
Immune Sensing of Candida albicans


Abstract
1. Introduction
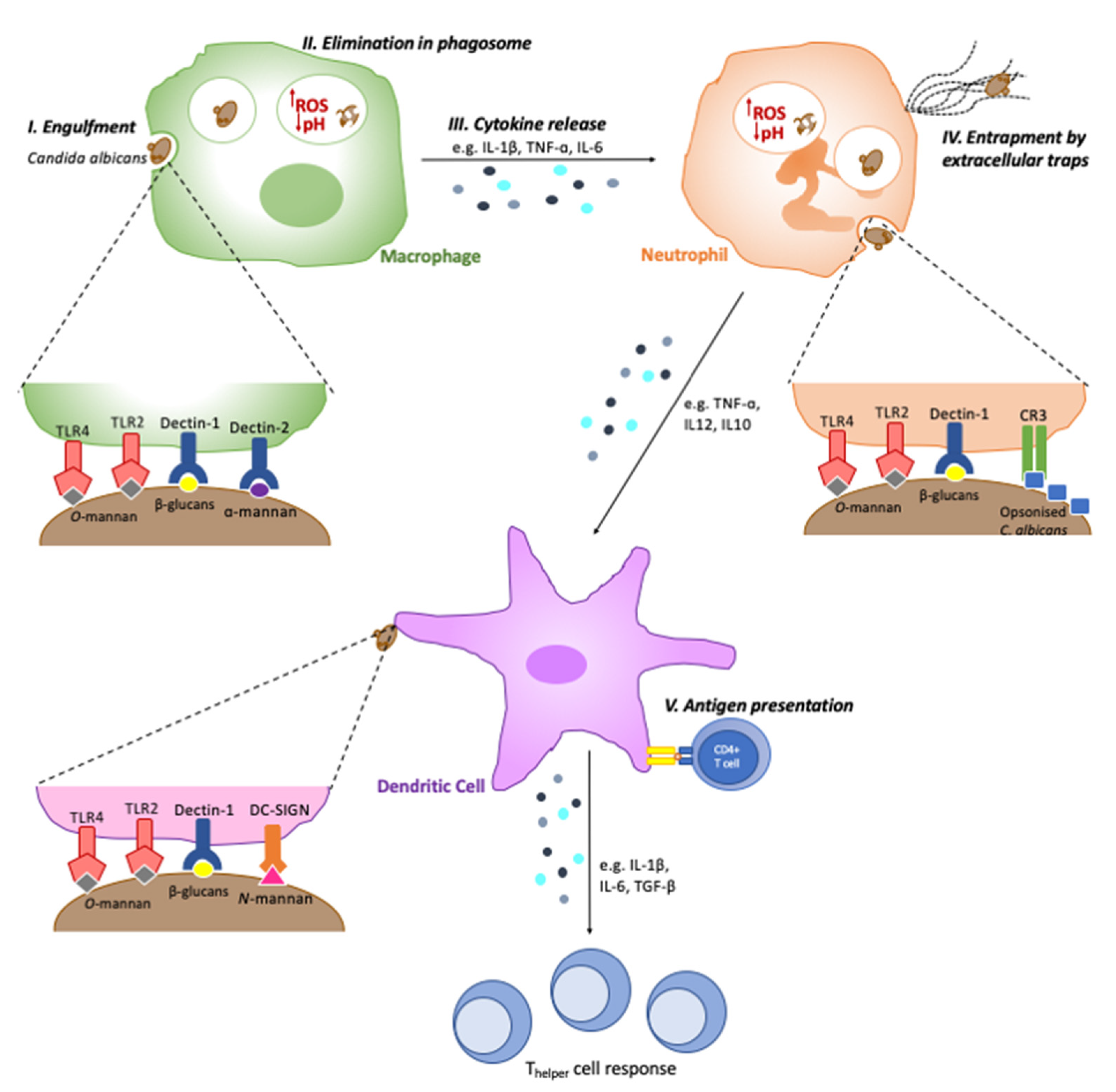
2. Recognition by Phagocytes
3. Niche-Specific Immune Sensing of C. albicans
3.1. Skin and Nail Infections
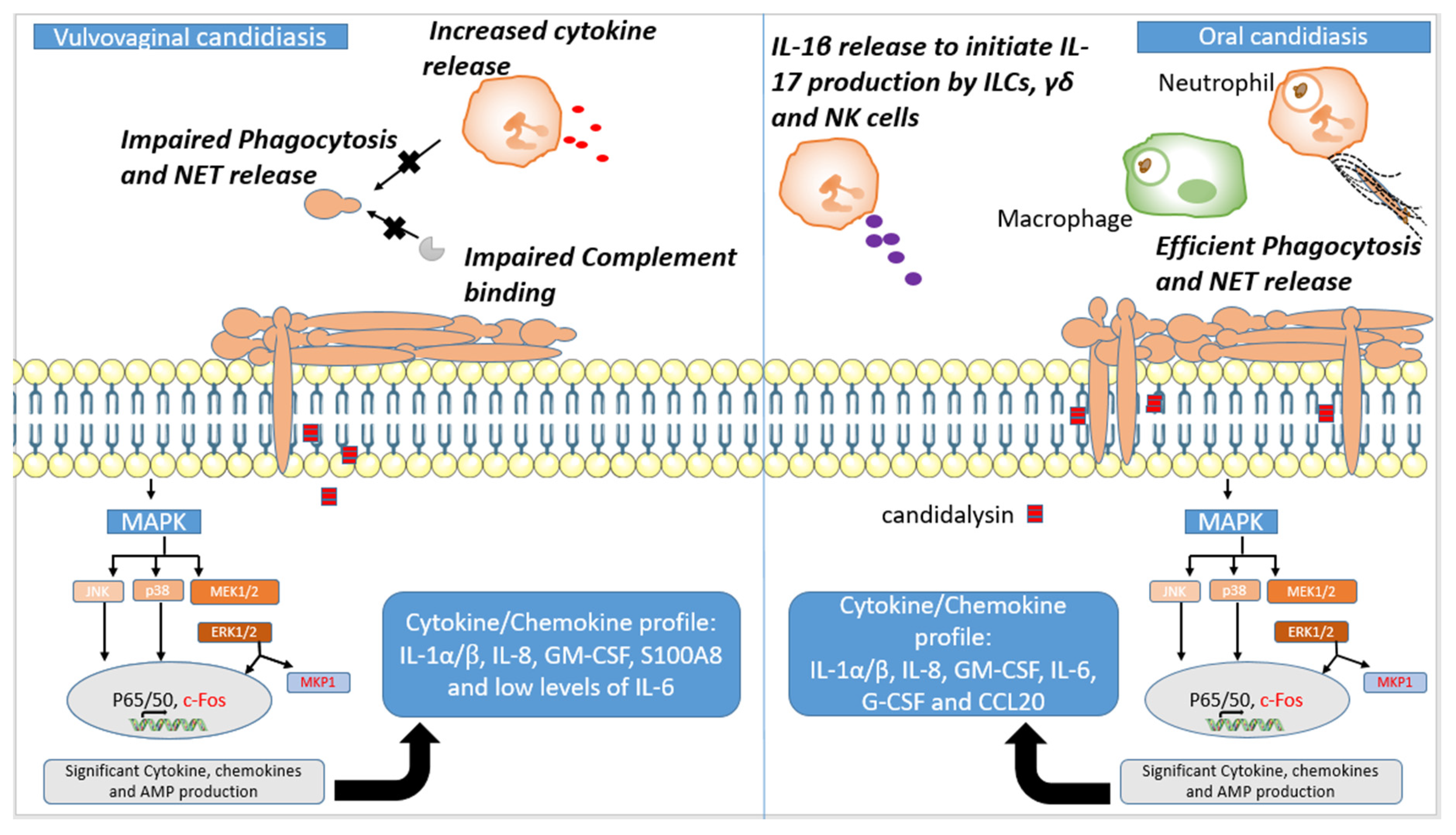
3.2. Oral Candidiasis
3.3. Vaginal Candidiasis
3.4. Candidemia
4. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ward, T.L.; Knights, D.; Gale, C.A. Infant fungal communities: Current knowledge and research opportunities. BMC Med. 2017, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Tso, G.H.W.; Reales-Calderon, J.A.; Pavelka, N. The elusive anti-Candida vaccine: Lessons from the past and opportunities for the future. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Moyes, D.L.; Wilson, D.; Richardson, J.P.; Mogavero, S.; Tang, S.X.; Wernecke, J.; Höfs, S.; Gratacap, R.L.; Robbins, J.; Runglall, M.; et al. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature 2016, 532, 64–68. [Google Scholar] [CrossRef]
- Zhu, W.; Filler, S.G. Interactions of Candida albicans with epithelial cells. Cell. Microbiol. 2010, 12, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Cerdeira, C.; Gregorio, M.C.; Molares-Vila, A.; López-Barcenas, A.; Fabbrocini, G.; Bardhi, B.; Sinani, A.; Sánchez-Blanco, E.; Arenas-Guzmán, R.; Hernandez-Castro, R. Biofilms and vulvovaginal candidiasis. Coll. Surf. B Biointerfaces 2019, 174, 110–125. [Google Scholar] [CrossRef]
- Du, Q.; Ren, B.; He, J.; Peng, X.; Guo, Q.; Zheng, L.; Li, J.; Dai, H.; Chen, V.; Zhang, L.; et al. Candida albicans promotes tooth decay by inducing oral microbial dysbiosis. ISME J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hall, R.A. Dressed to impress: Impact of environmental adaptation on the Candida albicans cell wall. Mol. Microbiol. 2015, 97, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Gow, N.A.R.; Latge, J.-P.; Munro, C.A. The Fungal cell wall: Structure, biosynthesis, and function. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef]
- Chen, S.M.; Shen, H.; Zhang, T.; Huang, X.; Liu, X.Q.; Guo, S.Y.; Zhao, J.J.; Wang, C.F.; Yan, L.; Xu, G.T.; et al. Dectin-1 plays an important role in host defense against systemic Candida glabrata infection. Virulence 2017, 8, 1643–1656. [Google Scholar] [CrossRef]
- Esteban, A.; Popp, M.W.; Vyas, V.K.; Strijbis, K.; Ploegh, H.L.; Fink, G.R. Fungal recognition is mediated by the association of dectin-1 and galectin-3 in macrophages. Proc. Nat. Acad. Sci. USA 2011, 108, 14270–14275. [Google Scholar] [CrossRef] [PubMed]
- Gantner, B.N.; Simmons, R.M.; Underhill, D.M. Dectin-1 mediates macrophage recognition of Candida albicans yeast but not filaments. EMBO J. 2005, 24, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Utomo, A.; Cullere, X.; Choi, M.M.; Milner, D.A., Jr.; Venkatesh, D.; Yun, S.H.; Mayadas, T.N. The β-glucan receptor Dectin-1 activates the integrin Mac-1 in neutrophils via Vav protein signaling to promote Candida albicans clearance. Cell Host Microbe 2011, 10, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Ifrim, D.C.; Quintin, J.; Courjol, F.; Verschueren, I.; van Krieken, J.H.; Koentgen, F.; Fradin, C.; Gow, N.A.; Joosten, L.A.; van der Meer, J.W.; et al. The role of dectin-2 for host defense against disseminated Candidiasis. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2016, 36, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Saijo, S.; Ikeda, S.; Yamabe, K.; Kakuta, S.; Ishigame, H.; Akitsu, A.; Fujikado, N.; Kusaka, T.; Kubo, S.; Chung, S.H.; et al. Dectin-2 recognition of alpha-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity 2010, 32, 681–691. [Google Scholar] [CrossRef]
- Zhu, L.L.; Zhao, X.Q.; Jiang, C.; You, Y.; Chen, X.P.; Jiang, Y.Y.; Jia, X.M.; Lin, X. C-type lectin receptors Dectin-3 and Dectin-2 form a heterodimeric pattern-recognition receptor for host defense against fungal infection. Immunity 2013, 39, 324–334. [Google Scholar] [CrossRef]
- Cambi, A.; Gijzen, K.; de Vries l, J.; Torensma, R.; Joosten, B.; Adema, G.J.; Netea, M.G.; Kullberg, B.J.; Romani, L.; Figdor, C.G. The C-type lectin DC-SIGN (CD209) is an antigen-uptake receptor for Candida albicans on dendritic cells. Eur. J. Immunol. 2003, 33, 532–538. [Google Scholar] [CrossRef]
- Cambi, A.; Netea, M.G.; Mora-Montes, H.M.; Gow, N.A.; Hato, S.V.; Lowman, D.W.; Kullberg, B.J.; Torensma, R.; Williams, D.L.; Figdor, C.G. Dendritic cell interaction with Candida albicans critically depends on N-linked mannan. J. Biol. Chem. 2008, 283, 20590–20599. [Google Scholar] [CrossRef]
- Wells, C.A.; Salvage-Jones, J.A.; Li, X.; Hitchens, K.; Butcher, S.; Murray, R.Z.; Beckhouse, A.G.; Lo, Y.L.; Manzanero, S.; Cobbold, C.; et al. The macrophage-inducible C-type lectin, mincle, is an essential component of the innate immune response to Candida albicans. J. immunol. 2008, 180, 7404–7413. [Google Scholar] [CrossRef]
- Netea, M.G.; Gow, N.A.; Munro, C.A.; Bates, S.; Collins, C.; Ferwerda, G.; Hobson, R.P.; Bertram, G.; Hughes, H.B.; Jansen, T.; et al. Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and Toll-like receptors. J. Clin. Investig. 2006, 116, 1642–1650. [Google Scholar] [CrossRef] [PubMed]
- van de Veerdonk, F.L.; Marijnissen, R.J.; Kullberg, B.J.; Koenen, H.J.; Cheng, S.C.; Joosten, I.; van den Berg, W.B.; Williams, D.L.; van der Meer, J.W.; Joosten, L.A.; et al. The macrophage mannose receptor induces IL-17 in response to Candida albicans. Cell Host Microbe 2009, 5, 329–340. [Google Scholar] [CrossRef]
- Blasi, E.; Mucci, A.; Neglia, R.; Pezzini, F.; Colombari, B.; Radzioch, D.; Cossarizza, A.; Lugli, E.; Volpini, G.; Del Giudice, G.; et al. Biological importance of the two Toll-like receptors, TLR2 and TLR4, in macrophage response to infection with Candida albicans. FEMS Immunol. Med. Microbiol. 2005, 44, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Tessarolli, V.; Gasparoto, T.H.; Lima, H.R.; Figueira, E.A.; Garlet, T.P.; Torres, S.A.; Garlet, G.P.; da Silva, J.S.; Campanelli, A.P. Absence of TLR2 influences survival of neutrophils after infection with Candida albicans. Med. Mycol. 2010, 48, 129–140. [Google Scholar] [CrossRef]
- Jouault, T.; Ibata-Ombetta, S.; Takeuchi, O.; Trinel, P.A.; Sacchetti, P.; Lefebvre, P.; Akira, S.; Poulain, D. Candida albicans phospholipomannan is sensed through toll-like receptors. J. Infect. Dis. 2003, 188, 165–172. [Google Scholar] [CrossRef]
- Netea, M.G.; van de Veerdonk, F.; Verschueren, I.; van der Meer, J.W.; Kullberg, B.J. Role of TLR1 and TLR6 in the host defense against disseminated candidiasis. FEMS Immunol. Med. Microbiol. 2008, 52, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Halder, L.D.; Jo, E.A.H.; Hasan, M.Z.; Ferreira-Gomes, M.; Krüger, T.; Westermann, M.; Palme, D.I.; Rambach, G.; Beyersdorf, N.; Speth, C.; et al. Immune modulation by complement receptor 3-dependent human monocyte TGF-β1-transporting vesicles. Nat. Commun. 2020, 11, 2331. [Google Scholar] [CrossRef] [PubMed]
- Levin, R.; Grinstein, S.; Canton, J. The life cycle of phagosomes: Formation, maturation, and resolution. Immunol. Rev. 2016, 273, 156–179. [Google Scholar] [CrossRef] [PubMed]
- Mukaremera, L.; Lee, K.K.; Mora-Montes, H.M.; Gow, N.A.R. Candida albicans yeast, pseudohyphal, and hyphal morphogenesis differentially affects immune recognition. Front. Immunol. 2017, 8, 629. [Google Scholar] [CrossRef] [PubMed]
- Hopke, A.; Nicke, N.; Hidu, E.E.; Degani, G.; Popolo, L.; Wheeler, R.T. Neutrophil Attack triggers extracellular trap-dependent Candida cell wall remodeling and altered immune recognition. PLoS Pathog. 2016, 12, e1005644. [Google Scholar] [CrossRef]
- Lewis, L.E.; Bain, J.M.; Lowes, C.; Gillespie, C.; Rudkin, F.M.; Gow, N.A.R.; Erwig, L.-P. Stage specific assessment of Candida albicans phagocytosis by macrophages identifies cell wall composition and morphogenesis as key determinants. PLoS Pathog. 2012, 8, e1002578. [Google Scholar] [CrossRef]
- D’Ostiani, C.F.; Del Sero, G.; Bacci, A.; Montagnoli, C.; Spreca, A.; Mencacci, A.; Ricciardi-Castagnoli, P.; Romani, L. Dendritic cells discriminate between yeasts and hyphae of the fungus Candida albicans. Implications for initiation of T helper cell immunity in vitro and in vivo. J. Exp. Med. 2000, 191, 1661–1674. [Google Scholar] [CrossRef] [PubMed]
- Hope, W.W.; Drusano, G.L.; Moore, C.B.; Sharp, A.; Louie, A.; Walsh, T.J.; Denning, D.W.; Warn, P.A. Effect of neutropenia and treatment delay on the response to antifungal agents in experimental disseminated candidiasis. Antimicrob. Agents Chemother. 2007, 51, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Ramy, N.; Hashim, M.; Abou Hussein, H.; Sawires, H.; Gaafar, M.; El Maghraby, A. Role of early onset neutropenia in development of Candidemia in premature infants. J. Trop. Pediatr. 2017, 64, 51–59. [Google Scholar] [CrossRef]
- Son, H.-J.; Kim, M.J.; Lee, S.; Choi, S.; Jung, K.H.; Jung, J.; Chong, Y.P.; Kim, S.-H.; Choi, S.-H.; Kim, Y.S.; et al. Risk factors and outcomes of patients with ocular involvement of candidemia. PLoS ONE 2019, 14, e0222356. [Google Scholar] [CrossRef]
- Fradin, C.; De Groot, P.; MacCallum, D.; Schaller, M.; Klis, F.; Odds, F.C.; Hube, B. Granulocytes govern the transcriptional response, morphology and proliferation of Candida albicans in human blood. Mol. Microbiol. 2005, 56, 397–415. [Google Scholar] [CrossRef]
- Newman, S.L.; Holly, A. Candida albicans is phagocytosed, killed, and processed for antigen presentation by human dendritic cells. Infect. Immun. 2001, 69, 6813–6822. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D.; Gordon, S. A new receptor for β-glucans. Nature 2001, 413, 36–37. [Google Scholar] [CrossRef] [PubMed]
- Erwig, L.P.; Gow, N.A.R. Interactions of fungal pathogens with phagocytes. Nat. Rev. Microbiol. 2016, 14, 163–176. [Google Scholar] [CrossRef]
- Thornton, B.P.; Vĕtvicka, V.; Pitman, M.; Goldman, R.C.; Ross, G.D. Analysis of the sugar specificity and molecular location of the beta-glucan-binding lectin site of complement receptor type 3 (CD11b/CD18). J. Immunol. 1996, 156, 1235–1246. [Google Scholar] [PubMed]
- Netea, M.G.; Brown, G.D.; Kullberg, B.J.; Gow, N.A. An integrated model of the recognition of Candida albicans by the innate immune system. Nat. Rev. Microbiol. 2008, 6, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Marie-Anaïs, F.; Mazzolini, J.; Herit, F.; Niedergang, F. Dynamin-actin cross talk contributes to phagosome formation and closure. Traffic 2016, 17, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Gold, E.S.; Underhill, D.M.; Morrissette, N.S.; Guo, J.; McNiven, M.A.; Aderem, A. Dynamin 2 is required for phagocytosis in macrophages. J. Exp. Med. 1999, 190, 1849–1856. [Google Scholar] [CrossRef] [PubMed]
- Campellone, K.G.; Welch, M.D. A nucleator arms race: Cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 2010, 11, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.L.; Harrison, R.E.; Grinstein, S. Phagocytosis by neutrophils. Microb. Infect. 2003, 5, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Nauclér, C.; Grinstein, S.; Sundler, R.; Tapper, H. Signaling to localized degranulation in neutrophils adherent to immune complexes. J. Leukoc. Biol. 2002, 71, 701–710. [Google Scholar] [CrossRef]
- Nordenfelt, P.; Tapper, H. Phagosome dynamics during phagocytosis by neutrophils. J. Leukoc. Biol. 2011, 90, 271–284. [Google Scholar] [CrossRef]
- Roberts, R.L.; Barbieri, M.A.; Ullrich, J.; Stahl, P.D. Dynamics of Rab5 activation in endocytosis and phagocytosis. J. Leukoc. Biol. 2000, 68, 627–632. [Google Scholar] [PubMed]
- Vieira, O.V.; Bucci, C.; Harrison, R.E.; Trimble, W.S.; Lanzetti, L.; Gruenberg, J.; Schreiber, A.D.; Stahl, P.D.; Grinstein, S. Modulation of Rab5 and Rab7 recruitment to phagosomes by phosphatidylinositol 3-kinase. Mol. Cell. Biol. 2003, 23, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Vylkova, S.; Lorenz, M.C. Modulation of phagosomal pH by Candida albicans promotes hyphal morphogenesis and requires Stp2p, a regulator of amino acid transport. PLoS Pathog. 2014, 10, e1003995. [Google Scholar] [CrossRef] [PubMed]
- Canton, J.; Khezri, R.; Glogauer, M.; Grinstein, S. Contrasting phagosome pH regulation and maturation in human M1 and M2 macrophages. Mol. Biol. Cell 2014, 25, 3330–3341. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, A.; Scott, C.C.; Grinstein, S. Determinants of the phagosomal pH in neutrophils. J. Biol. Chem. 2002, 277, 6059–6066. [Google Scholar] [CrossRef]
- Savina, A.; Jancic, C.; Hugues, S.; Guermonprez, P.; Vargas, P.; Moura, I.C.; Lennon-Duménil, A.M.; Seabra, M.C.; Raposo, G.; Amigorena, S. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 2006, 126, 205–218. [Google Scholar] [CrossRef]
- Chauhan, N.; Latge, J.P.; Calderone, R. Signalling and oxidant adaptation in Candida albicans and Aspergillus fumigatus. Nat. Rev. Microbiol. 2006, 4, 435–444. [Google Scholar] [CrossRef]
- Thompson, H.L.; Wilton, J.M. Interaction and intracellular killing of Candida albicans blastospores by human polymorphonuclear leucocytes, monocytes and monocyte-derived macrophages in aerobic and anaerobic conditions. Clin. Exp. Immunol. 1992, 87, 316–321. [Google Scholar] [CrossRef]
- Azevedo, E.P.; Rochael, N.C.; Guimarães-Costa, A.B.; de Souza-Vieira, T.S.; Ganilho, J.; Saraiva, E.M.; Palhano, F.L.; Foguel, D. A metabolic shift toward pentose phosphate pathway is necessary for amyloid fibril- and phorbol 12-myristate 13-acetate-induced Neutrophil Extracellular Trap (NET) Formation. J. Biol. Chem. 2015, 290, 22174–22183. [Google Scholar] [CrossRef] [PubMed]
- Jawale, C.V.; Ramani, K.; Li, D.-d.; Coleman, B.M.; Oberoi, R.S.; Kupul, S.; Lin, L.; Desai, J.V.; Delgoffe, G.M.; Lionakis, M.S.; et al. Restoring glucose uptake rescues neutrophil dysfunction and protects against systemic fungal infection in mouse models of kidney disease. Sci. Transl. Med. 2020, 12, eaay5691. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Ishihara, C.; Takeuchi, A.; Imanishi, T.; Xue, L.; Morris, S.W.; Inui, M.; Takai, T.; Shibuya, A.; Saijo, S.; et al. The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM-associated and Toll-like receptors. Nat. Immunol. 2007, 8, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Rogers, N.C.; Slack, E.C.; Edwards, A.D.; Nolte, M.A.; Schulz, O.; Schweighoffer, E.; Williams, D.L.; Gordon, S.; Tybulewicz, V.L.; Brown, G.D.; et al. Syk-dependent cytokine induction by Dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity 2005, 22, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Underhill, D.M.; Pearlman, E. Immune interactions with pathogenic and commensal fungi: A two-way street. Immunity 2015, 43, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Hernanz-Falcón, P.; Joffre, O.; Williams, D.L.; Reis e Sousa, C. Internalization of dectin-1 terminates induction of inflammatory responses. Eur. J. Immunol. 2009, 39, 507–513. [Google Scholar] [CrossRef]
- Westman, J.; Walpole, G.F.W.; Kasper, L.; Xue, B.Y.; Elshafee, O.; Hube, B.; Grinstein, S. Lysosome fusion maintains phagosome integrity during fungal infection. Cell Host Microbe 2020. [Google Scholar] [CrossRef]
- Aybay, C.; Imir, T. Tumor necrosis factor (TNF) induction from monocyte/macrophages by Candida species. Immunobiology 1996, 196, 363–374. [Google Scholar] [CrossRef]
- Ganesan, S.; Rathinam, V.A.K.; Bossaller, L.; Army, K.; Kaiser, W.J.; Mocarski, E.S.; Dillon, C.P.; Green, D.R.; Mayadas, T.N.; Levitz, S.M.; et al. Caspase-8 modulates dectin-1 and complement receptor 3-driven IL-1β production in response to β-glucans and the fungal pathogen, Candida albicans. J. Immunol. 2014, 193, 2519–2530. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Klein, T.W.; Friedman, H. Involvement of mannose receptor in cytokine interleukin-1beta (IL-1beta), IL-6, and granulocyte-macrophage colony-stimulating factor responses, but not in chemokine macrophage inflammatory protein 1beta (MIP-1beta), MIP-2, and KC responses, caused by attachment of Candida albicans to macrophages. Infect. Immun. 1997, 65, 1077–1082. [Google Scholar] [CrossRef] [PubMed]
- Chin, V.K.; Foong, K.J.; Maha, A.; Rusliza, B.; Norhafizah, M.; Chong, P.P. Early expression of local cytokines during systemic Candida albicans infection in a murine intravenous challenge model. Biomed. Rep. 2014, 2, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Romani, L.; Bistoni, F.; Puccetti, P. Initiation of T-helper cell immunity to Candida albicans by IL-12: The role of neutrophils. Chem. Immunol. 1997, 68, 110–135. [Google Scholar] [CrossRef] [PubMed]
- Maher, C.O.; Dunne, K.; Comerford, R.; O’Dea, S.; Loy, A.; Woo, J.; Rogers, T.R.; Mulcahy, F.; Dunne, P.J.; Doherty, D.G. Candida albicans stimulates IL-23 release by human dendritic cells and downstream IL-17 secretion by Vδ1 T cells. J. Immunol. 2015, 194, 5953–5960. [Google Scholar] [CrossRef]
- Mohammadi, F.; Ghasemi, Z.; Familsatarian, B.; Salehi, E.; Sharifynia, S.; Barikani, A.; mirzadeh, M.; Hosseini, M.A. Relationship between antifungal susceptibility profile and virulence factors in Candida albicans isolated from nail specimens. Rev. Soc. Bras. Med. Trop. 2020, 53, doi. [Google Scholar] [CrossRef] [PubMed]
- Naglik, J.; Albrecht, A.; Bader, O.; Hube, B. Candida albicans proteinases and host/pathogen interactions. Cell. Microbiol. 2004, 6, 915–926. [Google Scholar] [CrossRef]
- Schaller, M.; Borelli, C.; Korting, H.C.; Hube, B. Hydrolytic enzymes as virulence factors of Candida albicans. Mycoses 2005, 48, 365–377. [Google Scholar] [CrossRef]
- Hu, W.; Troutman, T.D.; Edukulla, R.; Pasare, C. Priming microenvironments dictate cytokine requirements for T helper 17 cell lineage commitment. Immunity 2011, 35, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Weindl, G.; Naglik, J.R.; Kaesler, S.; Biedermann, T.; Hube, B.; Korting, H.C.; Schaller, M. Human epithelial cells establish direct antifungal defense through TLR4-mediated signaling. J. Clin. Investig. 2007, 117, 3664–3672. [Google Scholar] [CrossRef] [PubMed]
- Tomalka, J.; Azodi, E.; Narra, H.P.; Patel, K.; O’Neill, S.; Cardwell, C.; Hall, B.A.; Wilson, J.M.; Hise, A.G. β-Defensin 1 plays a role in acute mucosal defense against Candida albicans. J. Immunol. 2015, 194, 1788–1795. [Google Scholar] [CrossRef]
- Byrd, A.S.; O’Brien, X.M.; Johnson, C.M.; Lavigne, L.M.; Reichner, J.S. An extracellular matrix-based mechanism of rapid neutrophil extracellular trap formation in response to Candida albicans. J. Immunol. 2013, 190, 4136–4148. [Google Scholar] [CrossRef] [PubMed]
- Gladiator, A.; Wangler, N.; Trautwein-Weidner, K.; LeibundGut-Landmann, S. Cutting edge: IL-17-secreting innate lymphoid cells are essential for host defense against fungal infection. J. Immunol. 2013, 190, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Conti, H.R.; Peterson, A.C.; Brane, L.; Huppler, A.R.; Hernández-Santos, N.; Whibley, N.; Garg, A.V.; Simpson-Abelson, M.R.; Gibson, G.A.; Mamo, A.J.; et al. Oral-resident natural Th17 cells and γδ T cells control opportunistic Candida albicans infections. J. Exp. Med. 2014, 211, 2075–2084. [Google Scholar] [CrossRef] [PubMed]
- Bishu, S.; Su, E.W.; Wilkerson, E.R.; Reckley, K.A.; Jones, D.M.; McGeachy, M.J.; Gaffen, S.L.; Levesque, M.C. Rheumatoid arthritis patients exhibit impaired Candida albicans-specific Th17 responses. Arthr. Res. Ther. 2014, 16, R50. [Google Scholar] [CrossRef]
- Conti, H.R.; Bruno, V.M.; Childs, E.E.; Daugherty, S.; Hunter, J.P.; Mengesha, B.G.; Saevig, D.L.; Hendricks, M.R.; Coleman, B.M.; Brane, L.; et al. IL-17 receptor signaling in oral epithelial cells is critical for protection against oropharyngeal candidiasis. Cell Host Microbe 2016, 20, 606–617. [Google Scholar] [CrossRef]
- Conti, H.R.; Shen, F.; Nayyar, N.; Stocum, E.; Sun, J.N.; Lindemann, M.J.; Ho, A.W.; Hai, J.H.; Yu, J.J.; Jung, J.W.; et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J. Exp. Med. 2009, 206, 299–311. [Google Scholar] [CrossRef]
- Yano, J.; Lilly, E.; Barousse, M.; Fidel, P.L., Jr. Epithelial cell-derived S100 calcium-binding proteins as key mediators in the hallmark acute neutrophil response during Candida vaginitis. Infect. Immun. 2010, 78, 5126–5137. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.; Palmer, G.E.; Eberle, K.E.; Peters, B.M.; Vogl, T.; McKenzie, A.N.; Fidel, P.L., Jr. Vaginal epithelial cell-derived S100 alarmins induced by Candida albicans via pattern recognition receptor interactions are sufficient but not necessary for the acute neutrophil response during experimental vaginal candidiasis. Infect. Immun. 2014, 82, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Wirnsberger, G.; Zwolanek, F.; Stadlmann, J.; Tortola, L.; Liu, S.W.; Perlot, T.; Järvinen, P.; Dürnberger, G.; Kozieradzki, I.; Sarao, R.; et al. Jagunal homolog 1 is a critical regulator of neutrophil function in fungal host defense. Nat. Genet. 2014, 46, 1028–1033. [Google Scholar] [CrossRef] [PubMed]
- Willcox, M.D.P.; Webb, B.C.; Thakur, A.; Harty, D.W.S. Interactions between Candida species and platelets. J. Med. Microbiol. 1998, 47, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Yeaman, M.R.; Soldan, S.S.; Ghannoum, M.A.; Edwards, J.E., Jr.; Filler, S.G.; Bayer, A.S. Resistance to platelet microbicidal protein results in increased severity of experimental Candida albicans endocarditis. Infect. Immun. 1996, 64, 1379–1384. [Google Scholar] [CrossRef] [PubMed]
- Alam, F.; Catlow, D.; Di Maio, A.; Blair, J.M.A.; Hall, R.A. Candida albicans enhances meropenem tolerance of Pseudomonas aeruginosa in a dual-species biofilm. J. Antimicrob. Chemother. 2020, 75, 925–935. [Google Scholar] [CrossRef]
- Ambe, N.F.; Longdoh, N.A.; Tebid, P.; Bobga, T.P.; Nkfusai, C.N.; Ngwa, S.B.; Nsai, F.S.; Cumber, S.N. The prevalence, risk factors and antifungal sensitivity pattern of oral candidiasis in HIV/AIDS patients in Kumba District Hospital, South West Region, Cameroon. Pan Afr. Med. J. 2020, 36, 23. [Google Scholar] [CrossRef] [PubMed]
- Kirti, Y.K. Prevalence of oral Candidiasis in Indian HIV sero-positive patients with CD4(+) cell count correlation. Indian J. Otolaryngol. Head Neck Surg. Off. Publ. Assoc. Otolaryngol. India 2019, 71, 124–127. [Google Scholar] [CrossRef]
- Naglik, J.R.; König, A.; Hube, B.; Gaffen, S.L. Candida albicans-epithelial interactions and induction of mucosal innate immunity. Curr. Opin. Microbiol. 2017, 40, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Nikou, S.A.; Kichik, N.; Brown, R.; Ponde, N.O.; Ho, J.; Naglik, J.R.; Richardson, J.P. Candida albicans interactions with mucosal surfaces during health and disease. Pathogens 2019, 8, 53. [Google Scholar] [CrossRef]
- Pavlova, A.; Sharafutdinov, I. Recognition of Candida albicans and role of innate type 17 immunity in oral candidiasis. Microorganisms 2020, 8, 1340. [Google Scholar] [CrossRef]
- Richardson, J.P.; Moyes, D.L.; Ho, J.; Naglik, J.R. Candida innate immunity at the mucosa. Semin. Cell Dev. Biol. 2019, 89, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Moyes, D.L.; Runglall, M.; Murciano, C.; Shen, C.; Nayar, D.; Thavaraj, S.; Kohli, A.; Islam, A.; Mora-Montes, H.; Challacombe, S.J.; et al. A biphasic innate immune MAPK response discriminates between the yeast and hyphal forms of Candida albicans in epithelial cells. Cell Host Microbe 2010, 8, 225–235. [Google Scholar] [CrossRef]
- Moyes, D.L.; Shen, C.; Murciano, C.; Runglall, M.; Richardson, J.P.; Arno, M.; Aldecoa-Otalora, E.; Naglik, J.R. Protection against epithelial damage during Candida albicans infection is mediated by PI3K/Akt and mammalian target of rapamycin signaling. J. Infect. Dis. 2014, 209, 1816–1826. [Google Scholar] [CrossRef] [PubMed]
- Swidergall, M.; Solis, N.V.; Lionakis, M.S.; Filler, S.G. EphA2 is an epithelial cell pattern recognition receptor for fungal β-glucans. Nat. Microbiol. 2018, 3, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.P.; Mogavero, S.; Moyes, D.L.; Blagojevic, M.; Krüger, T.; Verma, A.H.; Coleman, B.M.; De La Cruz Diaz, J.; Schulz, D.; Ponde, N.O.; et al. Processing of Candida albicans Ece1p Is critical for Candidalysin maturation and fungal virulence. mBio 2018, 9, e02178-17. [Google Scholar] [CrossRef]
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell. Microbiol. 2006, 8, 668–676. [Google Scholar] [CrossRef]
- Altmeier, S.; Toska, A.; Sparber, F.; Teijeira, A.; Halin, C.; LeibundGut-Landmann, S. IL-1 coordinates the neutrophil response to C. albicans in the oral mucosa. PLoS Pathog. 2016, 12, e1005882. [Google Scholar] [CrossRef] [PubMed]
- Hise, A.G.; Tomalka, J.; Ganesan, S.; Patel, K.; Hall, B.A.; Brown, G.D.; Fitzgerald, K.A. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe 2009, 5, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Conti, H.R.; Baker, O.; Freeman, A.F.; Jang, W.S.; Holland, S.M.; Li, R.A.; Edgerton, M.; Gaffen, S.L. New mechanism of oral immunity to mucosal candidiasis in hyper-IgE syndrome. Mucosal Immunol. 2011, 4, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Huppler, A.R.; Verma, A.H.; Conti, H.R.; Gaffen, S.L. Neutrophils do not express IL-17A in the context of acute oropharyngeal Candidiasis. Pathogens 2015, 4, 559–572. [Google Scholar] [CrossRef]
- Liu, L.; Okada, S.; Kong, X.-F.; Kreins, A.Y.; Cypowyj, S.; Abhyankar, A.; Toubiana, J.; Itan, Y.; Audry, M.; Nitschke, P.; et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J. Exp. Med. 2011, 208, 1635–1648. [Google Scholar] [CrossRef] [PubMed]
- Puel, A.; Cypowyj, S.; Bustamante, J.; Wright, J.F.; Liu, L.; Lim, H.K.; Migaud, M.; Israel, L.; Chrabieh, M.; Audry, M.; et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 2011, 332, 65–68. [Google Scholar] [CrossRef]
- Sobel, J.D. Pathogenesis and treatment of recurrent vulvovaginal candidiasis. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 1992, 14, S148–S153. [Google Scholar] [CrossRef]
- Sobel, J.D. Vaginitis. N. Eng. J. Med. 1997, 337, 1896–1903. [Google Scholar] [CrossRef]
- Gonçalves, B.; Ferreira, C.; Alves, C.T.; Henriques, M.; Azeredo, J.; Silva, S. Vulvovaginal candidiasis: Epidemiology, microbiology and risk factors. Crit. Rev. Microbiol. 2016, 42, 905–927. [Google Scholar] [CrossRef] [PubMed]
- Sobel, J.D. Vulvovaginal candidosis. Lancet 2007, 369, 1961–1971. [Google Scholar] [CrossRef]
- Giraldo, P.C.; Babula, O.; Gonçalves, A.K.; Linhares, I.M.; Amaral, R.L.; Ledger, W.J.; Witkin, S.S. Mannose-binding lectin gene polymorphism, vulvovaginal candidiasis, and bacterial vaginosis. Obstet. Gynecol. 2007, 109, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Hammad, N.M.; El Badawy, N.E.; Nasr, A.M.; Ghramh, H.A.; Al Kady, L.M. Mannose-binding lectin gene polymorphism and its association with susceptibility to recurrent vulvovaginal candidiasis. BioMed. Res. Int. 2018, 2018, 7648152. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, M.; Carvalho, A.; Cunha, C.; Plantinga, T.S.; van de Veerdonk, F.; Puccetti, M.; Galosi, C.; Joosten, L.A.; Dupont, B.; Kullberg, B.J.; et al. Association of a variable number tandem repeat in the NLRP3 gene in women with susceptibility to RVVC. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. European Soc. Clin. Microbiol. 2016, 35, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Rosentul, D.C.; Delsing, C.E.; Jaeger, M.; Plantinga, T.S.; Oosting, M.; Costantini, I.; Venselaar, H.; Joosten, L.A.; van der Meer, J.W.; Dupont, B.; et al. Gene polymorphisms in pattern recognition receptors and susceptibility to idiopathic recurrent vulvovaginal candidiasis. Front. Microbiol. 2014, 5, 483. [Google Scholar] [CrossRef] [PubMed]
- Fidel, P.L., Jr.; Lynch, M.E.; Sobel, J.D. Circulating CD4 and CD8 T cells have little impact on host defense against experimental vaginal candidiasis. Infect. Immun. 1995, 63, 2403–2408. [Google Scholar] [CrossRef] [PubMed]
- Leigh, J.E.; Barousse, M.; Swoboda, R.K.; Myers, T.; Hager, S.; Wolf, N.A.; Cutright, J.L.; Thompson, J.; Sobel, J.D.; Fidel, P.L., Jr. Candida-specific systemic cell-mediated immune reactivities in human immunodeficiency virus-positive persons with mucosal candidiasis. J. Infect. Dis. 2001, 183, 277–285. [Google Scholar] [CrossRef] [PubMed]
- White, M.H. Is vulvovaginal candidiasis an AIDS-related illness? Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 1996, 22, S124–S127. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Bernardis, F.; Graziani, S.; Tirelli, F.; Antonopoulou, S. Candida vaginitis: Virulence, host response and vaccine prospects. Med. Mycol. 2018, 56, 26–31. [Google Scholar] [CrossRef]
- Rosati, D.; Bruno, M.; Jaeger, M.; Ten Oever, J.; Netea, M.G. Recurrent vulvovaginal Candidiasis: An immunological perspective. Microorganisms 2020, 8, 144. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.; Peters, B.M.; Noverr, M.C.; Fidel, P.L., Jr. Novel mechanism behind the immunopathogenesis of vulvovaginal Candidiasis: “Neutrophil Anergy”. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.M.; Yano, J.; Noverr, M.C.; Fidel, P.L., Jr. Candida vaginitis: When opportunism knocks, the host responds. PLoS Pathog. 2014, 10, e1003965. [Google Scholar] [CrossRef]
- Fidel, P.L., Jr.; Barousse, M.; Espinosa, T.; Ficarra, M.; Sturtevant, J.; Martin, D.H.; Quayle, A.J.; Dunlap, K. An intravaginal live Candida challenge in humans leads to new hypotheses for the immunopathogenesis of vulvovaginal candidiasis. Infect. Immun. 2004, 72, 2939–2946. [Google Scholar] [CrossRef] [PubMed]
- Nomanbhoy, F.; Steele, C.; Yano, J.; Fidel, P.L., Jr. Vaginal and oral epithelial cell anti-Candida activity. Infect. Immun. 2002, 70, 7081–7088. [Google Scholar] [CrossRef] [PubMed]
- Soloviev, D.A.; Jawhara, S.; Fonzi, W.A. Regulation of innate immune response to Candida albicans infections by αMβ2-Pra1p interaction. Infect. Immun. 2011, 79, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.; Noverr, M.C.; Fidel, P.L., Jr. Vaginal heparan sulfate linked to neutrophil dysfunction in the acute inflammatory response associated with experimental vulvovaginal Candidiasis. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Hayashi, M.; Boutin, E.; Cunha, G.R.; Bernfield, M.; Trelstad, R.L. Hormonal modification of epithelial differentiation and expression of cell surface heparan sulfate proteoglycan in the mouse vaginal epithelium. An immunohistochemical and electron microscopic study. Lab. Investig. J. Tech. Methods Pathol. 1988, 58, 68–76. [Google Scholar]
- Hopke, A.; Brown, A.J.P.; Hall, R.A.; Wheeler, R.T. Dynamic fungal cell wall architecture in stress adaptation and immune evasion. Trends Microbiol. 2018, 26, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Ballou, E.R.; Avelar, G.M.; Childers, D.S.; Mackie, J.; Bain, J.M.; Wagener, J.; Kastora, S.L.; Panea, M.D.; Hardison, S.E.; Walker, L.A.; et al. Lactate signalling regulates fungal β-glucan masking and immune evasion. Nat. Microbiol. 2016, 2, 16238. [Google Scholar] [CrossRef]
- Ene, I.V.; Cheng, S.C.; Netea, M.G.; Brown, A.J. Growth of Candida albicans cells on the physiologically relevant carbon source lactate affects their recognition and phagocytosis by immune cells. Infect. Immun. 2013, 81, 238–248. [Google Scholar] [CrossRef]
- Sherrington, S.L.; Sorsby, E.; Mahtey, N.; Kumwenda, P.; Lenardon, M.D.; Brown, I.; Ballou, E.R.; MacCallum, D.M.; Hall, R.A. Adaptation of Candida albicans to environmental pH induces cell wall remodelling and enhances innate immune recognition. PLoS Pathog. 2017, 13, e1006403. [Google Scholar] [CrossRef] [PubMed]
- Cottier, F.; Sherrington, S.; Cockerill, S.; Del Olmo Toledo, V.; Kissane, S.; Tournu, H.; Orsini, L.; Palmer, G.E.; Pérez, J.C.; Hall, R.A. Remasking of Candida albicans β-glucan in response to environmental pH is regulated by quorum sensing. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Pericolini, E.; Perito, S.; Castagnoli, A.; Gabrielli, E.; Mencacci, A.; Blasi, E.; Vecchiarelli, A.; Wheeler, R.T. Epitope unmasking in vulvovaginal candidiasis is associated with hyphal growth and neutrophilic infiltration. PLoS ONE 2018, 13, e0201436. [Google Scholar] [CrossRef] [PubMed]
- Wisplinghoff, H.; Bischoff, T.; Tallent, S.M.; Seifert, H.; Wenzel, R.P.; Edmond, M.B. Nosocomial bloodstream infections in US hospitals: Analysis of 24,179 cases from a prospective nationwide surveillance study. Clin. Infect. Dis. 2004, 39, 309–317. [Google Scholar] [CrossRef]
- Spellberg, B.; Ibrahim, A.S.; Edwards, J.E., Jr.; Filler, S.G. Mice with disseminated candidiasis die of progressive sepsis. J. Infect. Dis. 2005, 192, 336–343. [Google Scholar] [CrossRef]
- Lionakis, M.S.; Fischer, B.G.; Lim, J.K.; Swamydas, M.; Wan, W.; Richard Lee, C.C.; Cohen, J.I.; Scheinberg, P.; Gao, J.L.; Murphy, P.M. Chemokine receptor Ccr1 drives neutrophil-mediated kidney immunopathology and mortality in invasive candidiasis. PLoS Pathog. 2012, 8, e1002865. [Google Scholar] [CrossRef] [PubMed]
- Swamydas, M.; Gao, J.L.; Break, T.J.; Johnson, M.D.; Jaeger, M.; Rodriguez, C.A.; Lim, J.K.; Green, N.M.; Collar, A.L.; Fischer, B.G.; et al. CXCR1-mediated neutrophil degranulation and fungal killing promote Candida clearance and host survival. Sci. Transl. Med. 2016, 8, 322ra310. [Google Scholar] [CrossRef] [PubMed]
- Jenne, C.N.; Kubes, P. Platelets in inflammation and infection. Platelets 2015, 26, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Arman, M.; Krauel, K.; Tilley, D.O.; Weber, C.; Cox, D.; Greinacher, A.; Kerrigan, S.W.; Watson, S.P. Amplification of bacteria-induced platelet activation is triggered by FcγRIIA, integrin αIIbβ3, and platelet factor 4. Blood 2014, 123, 3166–3174. [Google Scholar] [CrossRef]
- Seyoum, M.; Enawgaw, B.; Melku, M. Human blood platelets and viruses: Defense mechanism and role in the removal of viral pathogens. Thromb. J. 2018, 16, 16. [Google Scholar] [CrossRef] [PubMed]
- Ghuman, H.; Shepherd-Roberts, A.; Watson, S.; Zuidscherwoude, M.; Watson, S.P.; Voelz, K. Mucor circinelloides induces platelet aggregation through integrin αIIbβ3 and FcγRIIA. Platelets 2019, 30, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Eberl, C.; Speth, C.; Jacobsen, I.D.; Hermann, M.; Hagleitner, M.; Deshmukh, H.; Ammann, C.G.; Lass-Flörl, C.; Rambach, G. Candida: Platelet interaction and platelet activity in vitro. J. Innate Immun. 2019, 11, 52–62. [Google Scholar] [CrossRef]
- Watson, C.N.; Kerrigan, S.W.; Cox, D.; Henderson, I.R.; Watson, S.P.; Arman, M. Human platelet activation by Escherichia coli: Roles for FcγRIIA and integrin αIIbβ3. Platelets 2016, 27, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Bertling, A.; Niemann, S.; Uekötter, A.; Fegeler, W.; Lass-Flörl, C.; von Eiff, C.; Kehrel, B.E. Candida albicans and its metabolite gliotoxin inhibit platelet function via interaction with thiols. Thromb. Haemost. 2010, 104, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Kupfahl, C.; Ruppert, T.; Dietz, A.; Geginat, G.; Hof, H. Candida species fail to produce the immunosuppressive secondary metabolite gliotoxin in vitro. FEMS Yeast Res. 2007, 7, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Grubb, S.E.; Murdoch, C.; Sudbery, P.E.; Saville, S.P.; Lopez-Ribot, J.L.; Thornhill, M.H. Candida albicans-endothelial cell interactions: A key step in the pathogenesis of systemic candidiasis. Infect. Immun. 2008, 76, 4370–4377. [Google Scholar] [CrossRef]
- Phan, Q.T.; Fratti, R.A.; Prasadarao, N.V.; Edwards, J.E., Jr.; Filler, S.G. N-cadherin mediates endocytosis of Candida albicans by endothelial cells. J. Biol. Chem. 2005, 280, 10455–10461. [Google Scholar] [CrossRef] [PubMed]
- Phan, Q.T.; Belanger, P.H.; Filler, S.G. Role of hyphal formation in interactions of Candida albicans with endothelial cells. Infect. Immun. 2000, 68, 3485–3490. [Google Scholar] [CrossRef] [PubMed]
- Shintaku, T.; Glass, K.A.; Hirakawa, M.P.; Longley, S.J.; Bennett, R.J.; Bliss, J.M.; Shaw, S.K. Human endothelial cells internalize Candida parapsilosis via N-WASP-mediated endocytosis. Infect. Immun. 2013, 81, 2777–2787. [Google Scholar] [CrossRef] [PubMed]
- Gazendam, R.P.; van Hamme, J.L.; Tool, A.T.; van Houdt, M.; Verkuijlen, P.J.; Herbst, M.; Liese, J.G.; van de Veerdonk, F.L.; Roos, D.; van den Berg, T.K.; et al. Two independent killing mechanisms of Candida albicans by human neutrophils: Evidence from innate immunity defects. Blood 2014, 124, 590–597. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Family | PRR | PAMP | Expression | References |
|---|---|---|---|---|
| C-type lectin receptors | Dectin-1 | β-1,3-glucan | Dendritic cell, macrophage, neutrophil | [9,10,11,12] |
| Dectin-2 | α-mannan | Dendritic cell, macrophage | [13,14] | |
| Dectin-3 | α-mannan | Macrophage | [15] | |
| DC-SIGN | N-mannan | Dendritic cell | [16,17] | |
| Mincle | α-mannosyl residues | Macrophage | [18] | |
| Mannose receptor (MR) | N-mannan | Dendritic cell, macrophage | [17,19,20] | |
| Toll-like receptors | TLR2 | O-mannan | Dendritic cell, macrophage, neutrophil | [21,22] |
| TLR4 | O-mannan | Dendritic cell, macrophage, neutrophil | [19,21] | |
| TLR6 | Phospholipomannans | Macrophage | [23,24] | |
| Complement receptors | CR3 | β-glucans | Dendritic cell, neutrophil | [25] |
| Galectins | Galectin-3 | β-mannosides | Macrophage | [10] |
| Infection Type | Immune Sensing of Candida albicans | Inflammatory Response | References |
|---|---|---|---|
| Nail and Skin Infection | Dendritic cells | IL-12, IL-4, IL-1β, IL-6 | [30,35,70] |
| Oral Candidiasis | Oral epithelial cells | IL-1α/β, IL-8, G(M)-CSF, CCL20, CXCL2, β-defensins, S100A8/9 | [71,72] |
| Neutrophils | IL-1β, NET formation | [73] | |
| ILC3 | IL-17A/F | [74] | |
| γδ T-cells | IL-17A/F | [75] | |
| Natural Th17 cells | IL-17A/F | [76,77,78] | |
| Vaginal Candidiasis | Vaginal epithelial cells | IL-1β, S100A8 alarmin | [79,80] |
| Neutrophils | NET formation | [73] | |
| Candidemia | Neutrophils | ROS and NET formation, JAGN1 | [73,81] |
| Platelets | IL-8, platelet microbicidal protein | [82,83] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bojang, E.; Ghuman, H.; Kumwenda, P.; Hall, R.A. Immune Sensing of Candida albicans. J. Fungi 2021, 7, 119. https://doi.org/10.3390/jof7020119
Bojang E, Ghuman H, Kumwenda P, Hall RA. Immune Sensing of Candida albicans. Journal of Fungi. 2021; 7(2):119. https://doi.org/10.3390/jof7020119
Chicago/Turabian StyleBojang, Ebrima, Harlene Ghuman, Pizga Kumwenda, and Rebecca A. Hall. 2021. "Immune Sensing of Candida albicans" Journal of Fungi 7, no. 2: 119. https://doi.org/10.3390/jof7020119
APA StyleBojang, E., Ghuman, H., Kumwenda, P., & Hall, R. A. (2021). Immune Sensing of Candida albicans. Journal of Fungi, 7(2), 119. https://doi.org/10.3390/jof7020119