Targeted Delivery of Gene Silencing in Fungi Using Genetically Engineered Bacteria

 , , and
, , and 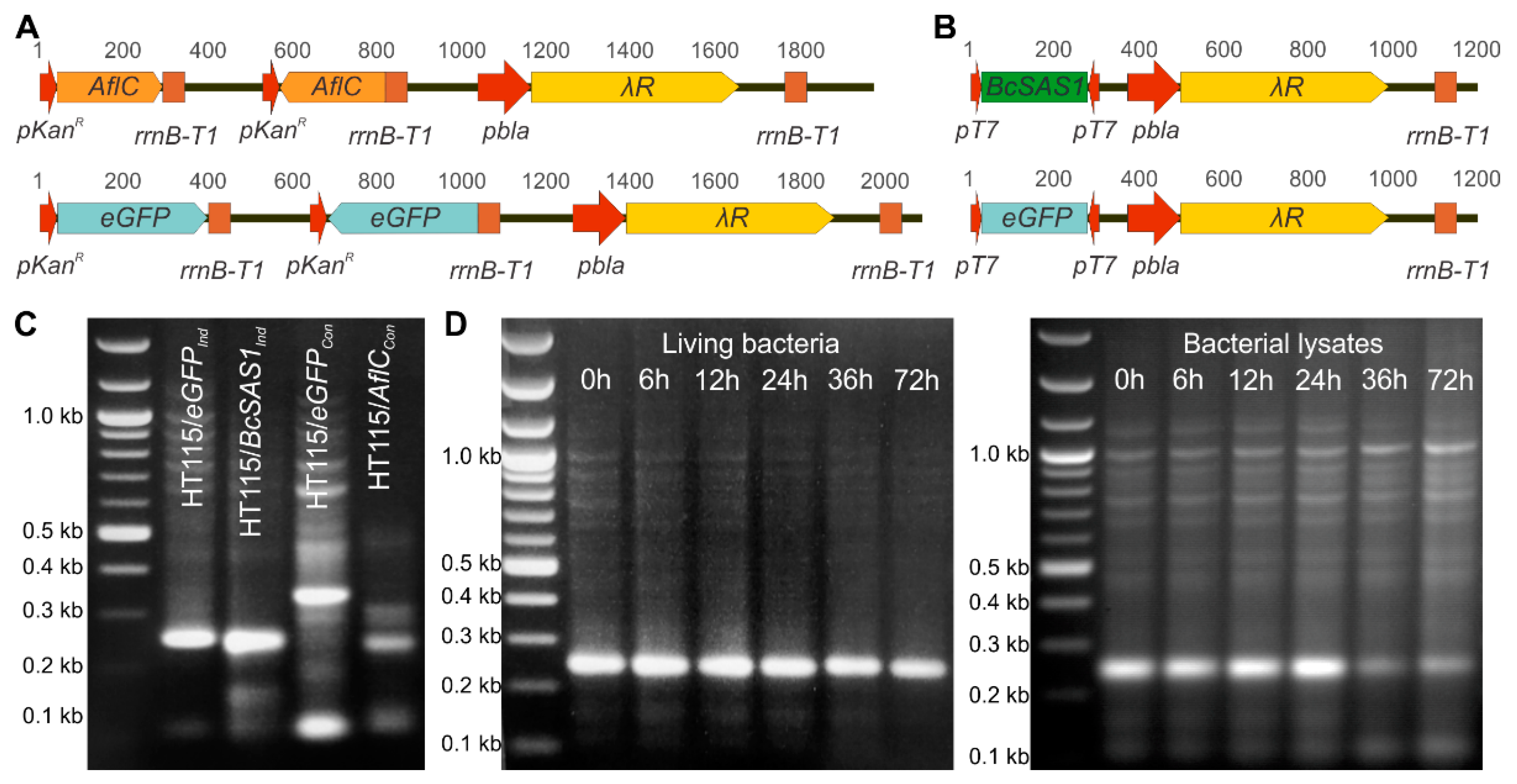{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmid Construction
2.2. Bacterial and Fungal Strains Used in This Study, Their Growth Conditions, and In Vitro Bioassays
2.3. Total RNA Extractions and RT-qPCR
2.4. dsRNA Isolation from E. coli HT115(DE3)
2.5. Bioassays
2.6. Analysis of Total Aflatoxins by ELISA
2.7. Statistical Analyses
3. Results
3.1. Bacteria Can Be Genetically Engineered to Produce dsRNAs against Target Genes in Fungi
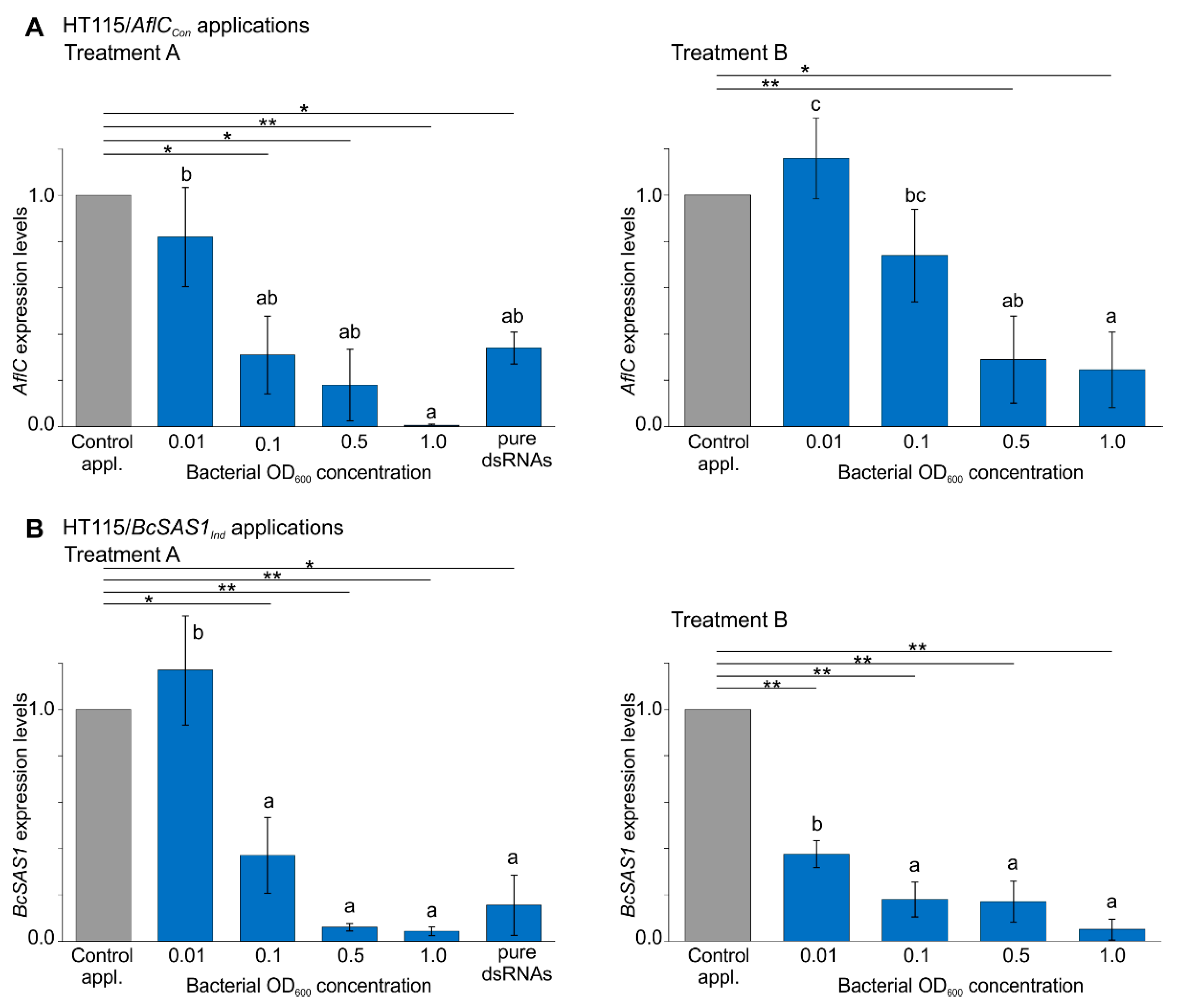
3.2. Bacterially-Produced dsRNAs Induce RNAi in Fungi in a Bacteria Concentration-Dependent Manner
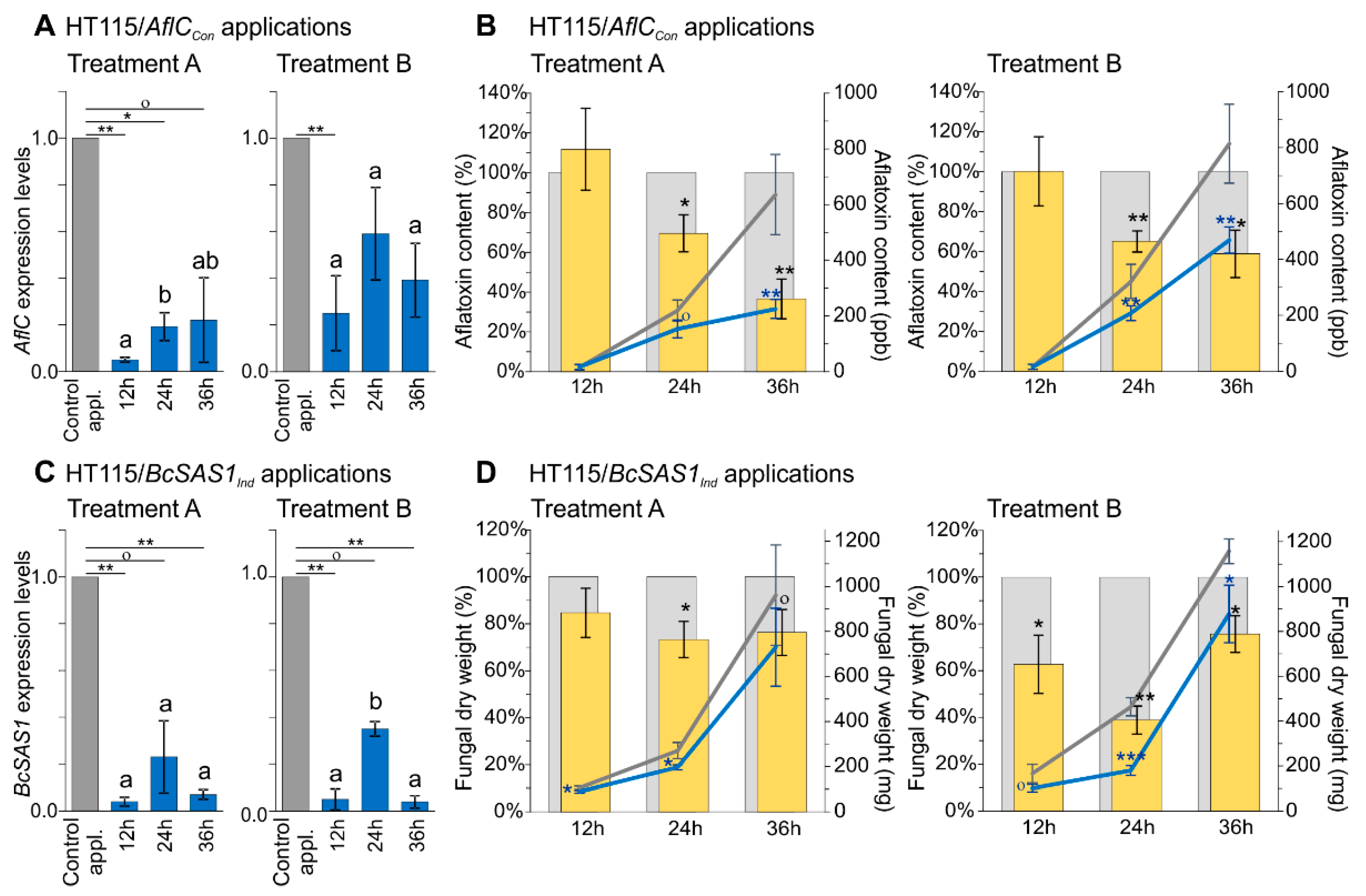
3.3. Increased Contact Times between Fungi and dsRNA-Producing Bacteria Have a Positive Effect on Reducing Production of Aflatoxins in A. flavus and Mycelial Growth in B. cinerea
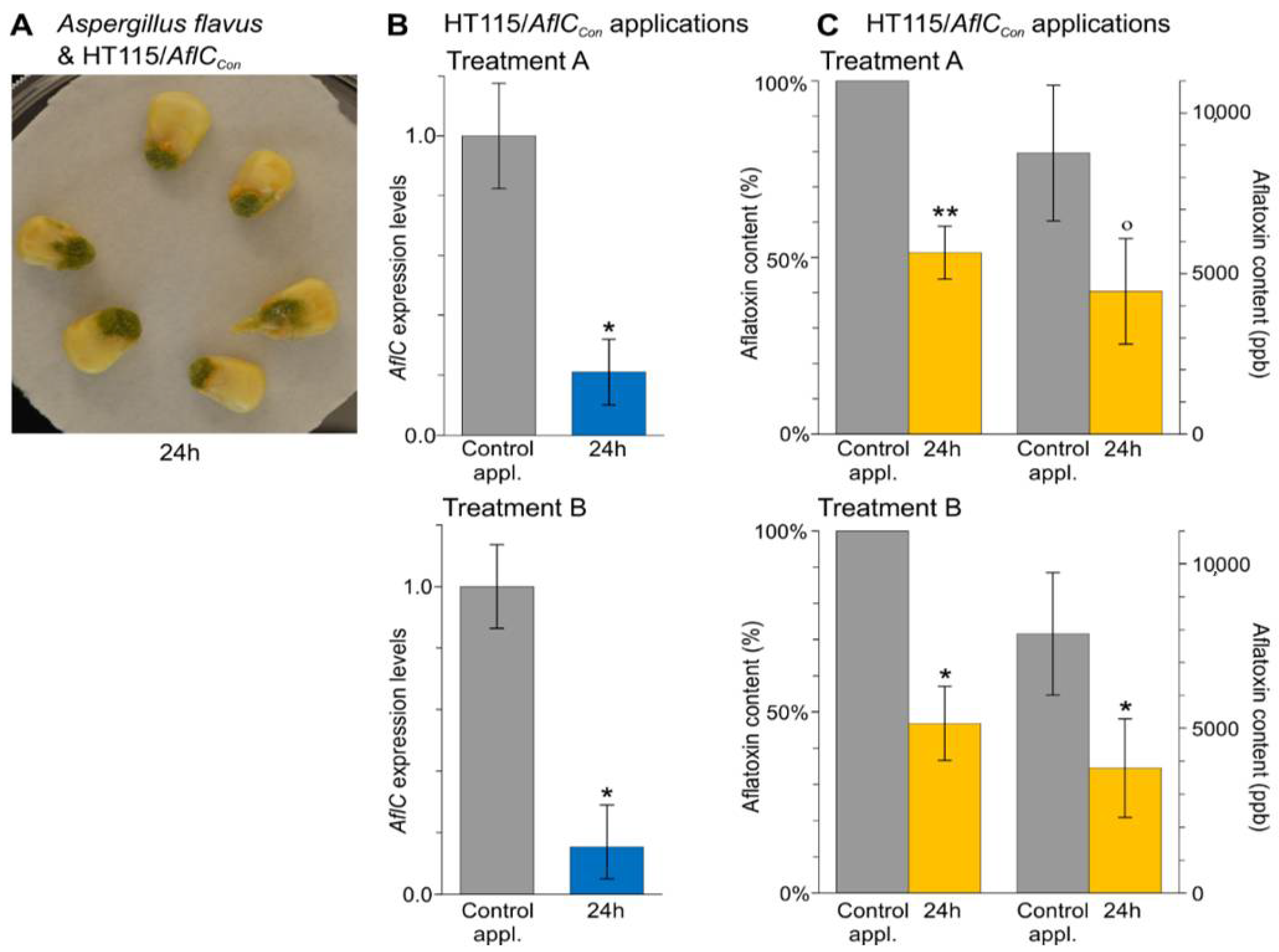
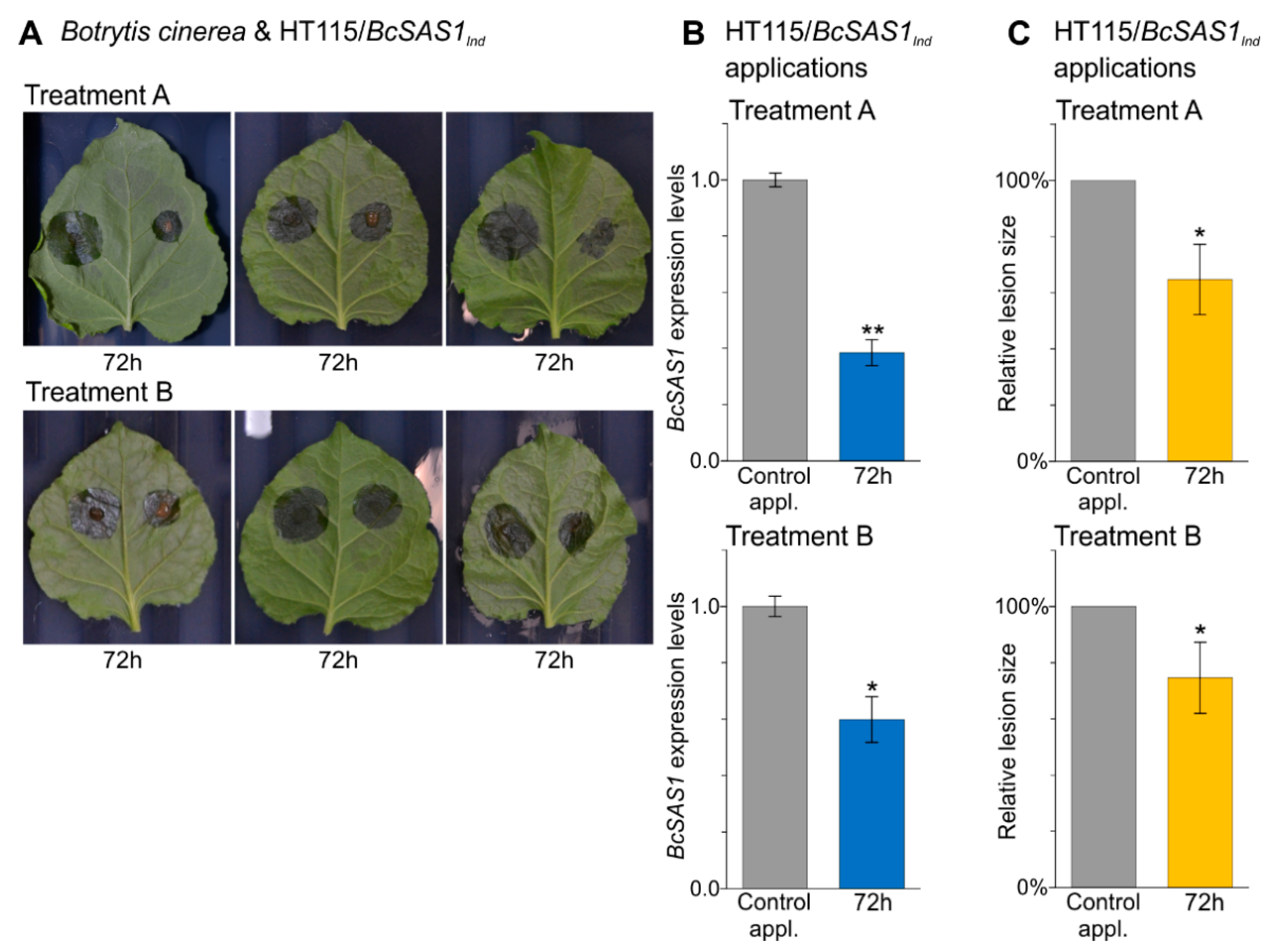
3.4. In Vivo Applications of dsRNA-Producing Bacteria Result in a Reduction of Aflatoxins Production by A. flavus and Disease Symptoms Caused by B. cinerea
4. Discussion
5. Conclusions
- The E. coli HT115(DE3) strain can produce biologically active dsRNAs against target genes in fungi.
- The biologically active bacterial dsRNAs were successfully delivered to fungi through two different approaches: (i) treatment with living bacteria and (ii) treatment with bacterial lysates following a programmable cell autolysis step.
- In vitro applications of dsRNA-producing bacteria show that the degree of the RNAi effect on fungi is positively correlated with the concentration of the bacteria, while the phenotypic changes (i.e., reduction in production of aflatoxins in A. flavus and reduction of mycelial growth in B. cinerea) are positively correlated with the contact time.
- In vivo applications of dsRNA-producing bacteria result in a reduction of aflatoxins production by A. flavus on corn seeds and a reduction of disease symptoms caused by B. cinerea on N. benthamiana leaves.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Agrawal, N.; Dasaradhi, P.; Mohmmed, A.; Malhotra, P.; Bhatnagar, R.K.; Mukherjee, S.K. RNA interference: Biology, mechanism, and applications. Microbiol. Mol. Biol. Rev. 2003, 67, 657–685. [Google Scholar] [CrossRef] [PubMed]
- Setten, R.L.; Rossi, J.J.; Han, S.P. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019, 18, 421–446. [Google Scholar] [CrossRef] [PubMed]
- Zotti, M.; dos Santos, E.A.; Cagliari, D.; Christiaens, O.; Taning, C.N.T.; Smagghe, G. RNA interference technology in crop protection against arthropod pests, pathogens and nematodes. Pest Manag. Sci. 2018, 74, 1239–1250. [Google Scholar] [CrossRef]
- Weng, Y.H.; Xiao, H.H.; Zhang, J.C.; Liang, X.J.; Huang, Y.Y. RNAi therapeutic and its innovative biotechnological evolution. Biotechnol. Adv. 2019, 37, 801–825. [Google Scholar] [CrossRef]
- Tiemann, K.; Rossi, J.J. RNAi-based therapeutics-current status, challenges and prospects. EMBO Mol. Med. 2009, 1, 142–151. [Google Scholar] [CrossRef]
- Aigner, A. Perspectives, issues and solutions in RNAi therapy: The expected and the less expected. Nanomedicine 2019, 14, 2777–2782. [Google Scholar] [CrossRef]
- Nunes, C.C.; Dean, R.A. Host-induced gene silencing: A tool for understanding fungal host interaction and for developing novel disease control strategies. Mol. Plant Pathol. 2012, 13, 519–529. [Google Scholar] [CrossRef]
- Machado, A.K.; Brown, N.A.; Urban, M.; Kanyuka, K.; Hammond-Kosack, K.E. RNAi as an emerging approach to control Fusarium head blight disease and mycotoxin contamination in cereals. Pest Manag. Sci. 2018, 74, 790–799. [Google Scholar] [CrossRef]
- Qi, T.; Guo, J.; Peng, H.; Liu, P.; Kang, Z.S.; Guo, J. Host-induced gene silencing: A powerful strategy to control diseases of wheat and barley. Int. J. Mol. Sci. 2019, 20, 206. [Google Scholar] [CrossRef] [PubMed]
- Gaffar, F.Y.; Koch, A. Catch me if you can! RNA Silencing-based improvement of antiviral plant immunity. Viruses 2019, 11, 673. [Google Scholar] [CrossRef]
- Majumdar, R.; Rajasekaran, K.; Cary, J.W. RNA Interference (RNAi) as a potential tool for control of mycotoxin contamination in crop plants: Concepts and considerations. Front. Plant. Sci. 2017, 8, 200. [Google Scholar] [CrossRef] [PubMed]
- Masanga, J.O.; Matheka, J.M.; Omer, R.A.; Ommeh, S.C.; Monda, E.O.; Alakonya, A.E. Downregulation of transcription factor aflR in Aspergillus flavus confers reduction to aflatoxin accumulation in transgenic maize with alteration of host plant architecture. Plant Cell Rep. 2015, 34, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- McDonald, T.; Brown, D.; Keller, N.P.; Hammond, T.M. RNA silencing of mycotoxin production in Aspergillus and Fusarium species. Mol. Plant Microbe Interact. 2005, 18, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Raruang, Y.; Omolehin, O.; Hu, D.; Wei, Q.; Han, Z.Q.; Rajasekaran, K.; Cary, J.W.; Wang, K.; Chen, Z.Y. Host induced gene silencing targeting Aspergillus flavus aflM reduced aflatoxin contamination in transgenic maize under field conditions. Front. Microbiol. 2020, 11, 754. [Google Scholar] [CrossRef]
- Thakare, D.; Zhang, J.; Wing, R.A.; Cotty, P.J.; Schmidt, M.A. Aflatoxin-free transgenic maize using host-induced gene silencing. Sci. Adv. 2017, 3, e1602382. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Mahato, D.K.; Kamle, M.; Mohanta, T.K.; Kang, S.G. Aflatoxins: A global concern for food safety, human health and their management. Front. Microbiol. 2017, 7, 2170. [Google Scholar] [CrossRef] [PubMed]
- Mahato, D.K.; Lee, K.E.; Kamle, M.; Devi, S.; Dewangan, K.N.; Kumar, P.; Kang, S.G. Aflatoxins in food and feed: An overview on prevalence, detection and control strategies. Front. Microbiol. 2019, 10, 2266. [Google Scholar] [CrossRef]
- Robens, J.F.; Richard, J.L. Aflatoxins in animal and human health. Rev. Environ. Contam. Toxicol. 1992, 127, 69–94. [Google Scholar]
- Huang, G.Z.; Allen, R.; Davis, E.L.; Baum, T.J.; Hussey, R.S. Engineering broad root-knot resistance in transgenic plants by RNAi silencing of a conserved and essential root-knot nematode parasitism gene. Proc. Natl. Acad. Sci. USA 2006, 103, 14302–14306. [Google Scholar] [CrossRef]
- Koch, A.; Kumar, N.; Weber, L.; Keller, H.; Imani, J.; Kogel, K.H. Host-induced gene silencing of cytochrome P450 lanosterol C14 alpha-demethylase-encoding genes confers strong resistance to Fusarium species. Proc. Natl. Acad. Sci. USA 2013, 110, 19324–19329. [Google Scholar] [CrossRef]
- Ramegowda, V.; Mysore, K.S.; Senthil-Kumar, M. Virus-induced gene silencing is a versatile tool for unraveling the functional relevance of multiple abiotic-stress-responsive genes in crop plants. Front. Plant Sci. 2014, 5, 323. [Google Scholar] [CrossRef]
- Dommes, A.B.; Gross, T.; Herbert, D.B.; Kivivirta, K.I.; Becker, A. Virus-induced gene silencing: Empowering genetics in non-model organisms. J. Exp. Bot. 2019, 70, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Burch-Smith, T.M.; Anderson, J.C.; Martin, G.B.; Dinesh-Kumar, S.P. Applications and advantages of virus-induced gene silencing for gene function studies in plants. Plant J. 2004, 39, 734–746. [Google Scholar] [CrossRef]
- Wang, M.; Weiberg, A.; Lin, F.-M.; Thomma, B.P.; Huang, H.-D.; Jin, H. Bidirectional cross-kingdom RNAi and fungal uptake of external RNAs confer plant protection. Nat. Plants 2016, 2, 16151. [Google Scholar] [CrossRef]
- Wang, M.; Jin, H. Spray-induced gene silencing: A powerful innovative strategy for crop protection. Trends Microbiol. 2017, 25, 4–6. [Google Scholar] [CrossRef]
- Dalakouras, A.; Wassenegger, M.; Dadami, E.; Ganopoulos, I.; Pappas, M.L.; Papadopoulou, K. Genetically modified organism-free RNA interference: Exogenous application of RNA molecules in plants. Plant Physiol. 2020, 182, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Cagliari, D.; Dias, N.P.; Galdeano, D.M.; dos Santos, E.A.; Smagghe, G.; Zotti, M.J. Management of pest insects and plant diseases by non-transformative RNAi. Front. Plant Sci. 2019, 10, 1319. [Google Scholar] [CrossRef] [PubMed]
- Mitter, N.; Worrall, E.A.; Robinson, K.E.; Li, P.; Jain, R.G.; Taochy, C.; Fletcher, S.J.; Carroll, B.J.; Lu, G.Q.; Xu, Z.P. Clay nanosheets for topical delivery of RNAi for sustained protection against plant viruses. Nat. Plants 2017, 3, 1–10. [Google Scholar] [CrossRef]
- Timmons, L.; Court, D.L.; Fire, A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene 2001, 263, 103–112. [Google Scholar] [CrossRef]
- Attasart, P.; Namramoon, O.; Kongphom, U.; Chimwai, C.; Panyim, S. Ingestion of bacteria expressing dsRNA triggers specific RNA silencing in shrimp. Virus Res. 2013, 171, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.; Chakraborty, T. Transfer of eukaryotic expression plasmids to mammalian host cells by bacterial carriers. Curr. Opin. Biotechnol. 2001, 12, 467–472. [Google Scholar] [CrossRef]
- Klich, M.A. Aspergillus flavus: The major producer of aflatoxin. Mol. Plant Pathol. 2007, 8, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Adeyeye, S.A.O. Aflatoxigenic fungi and mycotoxins in food: A review. Crit. Rev. Food Sci. 2020, 60, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Williamson, B.; Tudzynsk, B.; Tudzynski, P.; van Kan, J.A.L. Botrytis cinerea: The cause of grey mould disease. Mol. Plant Pathol. 2007, 8, 561–580. [Google Scholar] [CrossRef] [PubMed]
- Didovyk, A.; Tonooka, T.; Tsimring, L.; Hasty, J. Rapid and scalable preparation of bacterial lysates for cell-free gene expression. ACS Synth. Biol. 2017, 6, 2198–2208. [Google Scholar] [CrossRef]
- Weber, E.; Engler, C.; Gruetzner, R.; Werner, S.; Marillonnet, S. A modular cloning system for standardized assembly of multigene constructs. PLoS ONE 2011, 6, e16765. [Google Scholar] [CrossRef]
- Werner, S.; Engler, C.; Weber, E.; Gruetzner, R.; Marillonnet, S. Fast track assembly of multigene constructs using Golden Gate cloning and the MoClo system. Bioengineered 2012, 3, 38–43. [Google Scholar] [CrossRef]
- Miller, W.G.; Leveau, J.H.J.; Lindow, S.E. Improved gfp and inaZ broad-host-range promoter-probe vectors. Mol. Plant Microbe Interact. 2000, 13, 1243–1250. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Arias, R.S.; Dang, P.M.; Sobolev, V.S. RNAi-mediated control of aflatoxins in peanut: Method to analyze mycotoxin production and transgene expression in the peanut/Aspergillus pathosystem. J. Vis. Exp. JoVE 2015, 106, e53398. [Google Scholar] [CrossRef]
- Liu, M.J.; Duan, L.W.; Wang, M.F.; Zeng, H.M.; Liu, X.Q.; Qiu, D.W. Crystal structure analysis and the identification of distinctive functional regions of the protein elicitor Mohrip2. Front. Plant Sci. 2016, 7, 1103. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Chang, P.-K.; Ehrlich, K.C.; Cary, J.W.; Bhatnagar, D.; Cleveland, T.E.; Payne, G.A.; Linz, J.E.; Woloshuk, C.P.; Bennett, J.W. Clustered pathway genes in aflatoxin biosynthesis. Appl. Environ. Microbiol. 2004, 70, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Q.; Qin, G.Z.; Li, B.Q.; Tian, S.P. Knocking Out Bcsas1 in Botrytis cinerea Impacts Growth, Development, and secretion of extracellular proteins, which decreases virulence. Mol. Plant Microbe Interact. 2014, 27, 590–600. [Google Scholar] [CrossRef]
- Fisher, M.C.; Henk, D.A.; Briggs, C.J.; Brownstein, J.S.; Madoff, L.C.; McCraw, S.L.; Gurr, S.J. Emerging fungal threats to animal, plant and ecosystem health. Nature 2012, 484, 186. [Google Scholar] [CrossRef]
- Nelson, R.; Wiesner-Hanks, T.; Wisser, R.; Balint-Kurti, P. Navigating complexity to breed disease-resistant crops. Nat. Rev. Genet. 2018, 19, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Savary, S.; Willocquet, L.; Pethybridge, S.J.; Esker, P.; McRoberts, N.; Nelson, A. The global burden of pathogens and pests on major food crops. Nat. Ecol. Evol. 2019, 3, 430–439. [Google Scholar] [CrossRef]
- Strange, R.N.; Scott, P.R. Plant disease: A threat to global food security. Ann. Rev. Phytopath. 2005, 43, 83–116. [Google Scholar] [CrossRef]
- Goodfellow, S.; Zhang, D.; Wang, M.B.; Zhang, R. Bacterium-mediated RNA interference: Potential application in plant protection. Plants 2019, 8, 572. [Google Scholar] [CrossRef]
- Keates, A.C.; Fruehauf, J.; Xiang, S.L.; Li, C.J. TransKingdom RNA interference: A bacterial approach to challenges in RNAi therapy and delivery. Biotechnol. Genet. Eng. 2008, 25, 113–127. [Google Scholar] [CrossRef][Green Version]
- Whitten, M.M.A.; Facey, P.D.; Del Sol, R.; Fernandez-Martinez, L.T.; Evans, M.C.; Mitchell, J.J.; Bodger, O.G.; Dyson, P.J. Symbiont-mediated RNA interference in insects. Proc. R. Soc. B Biol. Sci. 2016, 283, 20160042. [Google Scholar] [CrossRef]
- Brautaset, T.; Lale, R.; Valla, S. Positively regulated bacterial expression systems. Microb. Biotechnol. 2009, 2, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Mascher, T. The applied side of antimicrobial peptide-inducible promoters from Firmicutes bacteria: Expression systems and whole-cell biosensors. Appl. Environ. Microbiol. 2016, 100, 4817–4829. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.J.; Donahue, K.; Koh, Y.; Martin, R.R.; Choi, M.Y. Microbial-based double-stranded RNA production to develop cost-effective RNA interference application for insect pest management. Int. J. Insect Sci. 2019, 11. [Google Scholar] [CrossRef]
- Zhong, C.; Smith, N.A.; Zhang, D.; Goodfellow, S.; Zhang, R.; Shan, W.; Wang, M.-B. Full-length hairpin RNA accumulates at high levels in yeast but not in bacteria and plants. Genes 2019, 10, 458. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.L.; Linsley, P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010, 9, 57–67. [Google Scholar] [CrossRef]
- Kanasty, R.L.; Whitehead, K.A.; Vegas, A.J.; Anderson, D.G. Action and reaction: The biological response to siRNA and its delivery vehicles. Mol. Ther. 2012, 20, 513–524. [Google Scholar] [CrossRef]
- McEwan, D.L.; Weisman, A.S.; Huntert, C.P. Uptake of extracellular double-stranded RNA by SID-2. Mol. Cell 2012, 47, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Hua, C.L.; Zhao, J.H.; Guo, H.S. Trans-kingdom RNA silencing in plant-fungal pathogen interactions. Mol. Plant 2018, 11, 235–244. [Google Scholar] [CrossRef]
- Cai, Q.; Qiao, L.L.; Wang, M.; He, B.Y.; Lin, F.M.; Palmquist, J.; Huang, S.N.D.; Jin, H.L. Plants send small RNAs in extracellular vesicles to fungal pathogen to silence virulence genes. Science 2018, 360, 1126–1129. [Google Scholar] [CrossRef]
- Timmons, L.; Fire, A. Specific interference by ingested dsRNA. Nature 1998, 395, 854. [Google Scholar] [CrossRef]
- Lecrivain, A.L.; Beckmann, B.M. Bacterial RNA in extracellular vesicles: A new regulator of host-pathogen interactions? BBA Gene Regul. Mech. 2020, 1863, 194519. [Google Scholar] [CrossRef] [PubMed]
- Tsatsaronis, J.A.; Franch-Arroyo, S.; Resch, U.; Charpentier, E. Extracellular vesicle RNA: A universal mediator of microbialcommunication? Trends Microbiol. 2018, 26, 401–410. [Google Scholar] [CrossRef]
- Cooper, A.M.; Silver, K.; Zhang, J.Z.; Park, Y.; Zhu, K.Y. Molecular mechanisms influencing efficiency of RNA interference in insects. Pest Manag. Sci. 2019, 75, 18–28. [Google Scholar] [CrossRef]
- Liu, S.S.; Jaouannet, M.; Dempsey, D.A.; Imani, J.; Coustau, C.; Kogel, K.H. RNA-based technologies for insect control in plant production. Biotechnol. Adv. 2020, 39, 107463. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.A.; Betel, D.; Miller, M.L.; Sander, C.; Leslie, C.S.; Marks, D.S. Transfection of small RNAs globally perturbs gene regulation by endogenous microRNAs. Nat. Biotechnol. 2009, 27, U549–U592. [Google Scholar] [CrossRef]
- Aleman, L.M.; Doench, J.; Sharp, P.A. Comparison of siRNA-induced off-target RNA and protein effects. RNA 2007, 13, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Beyer, A.; Aebersold, R. On the dependency of cellular protein levels on mRNA abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niño-Sánchez, J.; Chen, L.-H.; De Souza, J.T.; Mosquera, S.; Stergiopoulos, I. Targeted Delivery of Gene Silencing in Fungi Using Genetically Engineered Bacteria. J. Fungi 2021, 7, 125. https://doi.org/10.3390/jof7020125
Niño-Sánchez J, Chen L-H, De Souza JT, Mosquera S, Stergiopoulos I. Targeted Delivery of Gene Silencing in Fungi Using Genetically Engineered Bacteria. Journal of Fungi. 2021; 7(2):125. https://doi.org/10.3390/jof7020125
Chicago/Turabian StyleNiño-Sánchez, Jonatan, Li-Hung Chen, Jorge Teodoro De Souza, Sandra Mosquera, and Ioannis Stergiopoulos. 2021. "Targeted Delivery of Gene Silencing in Fungi Using Genetically Engineered Bacteria" Journal of Fungi 7, no. 2: 125. https://doi.org/10.3390/jof7020125
APA StyleNiño-Sánchez, J., Chen, L.-H., De Souza, J. T., Mosquera, S., & Stergiopoulos, I. (2021). Targeted Delivery of Gene Silencing in Fungi Using Genetically Engineered Bacteria. Journal of Fungi, 7(2), 125. https://doi.org/10.3390/jof7020125