.; Amann, R.; Ludwig, W.; Schleifer, K.-H. In situ probing of Gram-positive bacteria with high DNA G + C content using 23S rRNA-targeted oligonucleotides. Microbiology 1994, 140, 2849–2858. [Google Scholar] [CrossRef]
- Simek, K.; Pernthaler, J.; Weinbauer, M.G.; Hornák, K.; Dolan, J.R.; Nedoma, J.; Masín, M.; Amann, R. Changes in bacterial community composition and dynamics and viral mortality rates associated with enhanced flagellate grazing in a mesoeutrophic reservoir. Appl. Environ. Microbiol. 2001, 67, 2723–2733. [Google Scholar] [CrossRef] [PubMed]
- Gunasekera, T.S.; Dorsch, M.R.; Slade, M.B.; Veal, D.A. Specific detection of Pseudomonas spp. in milk by fluorescence in situ hybridization using ribosomal RNA directed probes. J. Appl. Microbiol. 2003, 94, 936–945. [Google Scholar] [CrossRef] [PubMed]
- Wallner, G.; Amann, R.; Beisker, W. Optimizing fluorescent in situ hybridization with rRNA-targeted oligonucleotide probes for flow cytometric identification of microorganisms. Cytometry 1993, 14, 136–143. [Google Scholar] [CrossRef]
- Cardinale, M.; Vieira de Castro, J., Jr.; Müller, H.; Berg, G.; Grube, M. In situ analysis of the bacterial community associated with the reindeer lichen Cladonia arbuscula reveals predominance of Alphaproteobacteria. FEMS Microbiol. Ecol. 2008, 66, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Terhonen, E.; Kasanen, R.; Asiegbu, F.O. Diversity and Community Structure of Primary Wood-Inhabiting Bacteria in Boreal Forest. Geomicrobiol. J. 2014, 31, 315–324. [Google Scholar] [CrossRef]
- Rintala, H.; Nevalainen, A.; Suutari, M. Diversity of streptomycetes in water-damaged building materials based on 16S rDNA sequences. Lett. Appl. Microbiol. 2002, 34, 439–443. [Google Scholar] [CrossRef]
- Suihko, M.L.; Priha, O.; Alakomi, H.L.; Thompson, P.; Mälarstig, B.; Stott, R.; Richardson, M. Detection and molecular characterization of filamentous actinobacteria and thermoactinomycetes present in water-damaged building materials. Indoor Air 2009, 19, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Amann, R.I.; Ludwig, W.; Schleifer, K.H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 1995, 59, 143–169. [Google Scholar] [CrossRef] [PubMed]
- Gohar, D.; Pent, M.; Põldmaa, K.; Bahram, M. Bacterial community dynamics across developmental stages of fungal fruiting bodies. FEMS Microbiol. Ecol. 2020, 96. [Google Scholar] [CrossRef]
- Sato, Y.; Narisawa, K.; Tsuruta, K.; Umezu, M.; Nishizawa, T.; Tanaka, K.; Yamaguchi, K.; Komatsuzaki, M.; Ohta, H. Detection of betaproteobacteria inside the mycelium of the fungus Mortierella elongata. Microbes Environ. 2010, 25, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Grosu, R.; Boiciuc, M.; Vladut, R. The influence of bacteria on the quality of wood and wood products. Ind. Lemn. 1973, 24, 143–147. [Google Scholar]
- Baum, S.P.; Pöhler, E.; Seubert Hunziker, H.; Weber, P.; Kupferschmid Albisetti, A.D. Holzkunde II—Teil 2 Holzchemie. Available online: https://doi.org/10.3929/ethz-a-004536640 (accessed on 29 January 2021).
- Pettersen, R.C. The Chemical Composition of Wood. In The Chemistry of Solid Wood; Advances in Chemistry; American Chemical Society: Washington, DC, USA, 1984; Volume 207, pp. 57–126. [Google Scholar]
- Fogarty, W.M.; Ward, O.P. Growth and enzyme production by Bacillus subtilis and Flavobacterium pectinovorum in Picea sitchensis. Wood Sci. Technol. 1973, 7, 261–270. [Google Scholar] [CrossRef]
- Cragg, S.M.; Beckham, G.T.; Bruce, N.C.; Bugg, T.D.H.; Distel, D.L.; Dupree, P.; Etxabe, A.G.; Goodell, B.S.; Jellison, J.; McGeehan, J.E.; et al. Lignocellulose degradation mechanisms across the Tree of Life. Curr. Opin. Chem. Biol. 2015, 29, 108–119. [Google Scholar] [CrossRef]
- Thayer, D.W. Facultative Wood-digesting Bacteria from the Hind-gut of the Termite Reticulitermes hesperus. Microbiology 1976, 95, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Knuth, D.T.; Mccoy, E. Bacterial deterioration of pine logs in pond storage. For. Prod. J. 1960, 12, 437–442. [Google Scholar]
- Kamath, R.; Schnoor, J.L.; Alvarez, P.J.J. Effect of Root-Derived Substrates on the Expression of nah-lux Genes in Pseudomonas fluorescens HK44: Implications for PAH Biodegradation in the Rhizosphere. Environ. Sci. Technol. 2004, 38, 1740–1745. [Google Scholar] [CrossRef]
- Leveau, J.H.J.; Uroz, S.; De Boer, W. The bacterial genus Collimonas: Mycophagy, weathering and other adaptive solutions to life in oligotrophic soil environments. Environ. Microbiol. 2010, 12, 281–292. [Google Scholar] [CrossRef]
- King, B.; Waite, J. Translocation of nitrogen to wood by fungi. Int. Biodeterior. Bull. 1979, 15, 29–35. [Google Scholar]
- Schroeckh, V.; Scherlach, K.; Nützmann, H.-W.; Shelest, E.; Schmidt-Heck, W.; Schuemann, J.; Martin, K.; Hertweck, C.; Brakhage, A.A. Intimate bacterial-fungal interaction triggers biosynthesis of archetypal polyketides in Aspergillus nidulans. Proc. Natl. Acad. Sci. USA 2009, 106, 14558–14563. [Google Scholar] [CrossRef] [PubMed]
- Oranusi, N.A.; Trinci, A.P. Growth of bacteria on chitin, fungal cell walls and fungal biomass, and the effect of extracellular enzymes produced by these cultures on the antifungal activity of amphotericin B. Microbios 1985, 43, 17–30. [Google Scholar] [PubMed]
- Korn-Wendisch, F.; Kutzner, H.J. The Family Streptomycetaceae; Springer: New York, NY, USA, 1992; pp. 921–995. [Google Scholar]
- Jag, V.; Poehlein, A.; Bengelsdorf, F.; Daniel, R.; Dürre, P. Genome sequencing and description of Oerskovia enterophila VJag, an agar- and cellulose-degrading bacterium. Stand. Genom. Sci. 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Liu, P.; Liao, W.; Miao, L. Complete Genome of the Chitin-Degrading Bacterium, Paenibacillus xylanilyticus W4. Genome Biol. Evol. 2019, 11, 3252–3255. [Google Scholar] [CrossRef]
- Someya, N.; Ikeda, S.; Morohoshi, T.; Noguchi Tsujimoto, M.; Yoshida, T.; Sawada, H.; Ikeda, T.; Tsuchiya, K. Diversity of Culturable Chitinolytic Bacteria from Rhizospheres of Agronomic Plants in Japan. Microbes Environ. 2009, 1011040239. [Google Scholar] [CrossRef] [PubMed]
- Freitas, M.A.; Medeiros, F.H.V.; Melo, I.S.; Pereira, P.F.; Peñaflor, M.F.G.V.; Bento, J.M.S.; Paré, P.W. Stem inoculation with bacterial strains Bacillus amyloliquefaciens (GB03) and Microbacterium imperiale (MAIIF2a) mitigates Fusarium root rot in cassava. Phytoparasitica 2019, 47, 135–142. [Google Scholar] [CrossRef]
- Balasundaram, S.V.; Hess, J.; Durling, M.B.; Moody, S.C.; Thorbek, L.; Progida, C.; LaButti, K.; Aerts, A.; Barry, K.; Grigoriev, I.V.; et al. The fungus that came in from the cold: Dry rot’s pre-adapted ability to invade buildings. ISME J. 2018, 12, 791–801. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Origin | Fruiting Body | Mycelia | Rhizomorph | Sampling Date | Bacterial Isolation | CFU Assay | FISH |
|---|---|---|---|---|---|---|---|---|
| 1 | Außerfern | 2 | 1 | 1 | 13 December 2018 | X | ||
| 2 | Brixen valley 1 | 0 | 2 | 0 | 19 October 2018 | X | ||
| 3 | Brixen valley 2 | 0 | 1 | 0 | 2 May 2019 | X | ||
| 4 | Brixen valley 2 | 1 | 1 | 0 | 19 June 2019 | X | ||
| 5 | Buchkirchen | 1 | 0 | 1 | 14 June 2019 | X | ||
| 6 | Grieskirchen | 1 | 1 | 1 | 13 June 2019 | X | ||
| 7 | Innsbruck 1 | 2 | 3 | 2 | 7 November 2018 | X | X | X |
| 8 | Innsbruck 2 | 2 | 0 | 0 | 22 March 2019 | X | X | |
| 9 | Innsbruck 2 | 1 | 0 | 0 | 2 July 2019 | X | X | |
| 10 | Innsbruck 2 | 1 | 1 | 1 | 16 August 2019 | X | X | |
| 11 | Innsbruck 2 | 1 | 0 | 1 | 6 September 2019 | X | X | |
| 12 | Innsbruck 3 | 1 | 1 | 1 | 30 October 2019 | X | X | |
| 13 | Innsbruck 4 | 1 | 0 | 0 | 18 November 2019 | X | X | X |
| 14 | Pitz valley | 1 | 1 | 1 | 16 May 2019 | X | X | |
| 15 | Salzburg | 1 | 0 | 0 | 31 October 2019 | X | X | X |
| 16 | Stubai valley 1 | 1 | 0 | 0 | 14 October 2019 | X | ||
| 17 | Stubai valley 2 | 0 | 1 | 1 | 28 November 2019 | X | X | |
| 18 | Wildschönau | 1 | 0 | 0 | 27 June 2018 | X | ||
| 19 | Traunkirchen | 0 | 0 | 1 | 26 June 2020 | X | ||
| 20 | Ziller valley | 0 | 1 | 0 | 2 July 2020 | X |
| Isolate No. | Taxonomy | Isolate No. | Taxonomy |
|---|---|---|---|
| 1 | Microbacterium sp. | 41 | Flavobacterium sp. |
| 17 | Microbacterium sp. | 46 | Flavobacterium sp. |
| 37 | Microbacterium sp. | 50 | Bordetella sp. |
| 45 | Microbacterium sp. | 52 | Alcaligenes sp. |
| 60 | Microbacterium sp. | 54 | Bacillus sp. |
| 95 | Microbacterium sp. | 71 | Bacillus sp. |
| 99 | Microbacterium sp. | 86 | Bacillus sp. |
| 5 | Rahnella sp. | 87 | Bacillus sp. |
| 7 | Brevibacterium sp. | 92 | Bacillus sp. |
| 70 | Brevibacterium sp. | 98 | Bacillus sp. |
| 10 | Staphylococcus sp. | 107 | Bacillus sp. |
| 40 | Staphylococcus sp. | 56 | Oerskovia sp. |
| 11 | Stenotrophomonas sp. | 106 | Oerskovia sp. |
| 35 | Stenotrophomonas sp. | 57 | Advenella sp. |
| 53 | Stenotrophomonas sp. | 64 | Cellulomonas sp. |
| 12 | Raoultella sp. | 69 | Arthrobacter sp. |
| 13 | Serratia sp. | 72 | Sporosarcina sp. |
| 77 | Serratia sp. | 74 | Pantoea sp. |
| 15 | Sphingobacterium sp. | 75 | Rhodococcus sp. |
| 36 | Paenibacillus sp. | 76a) | Sporosarcina sp. |
| 48 | Paenibacillus sp. | 81 | Erwinia sp. |
| 63 | Paenibacillus sp. | 88 | Agromyces sp. |
| 42 | Pseudomonas sp. | 101 | Agromyces sp. |
| 51 | Pseudomonas sp. | 105 | Chryseobacterium sp. |
| 78b) | Pseudomonas sp. | 111 | Streptomyces sp. |
| 44 | Paenisporosarcina sp. | 114 | Solibacillus sp. |
| Name | Sequence (5′-3′) | Target | Formamide Conc. [%] a | Fluorophore | NaCl Con. Washing Buffer [mM] | Reference |
|---|---|---|---|---|---|---|
| EUB338w b | gctgcctcccgtaggagt | Most bacteria | 10 | Cy3/Cy5 | 450 | [57] |
| EUB338IIw b | gcagccacccgtaggtgt | Planctomycetales | 10 | Cy3/Cy5 | 450 | [58] |
| EUB338IIIw b | gctgccacccgtaggtgt | Verrucomicrobiales | 10 | Cy3/Cy5 | 450 | [58] |
| ALF968 b | ggtaaggttctgcgcgtt | Alphaproteobacteria, except Rickettsiales | 40 | Cy3 | 56 | [59] |
| BET42aw b | gccttcccacttcgttt | Betaproteobacteria | 40 | 6FAM | 56 | [60] |
| GAM42aw b | gccttcccacatcgttt | Gammaproteobacteria | 40 | 6FAM | 56 | [60] |
| LGC354Aw b | tggaagattccctactgc | Firmicutes (low G + C Gram-positive bacteria) | 35 | Cy5 | 80 | [61] |
| LGC354Bw b | cggaagattccctactgc | Firmicutes (low G + C Gram-positive bacteria) | 35 | Cy5 | 80 | [61] |
| LGC354Cw b | ccgaagattccctactgc | Firmicutes (low G + C Gram-positive bacteria) | 35 | Cy5 | 80 | [61] |
| HGC69A | tatagttaccaccgccgt | Actinobacteria (high G + C Gram-positive bacteria) | 25 | Cy5 | 160 | [62] |
| R-FL615 | cactgcaatcgttgagcga | Bacteroidetes | 35 | Cy5 | 80 | [63] |
| PSE1284 | gatccggactacgatcggttt | Pseudomonadales | 30 | Cy5 | 220 | [64] |
| NONEUB | actcctacgggaggcagc | c | Cy3 | c | [65] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Embacher, J.; Neuhauser, S.; Zeilinger, S.; Kirchmair, M. Microbiota Associated with Different Developmental Stages of the Dry Rot Fungus Serpula lacrymans. J. Fungi 2021, 7, 354. https://doi.org/10.3390/jof7050354
Embacher J, Neuhauser S, Zeilinger S, Kirchmair M. Microbiota Associated with Different Developmental Stages of the Dry Rot Fungus Serpula lacrymans. Journal of Fungi. 2021; 7(5):354. https://doi.org/10.3390/jof7050354
Chicago/Turabian StyleEmbacher, Julia, Sigrid Neuhauser, Susanne Zeilinger, and Martin Kirchmair. 2021. "Microbiota Associated with Different Developmental Stages of the Dry Rot Fungus Serpula lacrymans" Journal of Fungi 7, no. 5: 354. https://doi.org/10.3390/jof7050354
APA StyleEmbacher, J., Neuhauser, S., Zeilinger, S., & Kirchmair, M. (2021). Microbiota Associated with Different Developmental Stages of the Dry Rot Fungus Serpula lacrymans. Journal of Fungi, 7(5), 354. https://doi.org/10.3390/jof7050354