
Structural Insights into the Azole Resistance of the Candida albicans Darlington Strain Using Saccharomyces cerevisiae Lanosterol 14α-Demethylase as a Surrogate



 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Yeast Strains
2.2. Construction of ScLDM6×His Encoding Mutants
2.3. Drug Susceptibility Assays
2.4. Agarose Disk Diffusion Assays
Purification of ScErg11p6×His
2.5. Quantitation of ScLDM6×His Expression
2.6. Spectral Characterization of Purified ScLDM6×His
2.7. In Vitro Binding Affinity of Test Azoles to ScLDM6×His
2.8. Crystallization of ScLDM6×His, X-Ray Data Collection and Molecular Model Building
3. Results
3.1. The ScLDM Y140H Mutation Confers Azole Resistance to FLC, VCZ, VT-1161 but Not ITC and Posaconazole (PCZ)
3.2. The ScLDM Y140H and I471T Mutations Synergistically Reduce Drug Susceptibility to FLC and VCZ but Not VT-1161, ITC or PCZ
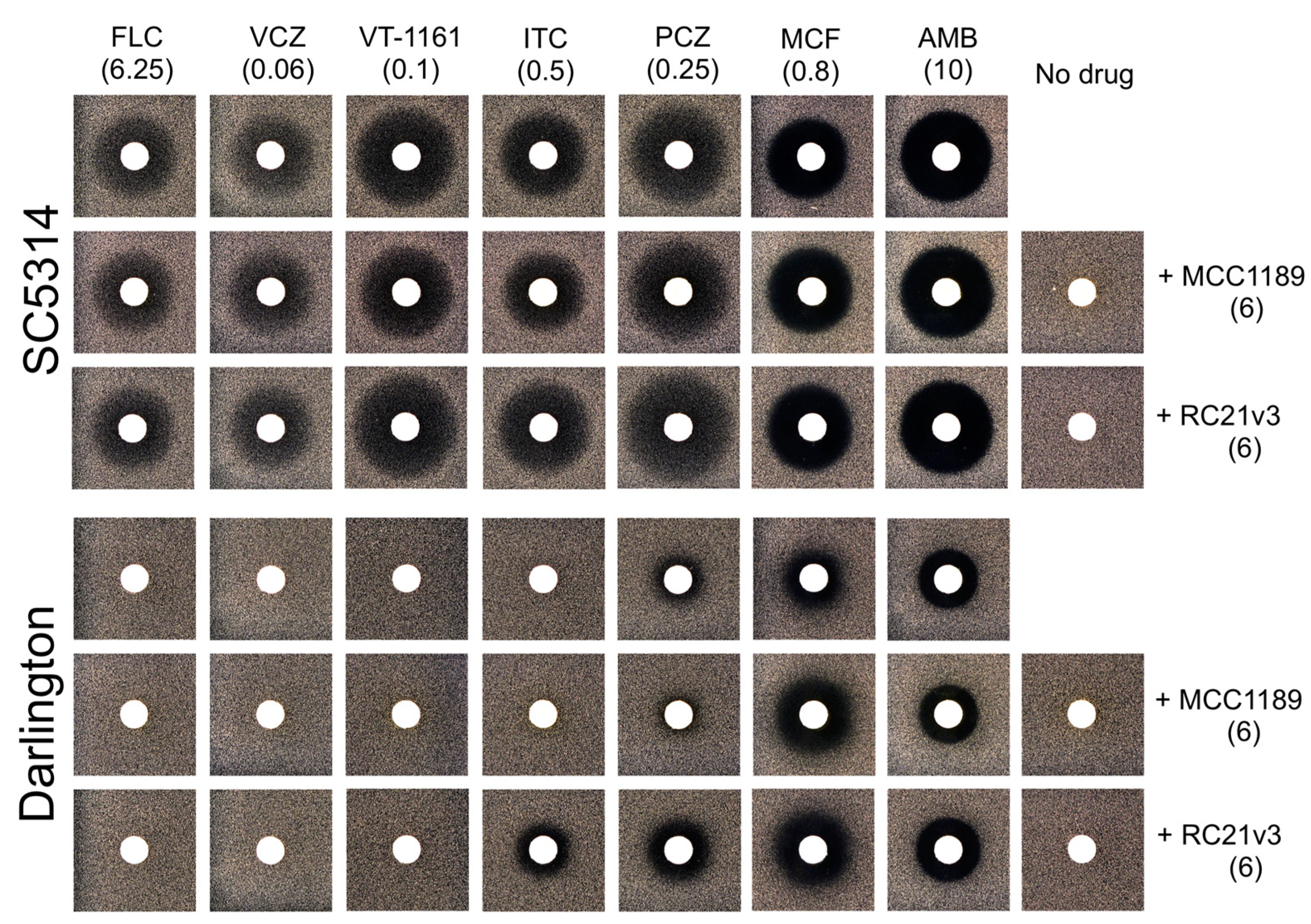
3.3. Role of Efflux Pumps in Azole Resistance in the Darlington Strain
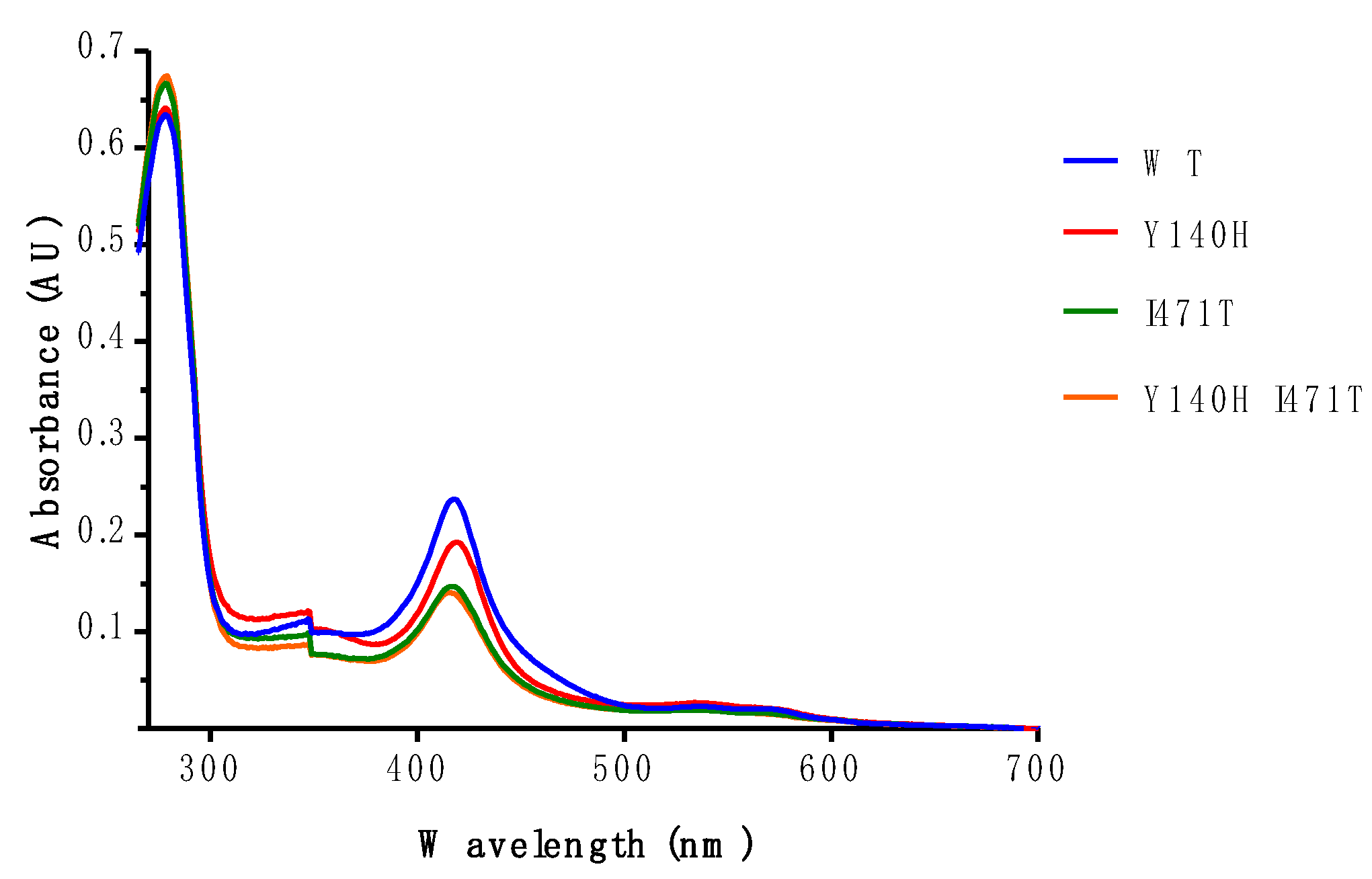
3.4. Electronic Properties of the Heme of Affinity Purified ScLDM6×His I471T and ScLDM6×His Y140H I471T
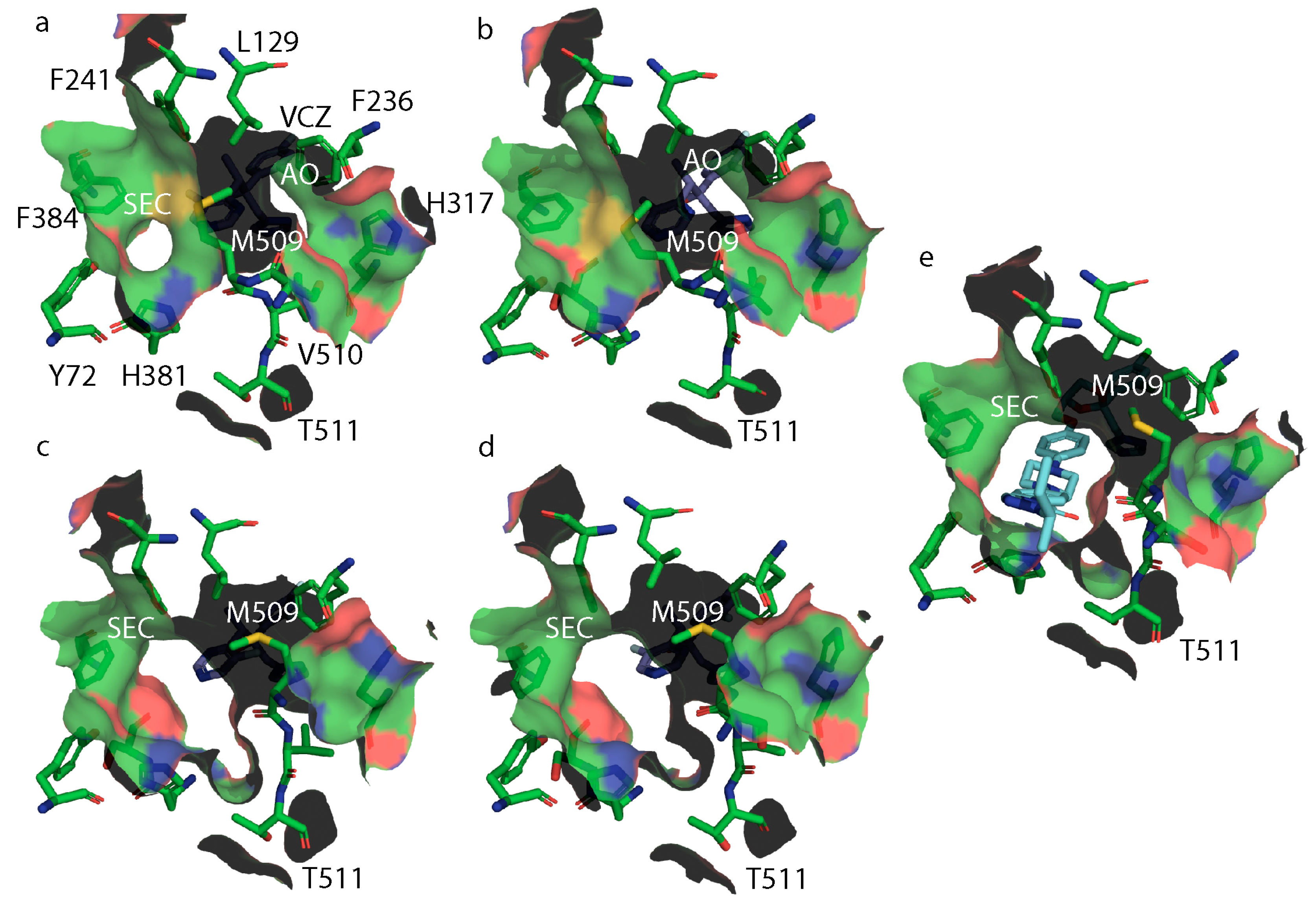
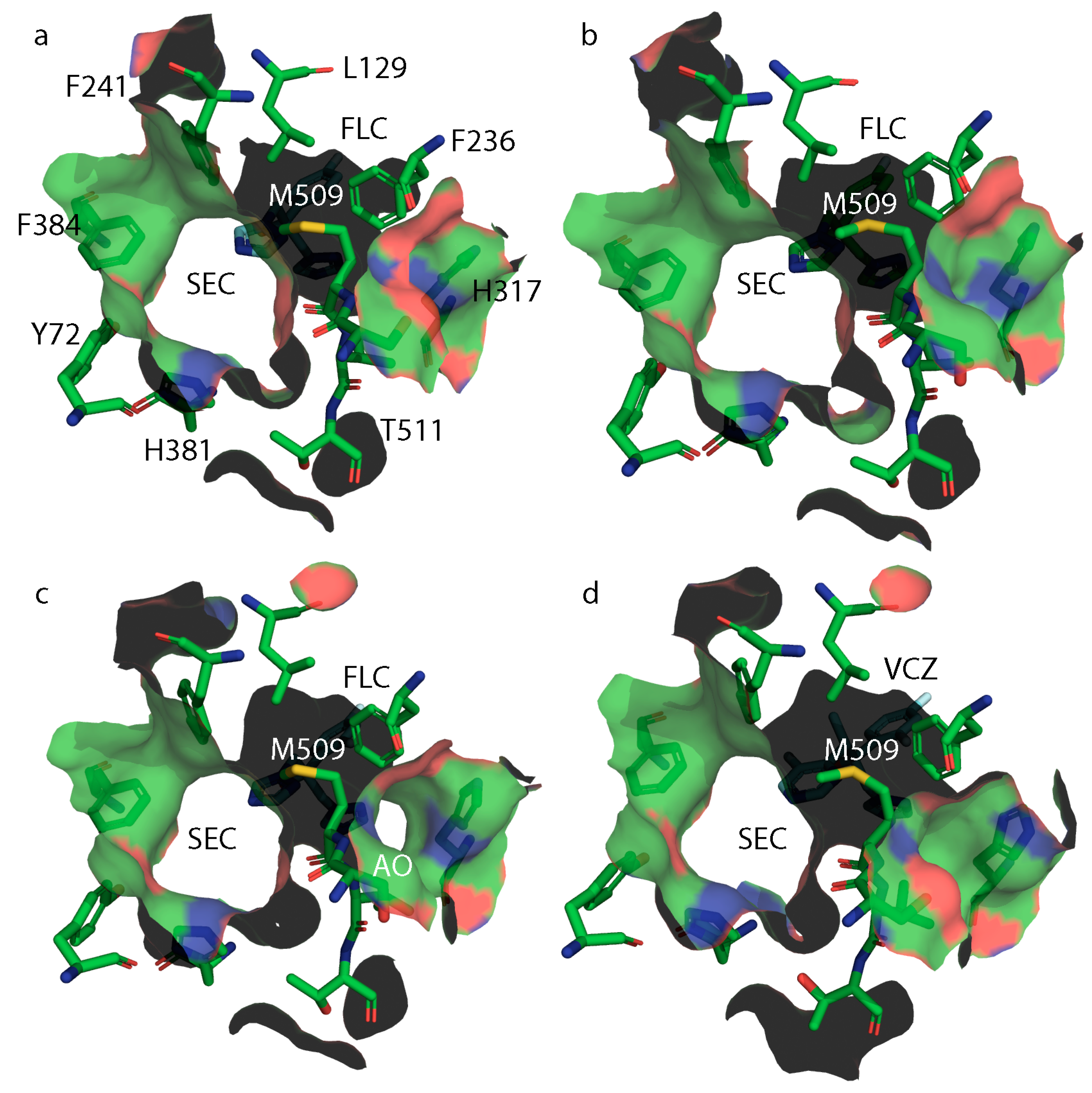
3.5. X-ray Crystal Structures of ScLDM6×His Y140H, ScLDM6×His I471T and ScLDM6×His Y140H I471T in Complex with Azole Drugs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brown, G.D.; Denning, D.W.; Gow, N.A.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4, 165rv13. [Google Scholar] [CrossRef] [Green Version]
- Becher, R.; Wirsel, S.G.R. Fungal cytochrome P450 sterol 14α-demethylase (CYP51) and azole resistance in plant and human pathogens. Appl. Microbiol. Biotechnol. 2012, 95, 825–840. [Google Scholar] [CrossRef]
- Price, C.L.; Parker, J.E.; Warrilow, A.G.; Kelly, D.E.; Kelly, S.L. Azole fungicides—Understanding resistance mechanisms in agricultural fungal pathogens. Pest Manag. Sci. 2015, 71, 1054–1058. [Google Scholar] [CrossRef]
- Schulze, J.; Sonnenborn, U. Yeasts in the gut: From commensals to infectious agents. Dtsch. Arztebl. Int. 2009, 106, 837–842. [Google Scholar] [PubMed]
- Mukherjee, P.K.; Sendid, B.; Hoarau, G.; Colombel, J.-F.; Poulain, D.; Ghannoum, M.A. Mycobiota in gastrointestinal diseases. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Clark, T.A.; Hajjeh, R.A. Recent trends in the epidemiology of invasive mycoses. Curr. Opin. Infect. Dis. 2002, 15, 569–574. [Google Scholar] [CrossRef]
- Maertens, J.; Vrebos, M.; Boogaerts, M. Assessing risk factors for systemic fungal infections. Eur. J. Cancer Care 2001, 10, 56–62. [Google Scholar] [CrossRef]
- Morgan, J.; Wannemuehler, K.; Marr, K.A.; Hadley, S.; Kontoyiannis, D.P.; Walsh, T.J.; Fridkin, S.K.; Pappas, P.G.; Warnock, D.W. Incidence of invasive aspergillosis following hematopoietic stem cell and solid organ transplantation: Interim results of a prospective multicenter surveillance program. Med. Mycol. 2005, 43, 49–58. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Sudbery, P. Candida albicans, a major human fungal pathogen. J. Microbiol. 2011, 49, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Pfaller, M.A.; Diekema, D. Epidemiology of invasive candidiasis: A persistent public health problem. Clin. Microbiol. Rev. 2007, 20, 133–163. [Google Scholar] [CrossRef] [Green Version]
- Daum, G.; Lees, N.D.; Bard, M.; Dickson, R. Biochemistry, cell biology and molecular biology of lipids of Saccharomyces cerevisiae. Yeast 1998, 14, 1471–1510. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Esquivel, B.D.; White, T.C. Overexpression or deletion of ergosterol biosynthesis genes alters doubling time, response to stress agents, and drug susceptibility in Saccharomyces cerevisiae. Mbio 2018, 9, e01291-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorda, T.; Puig, S. Regulation of ergosterol biosynthesis in Saccharomyces cerevisiae. Genes 2020, 11, 795. [Google Scholar] [CrossRef]
- Wagner, C.; Graninger, W.; Presterl, E.; Joukhadar, C. The echinocandins: Comparison of their pharmaco-kinetics, pharmacodynamics and clinical applications. Pharmacology 2006, 78, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Denning, D.W. Echinocandins: A new class of antifungal. J. Antimicrob. Chemother. 2002, 49, 889–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denning, D. Echinocandins and pneumocandins—A new antifungal class with a novel mode of action. J. Antimicrob. Chemother. 1997, 40, 611–614. [Google Scholar] [CrossRef] [PubMed]
- Wood, A.J.; Como, J.A.; Dismukes, W.E. Oral azole drugs as systemic antifungal therapy. N. Engl. J. Med. 1994, 330, 263–272. [Google Scholar] [CrossRef]
- E Verweij, P.; Kema, G.H.; Zwaan, B.; Melchers, W.J. Triazole fungicides and the selection of resistance to medical triazoles in the opportunistic mould Aspergillus fumigatus. Pest Manag. Sci. 2013, 69, 165–170. [Google Scholar] [CrossRef]
- Richardson, M. Changing patterns and trends in systemic fungal infections. J. Antimicrob. Chemother. 2005, 56, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Sims, C.R.; Ostrosky-Zeichner, L.; Rex, J.H. Invasive candidiasis in immunocompromised hospitalized patients. Arch. Med. Res. 2005, 36, 660–671. [Google Scholar] [CrossRef]
- Morio, F.; Loge, C.; Besse, B.; Hennequin, C.; Le Pape, P. Screening for amino acid substitutions in the Candida albicans Erg11 protein of azole-susceptible and azole-resistant clinical isolates: New substitutions and a review of the literature. Diagn. Microbiol. Infect. Dis. 2010, 66, 373–384. [Google Scholar] [CrossRef]
- Chau, A.S.; Mendrick, C.A.; Sabatelli, F.J.; Loebenberg, D.; McNicholas, P.M. Application of real-time quantitative PCR to molecular analysis of Candida albicans strains exhibiting reduced susceptibility to azoles. Antimicrob. Agents Chemother. 2004, 48, 2124–2131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, G.H.; da Silva Ferreira, M.E.; dos Reis Marques, E.; Savoldi, M.; Perlin, D.; Park, S.; Godoy Martinez, P.C.; Goldman, M.H.; Colombo, A.L. Evaluation of fluconazole resistance mechanisms in Candida albicans clinical isolates from HIV-infected patients in Brazil. Diagn. Microbiol. Infect. Dis. 2004, 50, 25–32. [Google Scholar] [CrossRef]
- Perea, S.; Lopez-Ribot, J.L.; Kirkpatrick, W.R.; McAtee, R.K.; Santillan, R.A.; Martinez, M.; Calabrese, D.; Sanglard, D.; Patterson, T.F. Prevalence of molecular mechanisms of resistance to azole antifungal agents in Candida albicans strains displaying high-level fluconazole resistance isolated from human immunodeficiency virus infected patients. Antimicrob. Agents Chemother. 2001, 45, 2676–2684. [Google Scholar] [CrossRef] [Green Version]
- Favre, B.; Ryder, N.S.; Didmon, M. Multiple amino acid substitutions in lanosterol 14α-demethylase contribute to azole resistance in Candida albicans. Microbiology 1999, 145, 2715–2725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakeya, H.; Miyazaki, Y.; Miyazaki, H.; Nyswaner, K.; Grimberg, B.; Bennett, J.E. Genetic analysis of azole resistance in the darlington strain of Candida albicans. Antimicrob. Agents Chemother. 2000, 44, 2985–2990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marichal, P.; Koymans, L.; Willemsens, S.; Bellens, D.; Verhasselt, P.; Luyten, W.; Borgers, M.; Ramaekers, F.C.S.; Odds, F.C.; Bossche, H.V. Contribution of mutations in the cytochrome P450 14α-demethylase (Erg11p, Cyp51p) to azole resistance in Candida albicans. Microbiology 1999, 145, 2701–2713. [Google Scholar] [CrossRef] [Green Version]
- Sanglard, D.; Ischer, F.; Koymans, L.; Bille, J. Amino acid substitutions in the cytochrome P-450 lanosterol 14alpha-demethylase (CYP51A1) from azole-resistant Candida albicans clinical isolates contribute to resistance to azole antifungal agents. Antimicrob. Agents Chemother. 1998, 42, 241–253. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Chen, L.; Li, C. Susceptibility of clinical isolates of Candida species to fluconazole and detection of Candida albicans ERG11 mutations. J. Antimicrob. Chemother. 2008, 61, 798–804. [Google Scholar] [CrossRef] [Green Version]
- Flowers, S.A.; Colón, B.; Whaley, S.G.; Schuler, M.A.; Rogers, P.D. Contribution of clinically derived mutations in ERG11 to azole resistance in Candida albicans. Antimicrob. Agents Chemother. 2014, 59, 450–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, S.L.; Lamb, D.C.; Kelly, D.E. Y132H substitution in Candida albicans sterol 14alpha-demethylase confers fluconazole resistance by preventing binding to haem. FEMS Microbiol. Lett. 1999, 180, 171–175. [Google Scholar] [PubMed] [Green Version]
- Rosam, K.; Monk, B.C.; Lackner, M. Sterol 14α-demethylase ligand-binding pocket-mediated acquired and intrinsic azole resistance in fungal pathogens. J. Fungi. 2020, 7, 1. [Google Scholar] [CrossRef]
- Warnock, D.; Johnson, E.; Richardson, M.; Vickers, C. Modified response to ketoconazole of Candida albicans from a treatment failure. Lancet 1983, 321, 642–643. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Geber, A.; Miyazaki, H.; Falconer, D.; Parkinson, T.; Hitchcock, C.; Grimberg, B.; Nyswaner, K.; Bennett, J.E. Cloning, sequencing, expression and allelic sequence diversity of ERG3 (C-5 sterol desaturase gene) in Candida albicans. Gene 1999, 236, 43–51. [Google Scholar] [CrossRef]
- Howell, S.A.; Mallet, A.I.; Noble, W.C. A comparison of the sterol content of multiple isolates of the Candida albicans Darlington strain with other clinically azole-sensitive and resistant strains. J. Appl. Bacteriol. 1990, 69, 692–696. [Google Scholar] [CrossRef]
- Albertson, G.D.; Niimi, M.; Cannon, R.D.; Jenkinson, H.F. Multiple efflux mechanisms are involved in Candida albicans fluconazole resistance. Antimicrob. Agents Chemother. 1996, 40, 2835–2841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flowers, S.A.; Barker, K.S.; Berkow, E.L.; Toner, G.; Chadwick, S.G.; Gygax, S.E.; Morschhäuser, J.; Rogers, P.D. Gain-of-function mutations in UPC2 are a frequent cause of ERG11 upregulation in azole-resistant clinical isolates of Candida albicans. Eukaryot. Cell 2012, 11, 1289–1299. [Google Scholar] [CrossRef] [Green Version]
- Morio, F.; Pagniez, F.; Besse, M.; Gay-andrieu, F.; Miegeville, M.; Le Pape, P. Deciphering azole resistance mechanisms with a focus on transcription factor-encoding genes TAC1, MRR1 and UPC2 in a set of fluconazole-resistant clinical isolates of Candida albicans. Int. J. Antimicrob. Agents 2013, 42, 410–415. [Google Scholar] [CrossRef]
- Selmecki, A.; Forche, A.; Berman, J. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science 2006, 313, 367–370. [Google Scholar] [CrossRef] [Green Version]
- Warrilow, A.G.S.; Martel, C.M.; Parker, J.E.; Melo, N.; Lamb, D.C.; Nes, W.D.; Kelly, D.E.; Kelly, S.L. Azole binding properties of Candida albicans sterol 14-α demethylase (CaCYP51). Antimicrob. Agents Chemother. 2010, 54, 4235–4245. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, H.K.; Medeiros, C.-A.; Craig, I.R.; Stammler, G. Sensitivity of Phakopsora pachyrhizi towards quinone-outside-inhibitors and demethylation-inhibitors, and corresponding resistance mechanisms. Pest Manag. Sci. 2014, 70, 378–388. [Google Scholar] [CrossRef]
- Sagatova, A.; Keniya, M.V.; Wilson, R.K.; Monk, B.C.; Tyndall, J.D.A. Structural insights into binding of the antifungal drug fluconazole to Saccharomyces cerevisiae lanosterol 14α-demethylase. Antimicrob. Agents Chemother. 2015, 59, 4982–4989. [Google Scholar] [CrossRef] [Green Version]
- Sagatova, A.; Keniya, M.V.; Wilson, R.K.; Sabherwal, M.; Tyndall, J.D.A.; Monk, B.C. Triazole resistance mediated by mutations of a conserved active site tyrosine in fungal lanosterol 14α-demethylase. Sci. Rep. 2016, 6, 26213. [Google Scholar] [CrossRef] [Green Version]
- Monk, B.C.; Keniya, M.V.; Sabherwal, M.; Wilson, R.K.; Graham, D.O.; Hassan, H.F.; Chen, D.; Tyndall, J.D.A. Azole resistance reduces susceptibility to the tetrazole antifungal VT-1161. Antimicrob. Agents Chemother. 2019, 63, 2114–2118. [Google Scholar] [CrossRef] [Green Version]
- Keniya, M.V.; Sabherwal, M.; Wilson, R.K.; Woods, M.A.; Sagatova, A.A.; Tyndall, J.D.A.; Monk, B.C. Crystal structures of full-length lanosterol 14alpha-demethylases of prominent fungal pathogens Candida albicans and Candida glabrata provide tools for antifungal discovery. Antimicrob. Agents Chemother. 2018, 62, e01134-18. [Google Scholar] [PubMed] [Green Version]
- Hargrove, T.Y.; Wawrzak, Z.; Fisher, P.M.; Child, S.A.; Nes, W.D.; Guengerich, F.P.; Waterman, M.R.; Lepesheva, G.I. Binding of a physiological substrate causes large-scale conformational reorganization in cytochrome P450 51. J. Biol. Chem. 2018, 293, 19344–19353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hargrove, T.Y.; Wawrzak, Z.; Guengerich, F.P.; Lepesheva, G.I. A requirement for an active proton delivery network supports a compound I-mediated C-C bond cleavage in CYP51 catalysis. J. Biol. Chem. 2020, 295, 9998–10007. [Google Scholar] [CrossRef]
- Lamping, E.; Monk, B.C.; Niimi, K.; Holmes, A.R.; Tsao, S.; Tanabe, K.; Niimi, M.; Uehara, Y.; Cannon, R.D. Characterization of three classes of membrane proteins involved in fungal azole resistance by functional hyperexpression in Saccharomyces cerevisiae. Eukaryot. Cell 2007, 6, 1150–1165. [Google Scholar] [CrossRef] [Green Version]
- Keniya, M.V.; Fleischer, E.; Klinger, A.; Cannon, R.D.; Monk, B.C. Inhibitors of the Candida albicans major facilitator superfamily transporter Mdr1p responsible for fluconazole resistance. PLoS ONE 2015, 10, e0126350. [Google Scholar] [CrossRef] [PubMed]
- Niimi, K.; Harding, D.R.K.; Holmes, A.R.; Lamping, E.; Niimi, M.; Tyndall, J.D.A.; Cannon, R.D.; Monk, B.C. Specific interactions between the Candida albicans ABC transporter Cdr1p ectodomain and a D-octapeptide derivative inhibitor. Mol. Microbiol. 2012, 85, 747–767. [Google Scholar] [CrossRef] [Green Version]
- Keniya, M.V.; Ruma, Y.N.; Tyndall, J.D.A.; Monk, B.C. Heterologous expression of full-length lanosterol 14α-demethylases of prominent fungal pathogens Candida albicans and Candida glabrata provides tools for antifungal discovery. Antimicrob. Agents Chemother. 2018, 62, e01131-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monk, B.C.; Tomasiak, T.M.; Keniya, M.V.; Huschmann, F.U.; Tyndall, J.D.; O’Connell, J.D., 3rd; Cannon, R.D.; McDonald, J.G.; Rodriguez, A.; Finer-Moore, J.S.; et al. Architecture of a single membrane spanning cytochrome P450 suggests constraints that orient the catalytic domain relative to a bilayer. Proc. Natl. Acad. Sci. USA 2014, 111, 3865–3870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Guengerich, F.P.; Martin, M.V.; Sohl, C.D.; Cheng, Q. Measurement of cytochrome P450 and NADPH–cytochrome P450 reductase. Nat. Protoc. 2009, 4, 1245–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battye, T.G.; Kontogiannis, L.; Johnson, O.; Powell, H.R.; Leslie, A.G. iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. Phenix: A comprehensive python-based system for macromolecular structure solution. Acta Cryst. D Biol. Cryst. 2019, 66, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.; Cowtan, K.D. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Hoekstra, W.J.; Garvey, E.P.; Moore, W.R.; Rafferty, S.W.; Yates, C.M.; Schotzinger, R.J. Design and optimization of highly-selective fungal CYP51 inhibitors. Bioorganic Med. Chem. Lett. 2014, 24, 3455–3458. [Google Scholar] [CrossRef]
- Warrilow, A.G.S.; Parker, J.E.; Price, C.; Nes, W.D.; Garvey, E.P.; Hoekstra, W.J.; Schotzinger, R.J.; Kelly, D.E.; Kelly, S.L. The investigational drug VT-1129 is a highly potent inhibitor of Cryptococcus species CYP51 but only weakly inhibits the human enzyme. Antimicrob. Agents Chemother. 2016, 60, 4530–4538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hargrove, T.Y.; Wawrzak, Z.; Lamb, D.C.; Guengerich, F.P.; Lepesheva, G.I. Structure-functional characterization of cytochrome P450 sterol 14alpha-demethylase (CYP51B) from Aspergillus fumigatus and molecular basis for the development of antifungal drugs. J. Biol. Chem. 2015, 290, 23916–23934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, J.; Brand, S.; Voice, M.; Caballero, I.; Calvo, D.; Read, K.D. Development of a fluorescence-based Trypanosoma cruzi CYP51 inhibition assay for effective compound triaging in drug discovery programmes for Chagas Disease. PLoS Negl. Trop. Dis. 2015, 9, e0004014. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Guerra, T.M.; Mellado, E.; Cuenca-Estrella, M.; Rodriguez-Tudela, J.L. A point mutation in the 14α-sterol demethylase gene cyp51A contributes to itraconazole resistance in Aspergillus fumigatus. Antimicrob. Agents Chemother. 2003, 47, 1120–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snelders, E.; Karawajczyk, A.; Schaftenaar, G.; Verweij, P.; Melchers, W.J.G. Azole resistance profile of amino acid changes in Aspergillus fumigatus CYP51A based on protein homology modeling. Antimicrob. Agents Chemother. 2010, 54, 2425–2430. [Google Scholar] [CrossRef] [Green Version]
- McPhillips, T.M.; McPhillips, S.E.; Chiu, H.J.; Cohen, A.E.; Deacon, A.M.; Ellis, P.J.; Garman, E.; Gonzalez, A.; Sauter, N.K.; Phizackerley, R.P.; et al. Blu-Ice and the distributed control system: Software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Rad. 2002, 9, 401–406. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | MIC80 | ||||||
|---|---|---|---|---|---|---|---|
| FLC (µM) | VCZ (nM) | VT-1161 (nM) | ITC (nM) | PCZ (nM) | MCF (nM) | AMB (μM) | |
| AD2Δ | 1.33 (0.06) | 37.50 (0.50) | 28.75 (3.92) | 73.50 (0.65) | 160.40 (21.40) | 173.80 (11.07) | 2.18 (0.27) |
| AD3Δ | 3.64 (0.37) | 93.33 (5.83) | 75.40 (2.91) | 143.40 (0.46) | 423.30 (29.91) | 145.80 (17.05) | 2.34 (0.28) |
| Y140H | 7.40 (0.48) | 163.70 (5.24) | 115.60 (10.59) | 112.10 (3.03) | 330.00 (15.10) | 166.00 (12.80) | 2.19 (0.28) |
| I471T | 1.88 (0.60) | 74.00 (0.58) | 34.63 (0.55) | 102.10 (4.87) | 269.50 (22.86) | 164.40 (13.03) | 2.26 (0.34) |
| Y140H + I471T | 11.50 (0.70) | 249.60 (3.25) | 67.60 (1.63) | 107.90 (1.07) | 264.80 (23.13) | 162.40 (13.03) | 2.14 (0.25) |
| MIC80/Relative Amount of ScLDM6×His | |||||
|---|---|---|---|---|---|
| Strain (Relative Expression) | FLC (µM) | VCZ (nM) | VT-1161 (nM) | ITC (nM) | PCZ (nM) |
| AD3Δ (1) | 3.64 | 93.3 | 75.4 | 143 | 423 |
| Y140H (0.74) | 10 (2.8×) | 221 (2.4×) | 157 (2.1×) | 151 (1.1×) | 445 (1.1×) |
| I471T (0.51) | 3.69 (1.0×) | 145 (1.6×) | 67.9 (0.90×) | 200 (1.4×) | 527 (1.3×) |
| Y140H+I471T (0.41) | 28.0 (7.7×) | 609 (6.5×) | 165 (2.2×) | 263 (1.8×) | 646 (1.5×) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Graham, D.O.; Wilson, R.K.; Ruma, Y.N.; Keniya, M.V.; Tyndall, J.D.A.; Monk, B.C. Structural Insights into the Azole Resistance of the Candida albicans Darlington Strain Using Saccharomyces cerevisiae Lanosterol 14α-Demethylase as a Surrogate. J. Fungi 2021, 7, 897. https://doi.org/10.3390/jof7110897
Graham DO, Wilson RK, Ruma YN, Keniya MV, Tyndall JDA, Monk BC. Structural Insights into the Azole Resistance of the Candida albicans Darlington Strain Using Saccharomyces cerevisiae Lanosterol 14α-Demethylase as a Surrogate. Journal of Fungi. 2021; 7(11):897. https://doi.org/10.3390/jof7110897
Chicago/Turabian StyleGraham, Danyon O., Rajni K. Wilson, Yasmeen N. Ruma, Mikhail V. Keniya, Joel D. A. Tyndall, and Brian C. Monk. 2021. "Structural Insights into the Azole Resistance of the Candida albicans Darlington Strain Using Saccharomyces cerevisiae Lanosterol 14α-Demethylase as a Surrogate" Journal of Fungi 7, no. 11: 897. https://doi.org/10.3390/jof7110897
APA StyleGraham, D. O., Wilson, R. K., Ruma, Y. N., Keniya, M. V., Tyndall, J. D. A., & Monk, B. C. (2021). Structural Insights into the Azole Resistance of the Candida albicans Darlington Strain Using Saccharomyces cerevisiae Lanosterol 14α-Demethylase as a Surrogate. Journal of Fungi, 7(11), 897. https://doi.org/10.3390/jof7110897