
Regulation of Pkc1 Hyper-Phosphorylation by Genotoxic Stress

 , ,
, , 
Abstract
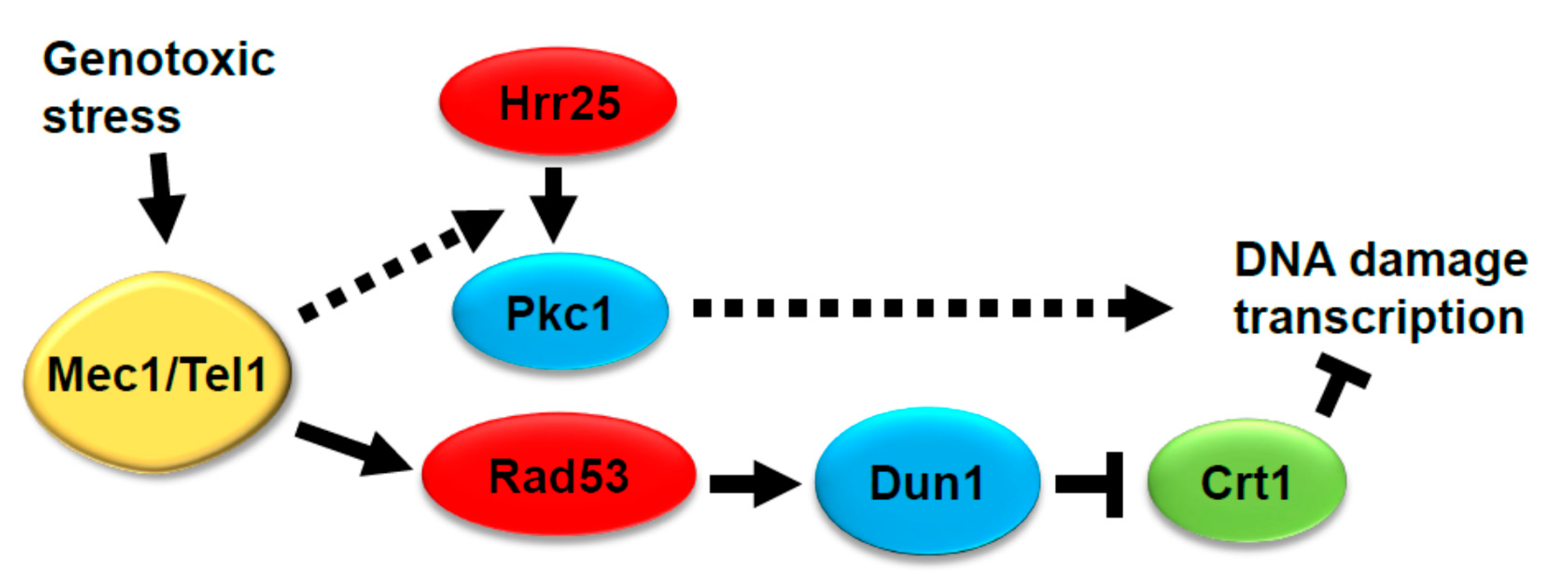
1. Introduction
2. Materials and Methods
2.1. Strains, Growth Conditions, and Transformations
2.2. Chromosomal Deletions and Strain Construction
2.3. Plasmid Construction and Mutagenesis
2.4. Protein Extraction
2.5. β-galactosidase Measurements
2.6. Dephosphorylation Assay
2.7. Co-immunoprecipitation
2.8. SDS-PAGE Electrophoresis and Immunoblot Analysis
2.9. Mass Spectrometric Analysis of Pkc1 Co-Immunoprecipitates
2.10. Mass Spectrometric Analysis of Pkc1 Phospho-Sites
2.11. Notes on Reproducibility
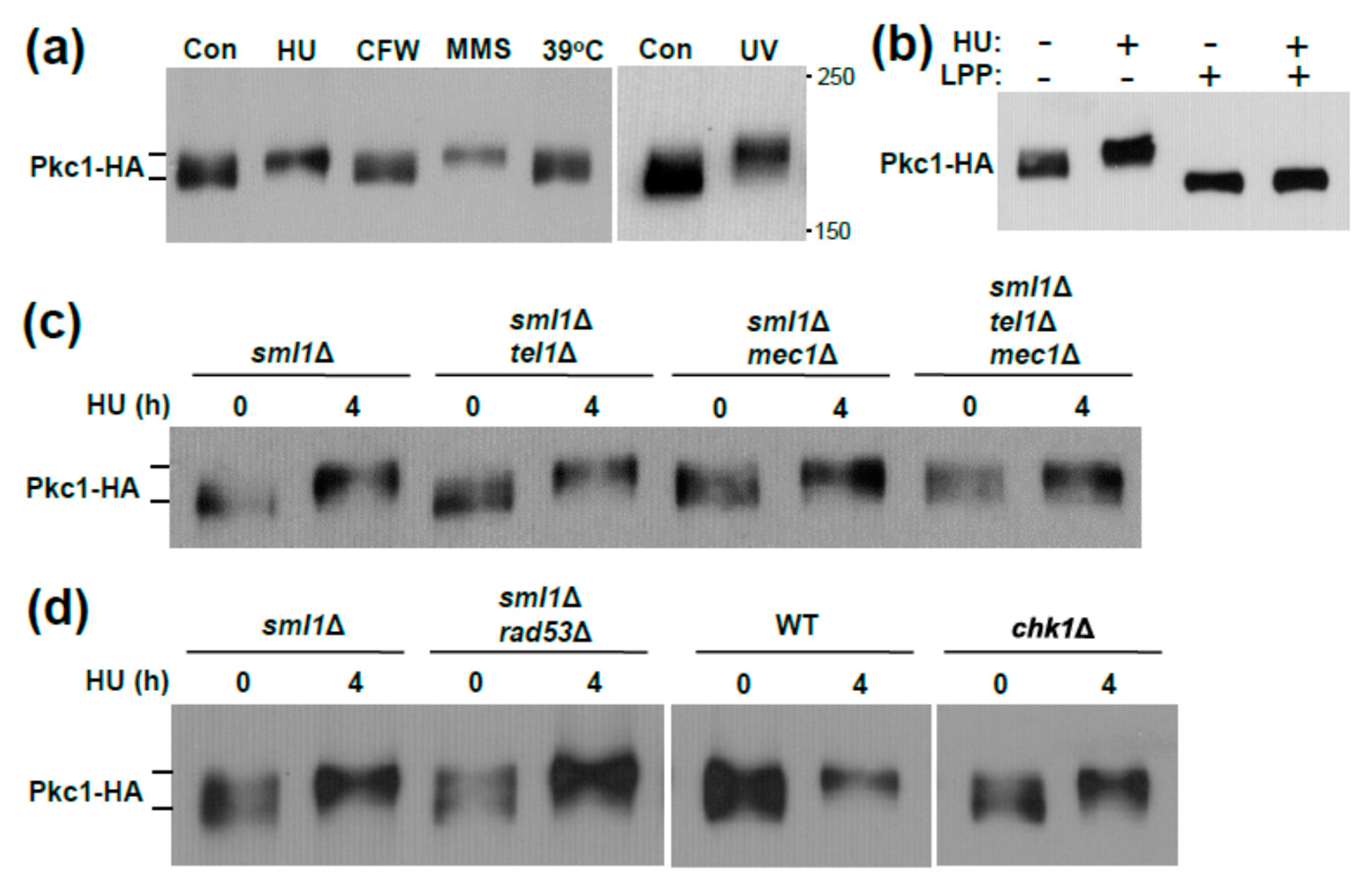
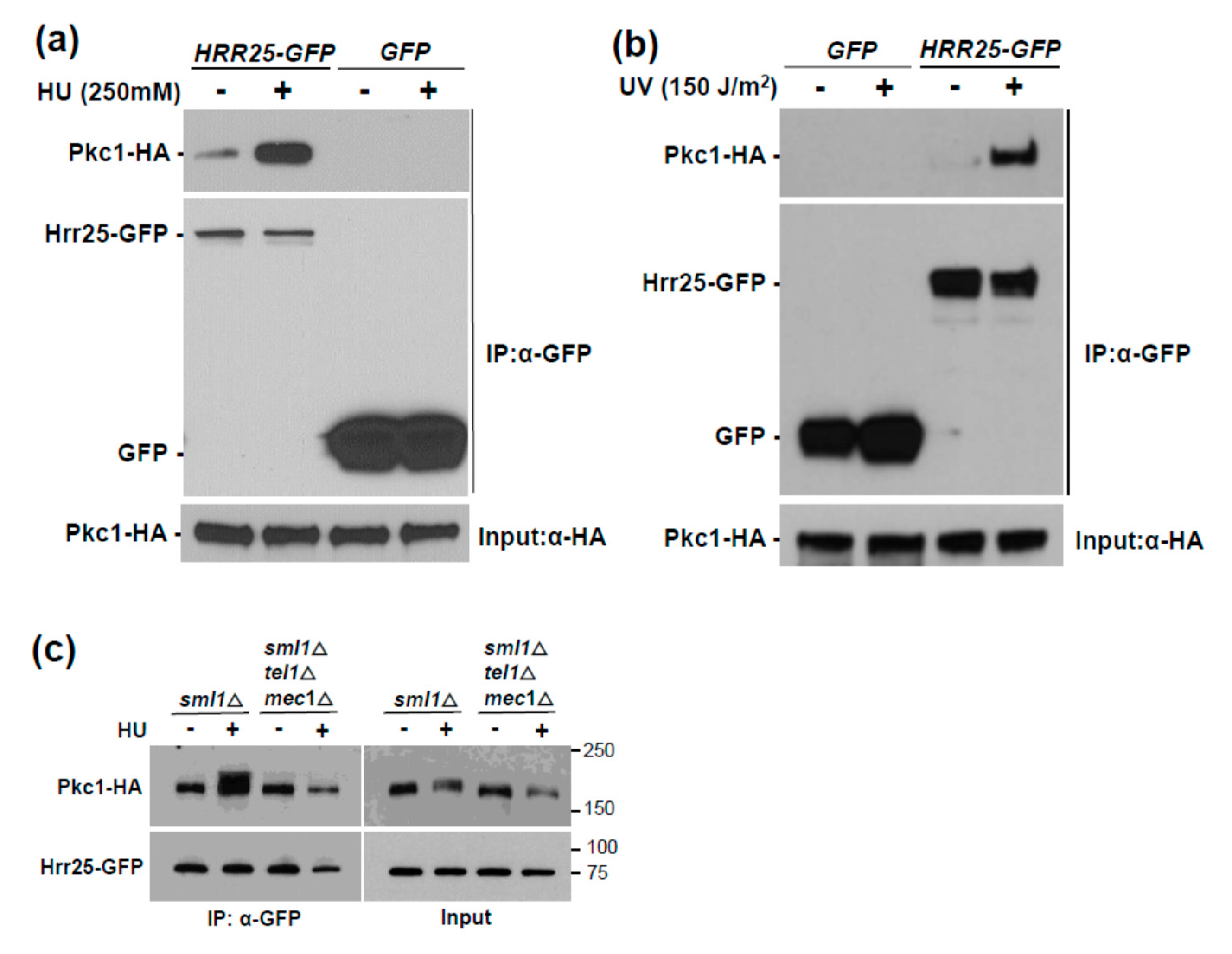
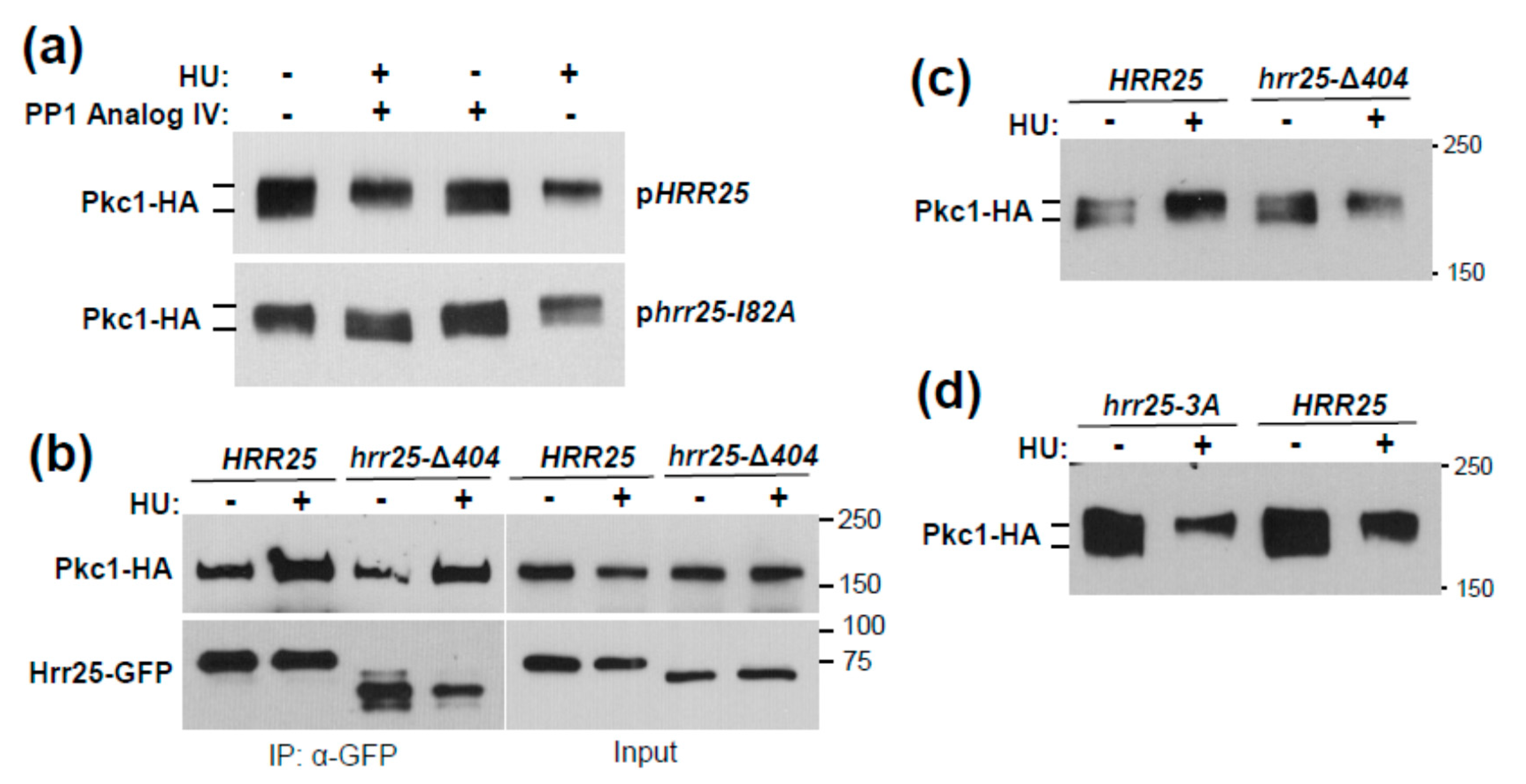
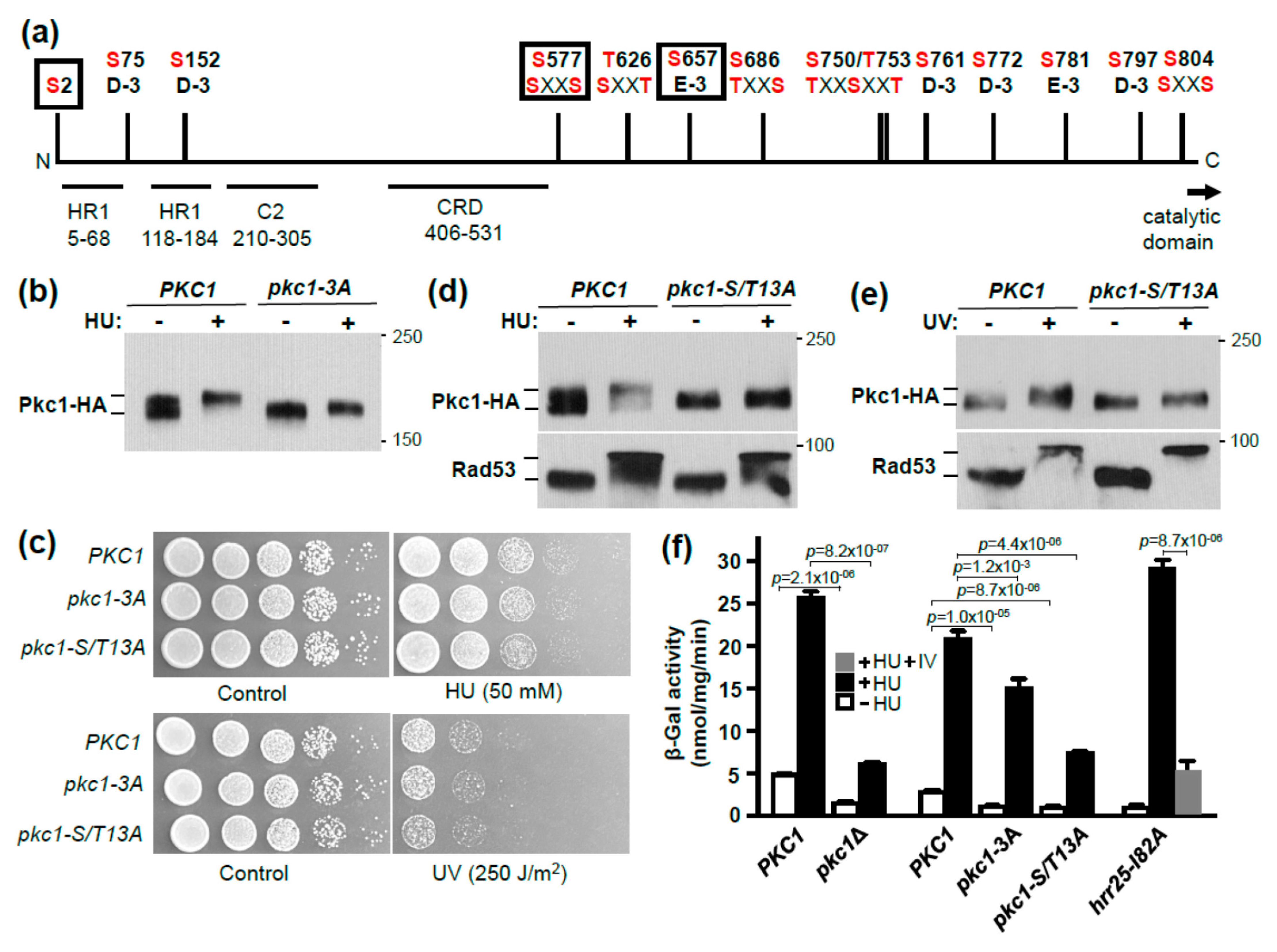
3. Results and Discussion
3.1. Genotoxic Stress Induces Hrr25 Association with Pkc1
3.2. Identification of Pkc1 Phospho-Sites in Response to HU Treatment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klis, F.M.; Mol, P.; Hellingwerf, K.; Brul, S. Dynamics of cell wall structure in Saccharomyces cerevisiae. FEMS Microbiol. Rev. 2002, 26, 239–256. [Google Scholar] [CrossRef]
- Levin, D.E. Cell Wall Integrity Signaling in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2005, 69, 262–291. [Google Scholar] [CrossRef]
- Levin, D.E. Regulation of cell wall biosynthesis in Saccharomyces cerevisiae: The cell wall integrity signaling pathway. Genetics 2011, 189, 1145–1175. [Google Scholar] [CrossRef]
- Lesage, G.; Bussey, H. Cell Wall Assembly in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2006, 70, 317–343. [Google Scholar] [CrossRef] [PubMed]
- Jung, U.S.; Levin, D. Genome-wide analysis of gene expression regulated by the yeast cell wall integrity signalling pathway. Mol. Microbiol. 1999, 34, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.J.; Nelson, B.; Marton, M.J.; Stoughton, R.; Meyer, M.R.; Bennett, H.A.; He, Y.D.; Dai, H.; Walker, W.L.; Hughes, T.R.; et al. Signaling and Circuitry of Multiple MAPK Pathways Revealed by a Matrix of Global Gene Expression Profiles. Science 2000, 287, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Jung, U.S.; Sobering, A.K.; Romeo, M.J.; Levin, D.E. Regulation of the yeast Rlm1 transcription factor by the Mpk1 cell wall integrity MAP kinase. Mol. Microbiol. 2002, 46, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Garcia, R.; Bermejo, C.; Grau, C.; Pérez, R.; Peña, J.M.R.; François, J.M.; Nombela, C.; Arroyo, J. The Global Transcriptional Response to Transient Cell Wall Damage in Saccharomyces cerevisiae and Its Regulation by the Cell Integrity Signaling Pathway. J. Biol. Chem. 2004, 279, 15183–15195. [Google Scholar] [CrossRef]
- Boorsma, A.; De Nobel, H.; Ter Riet, B.; Bargmann, B.; Brul, S.; Hellingwerf, K.J.; Klis, F.M. Characterization of the transcriptional response to cell wall stress in Saccharomyces cerevisiae. Yeast 2004, 21, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Dodou, E.; Treisman, R. The Saccharomyces cerevisiae MADS-box transcription factor Rlm1 is a target for the Mpk1 mitogen-activated protein kinase pathway. Mol. Cell. Biol. 1997, 17, 1848–1859. [Google Scholar] [CrossRef]
- Watanabe, Y.; Takaesu, G.; Hagiwara, M.; Irie, K.; Matsumoto, K. Characterization of a serum response factor-like protein in Saccharomyces cerevisiae, Rlm1, which has transcriptional activity regulated by the Mpk1 (Slt2) mitogen-activated protein kinase pathway. Mol. Cell. Biol. 1997, 17, 2615–2623. [Google Scholar] [CrossRef]
- Madden, K.; Sheu, Y.-J.; Baetz, K.; Andrews, B.; Snyder, M. SBF Cell Cycle Regulator as a Target of the Yeast PKC-MAP Kinase Pathway. Science 1997, 275, 1781–1784. [Google Scholar] [CrossRef]
- Baetz, K.; Moffat, J.; Haynes, J.; Chang, M.; Andrews, B. Transcriptional Coregulation by the Cell Integrity Mitogen-Activated Protein Kinase Slt2 and the Cell Cycle Regulator Swi4. Mol. Cell. Biol. 2001, 21, 6515–6528. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Truman, A.W.; Levin, D.E. Yeast Mpk1 mitogen-activated protein kinase activates transcription through Swi4/Swi6 by a non-catalytic mechanism that requires upstream signal. Mol. Cell. Biol. 2008, 28, 2579–2589. [Google Scholar] [CrossRef]
- Kim, K.-Y.; Levin, D.E. Mpk1 MAPK Association with the Paf1 Complex Blocks Sen1-Mediated Premature Transcription Termination. Cell 2011, 144, 745–756. [Google Scholar] [CrossRef] [PubMed]
- LeDuc, A.; He, C.H.; Ramotar, D. Disruption of the Saccharomyces cerevisiae cell-wall pathway gene SLG1 causes hypersensitivity to the antitumor drug bleomycin. Mol. Genet. Genom. 2003, 269, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Queralt, E.; Igual, J.C. Functional Connection Between the Clb5 Cyclin, the Protein Kinase C Pathway and the Swi4 Transcription Factor in Saccharomyces cerevisiae. Genetics 2005, 171, 1485–1498. [Google Scholar] [CrossRef]
- Zu, T.; Verna, J.; Ballester, R. Mutations in WSC genes for putative stress receptors result in sensitivity to multiple stress conditions and impairment of Rlm1-dependent gene expression in Saccharomyces cerevisiae. Mol. Genet. Genom. 2001, 266, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Soriano-Carot, M.; Bañó, M.C.; Igual, J.C. The yeast mitogen-activated protein kinase Slt2 is involved in the cellular response to genotoxic stress. Cell Div. 2012, 7, 1. [Google Scholar] [CrossRef]
- Jiménez-Gutiérrez, E.; Alegría-Carrasco, E.; Sellers-Moya, A.; Molina, M.; Martín, H. Not just the wall: The other ways to turn the yeast CWI pathway on. Int. Microbiol. 2020, 23, 107–119. [Google Scholar] [CrossRef]
- Albuquerque, C.P.; Smolka, M.B.; Payne, S.; Bafna, V.; Eng, J.; Zhou, H. A Multidimensional Chromatography Technology for In-depth Phosphoproteome Analysis. Mol. Cell. Proteom. 2008, 7, 1389–1396. [Google Scholar] [CrossRef]
- Truman, A.; Kim, K.-Y.; Levin, D.E. Mechanism of Mpk1 Mitogen-Activated Protein Kinase Binding to the Swi4 Transcription Factor and Its Regulation by a Novel Caffeine-Induced Phosphorylation. Mol. Cell. Biol. 2009, 29, 6449–6461. [Google Scholar] [CrossRef]
- Gobbini, E.; Cesena, D.; Galbiati, A.; Lockhart, A.; Longhese, M.P. Interplays between ATM/Tel1 and ATR/Mec1 in sensing and signaling DNA double-strand breaks. DNA Repair 2013, 12, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Nakada, D.; Shimomura, T.; Matsumoto, K.; Sugimoto, K. The ATM-related Tel1 protein of Saccharomyces cerevisiae controls a checkpoint response following phleomycin treatment. Nucleic Acids Res. 2003, 31, 1715–1724. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA Damage Through ATRIP Recognition of RPA-ssDNA Complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Zhou, Z.; Elledge, S.J. The DNA Replication and Damage Checkpoint Pathways Induce Transcription by Inhibition of the Crt1 Repressor. Cell 1998, 94, 595–605. [Google Scholar] [CrossRef]
- Melo, J.; Toczyski, D. A unified view of the DNA-damage checkpoint. Curr. Opin. Cell Biol. 2002, 14, 237–245. [Google Scholar] [CrossRef]
- Sidorova, J.M.; Breeden, L.L. Precocious G1/S transitions and genomic instability: The origin connection. Mutat. Res. 2003, 532, 5–19. [Google Scholar] [CrossRef]
- Liang, F.; Wang, Y. DNA Damage Checkpoints Inhibit Mitotic Exit by Two Different Mechanisms. Mol. Cell. Biol. 2007, 27, 5067–5078. [Google Scholar] [CrossRef] [PubMed]
- Soriano-Carot, M.; Quilis, I.; Bañó, C.; Igual, J.C. Protein kinase C controls activation of the DNA integrity checkpoint. Nucleic Acids Res. 2014, 42, 7084–7095. [Google Scholar] [CrossRef]
- Liu, L.; Levin, D.E. Intracellular mechanism by which genotoxic stress activates yeast SAPK Mpk1. Mol. Biol. Cell 2018, 29, 2898–2909. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.N.; Symington, L.S. Mutation of the gene encoding protein kinase C 1 stimulates mitotic recombination in Saccharomyces cerevisiae. Mol. Cell. Biol. 1994, 14, 6039–6045. [Google Scholar] [CrossRef][Green Version]
- Li, X.; Heyer, W.-D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef]
- Yang, W.-L.; Bruno, M.E.C.; Carman, G.M. Regulation of Yeast CTP Synthetase Activity by Protein Kinase C. J. Biol. Chem. 1996, 271, 11113–11119. [Google Scholar] [CrossRef]
- Lee, J.; Liu, L.; Levin, D.E. Stressing out or stressing in: Intracellular pathways for SAPK activation. Curr. Genet. 2019, 65, 417–421. [Google Scholar] [CrossRef]
- Siliciano, P.G.; Tatchell, K. Transcription and regulatory signals at the mating type locus in yeast. Cell 1984, 37, 969–978. [Google Scholar] [CrossRef]
- Kafadar, K.A.; Zhu, H.; Snyder, M.; Cyert, M.S. Negative regulation of calcineurin signaling by Hrr25p, a yeast homolog of casein kinase I. Genes Dev. 2003, 17, 2698–2708. [Google Scholar] [CrossRef]
- Gietz, R.D.; Schiestl, R.H.; Willems, A.R.; Woods, R.A. Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast 1995, 11, 355–360. [Google Scholar] [CrossRef]
- Sikorski, R.S.; Hieter, P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 1989, 122, 19–27. [Google Scholar] [CrossRef]
- Hill, J.E.; Myers, A.; Koerner, T.J.; Tzagoloff, A. Yeast/E. coli shuttle vectors with multiple unique restriction sites. Yeast 1986, 2, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Denis, V.; Cyert, M.S. Molecular Analysis Reveals Localization of Saccharomyces cerevisiae Protein Kinase C to Sites of Polarized Growth and Pkc1p Targeting to the Nucleus and Mitotic Spindle. Eukaryot. Cell 2005, 4, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Reiter, W.; Dohnal, I.; Gregori, C.; Beese-Sims, S.; Kuchler, K.; Ammerer, G.; Levin, D.E. MAPK Hog1 closes the S. cerevisiae glycerol channel Fps1 by phosphorylating and displacing its positive regulators. Genes Dev. 2013, 27, 2590–2601. [Google Scholar] [CrossRef] [PubMed]
- Reiter, W.; Anrather, R.; Dohnal, I.; Pichler, P.; Veis, J.; Grøtli, M.; Posas, F.; Ammerer, G. Validation of regulated protein phosphorylation events in yeast by quantitative mass spectrometry analysis of purified proteins. Proteomics 2012, 12, 3030–3043. [Google Scholar] [CrossRef]
- Kamada, Y.; Jung, U.S.; Piotrowski, J.; Levin, D. The protein kinase C-activated MAP kinase pathway of Saccharomyces cerevisiae mediates a novel aspect of the heat shock response. Genes Dev. 1995, 9, 1559–1571. [Google Scholar] [CrossRef] [PubMed]
- Kushnirov, V.V. Rapid and reliable protein extraction from yeast. Yeast 2000, 16, 857–860. [Google Scholar] [CrossRef]
- Zhao, C.; Jung, U.S.; Garrett-Engele, P.; Roe, T.; Cyert, M.S.; Levin, D.E. Temperature-Induced Expression of Yeast FKS2 Is under the Dual Control of Protein Kinase C and Calcineurin. Mol. Cell. Biol. 1998, 18, 1013–1022. [Google Scholar] [CrossRef]
- Ong, S.-E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Nyberg, K.A.; Michelson, R.J.; Putnam, C.W.; Weinert, T.A. Toward Maintaining the Genome: DNA Damage and Replication Checkpoints. Annu. Rev. Genet. 2002, 36, 617–656. [Google Scholar] [CrossRef]
- Harper, J.W.; Elledge, S.J. The DNA Damage Response: Ten Years After. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef]
- Hoekstra, M.F.; Liskay, R.M.; Ou, A.C.; DeMaggio, A.J.; Burbee, D.G.; Heffron, F. HRR25, a putative protein kinase from budding yeast: Association with repair of damaged DNA. Science 1991, 253, 1031–1034. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.; Mason, S.; Kobayashi, R.; Hoekstra, M.; Andrews, B. Role of the casein kinase I isoform, Hrr25, and the cell cycle-regulatory transcription factor, SBF, in the transcriptional response to DNA damage in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1997, 94, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.S.; Kliegman, J.I.; Shokat, K.M. The Logic and Design of Analog-Sensitive Kinases and Their Small Molecule Inhibitors. Methods Enz. 2014, 548, 189–213. [Google Scholar] [CrossRef]
- Peng, Y.; Grassart, A.; Lu, R.; Wong, C.C.; Yates, J.; Barnes, G.; Drubin, D.G. Casein Kinase 1 Promotes Initiation of Clathrin-Mediated Endocytosis. Dev. Cell 2015, 32, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Flotow, H.; Roach, P.J. Synergistic phosphorylation of rabbit muscle glycogen synthase by cyclic AMP-dependent protein kinase and casein kinase I. Implications for hormonal regulation of glycogen synthase. J. Biol. Chem. 1989, 264, 9126–9128. [Google Scholar] [CrossRef]
- Flotow, H.; Roach, P.J. Role of acidic residues as substrate determinants for casein kinase I. Role of acidic residues as substrate determinants for casein kinase I. J. Biol. Chem. 1991, 266, 3724–3727. [Google Scholar] [CrossRef]
- Flotow, H.; Graves, P.; Wang, A.; Fiol, C.; Roeske, R.; Roach, P. Phosphate groups as substrate determinants for casein kinase I action. J. Biol. Chem. 1990, 265, 14264–14269. [Google Scholar] [CrossRef]
- Pulgar, V.; Marin, O.; Meggio, F.; Allende, C.C.; Allende, J.E.; Pinna, L.A. Optimal sequences for non-phosphate-directed phoshorylation by protein kinase CK1 (casein kinase-1)—A re-evaluation. Eur. J. Biochem. 1999, 260, 520–526. [Google Scholar] [CrossRef]
- Swaney, D.L.; Beltrao, P.; Starita, L.; Guo, A.; Rush, J.; Fields, S.; Krogan, N.J.; Villen, J. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat. Methods 2013, 10, 676–682. [Google Scholar] [CrossRef]
- MacGilvray, M.E.; Shishkova, E.; Place, M.; Wagner, E.R.; Coon, J.J.; Gasch, A.P. Phosphoproteome Response to Dithiothreitol Reveals Unique Versus Shared Features of Saccharomyces cerevisiae Stress Responses. J. Proteome Res. 2020, 19, 3405–3417. [Google Scholar] [CrossRef]
- Lanz, M.C.; Yugandhar, K.; Gupta, S.; Sanford, E.J.; Faça, V.M.; Vega, S.; Joiner, A.M.N.; Fromme, J.C.; Yu, H.; Smolka, M.B. In-depth and 3-dimensional exploration of the budding yeast phosphoproteome. EMBO Rep. 2021, 22, e51121. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Relevant Genotype | Source or Reference |
|---|---|---|
| DL100 | MATa EG123 ura3-52 leu2-3,112 trp1-1 his4 can1r | [36] |
| DL376 | MATa EG123 ura3-52 leu2-3,112 trp1-1 his4 pkc1∆::LEU2 | David Levin |
| DL1021 | MATa SEY6210 leu2-3,112 ura3-52 his3-200 trp1-901 ede2-101 pkc1∆::HIS3 suc2-9 (GPY1115) | Gerhard Paravicini |
| DL2772 | MATα S288c (BY4742) his3 leu2 lys2 ura3 | Research Genetics |
| DL3950 | MATα MBS62 sml1∆::TRP1 | Marcus Smolka |
| DL3951 | MATα MBS103 sml1∆::TRP1 tel1∆::URA3 | Marcus Smolka |
| DL3952 | MATα MBS104 sml1∆::TRP1 mec1∆::KanMX | Marcus Smolka |
| DL3953 | MATα MBS72 sml1∆::TRP1 rad53∆::HIS3 | Marcus Smolka |
| DL3954 | MATa/α MBS115 SML1/sml1∆::TRP1 MEC1/mec1∆::HIS3 TEL1/tel1∆::URA3 | Marcus Smolka |
| DL4206 | MATa W303 ade2 trp1 leu2 his3 ura3 can1 | Juan Carlos Igual |
| DL4277 | MATα MBS sml1∆::TRP1 mec1∆::HIS3 tel1∆::URA3 | This study |
| DL4286 | MATα BY4742 chk1∆::KanMX | Research Genetics |
| DL4290 | MATa ura3-52 lys2-801 ade2-101 trp1-∆63 his3-∆200 leu2-∆1 hrr25∆::loxP-kanMX-loxP pGAL1-3HA-HRR25degron (KKY387) | [37] |
| DL4503 | MATα MBS sml1∆::TRP1 mec1∆::KanMX tel1∆::URA3 | This study |
| DL4515 | MATa W303 hrr25∆::HPHMX4 (pHRR25-HA; p3484, LEU2 2µ) | This study |
| DL4527 | MATa W303 hrr25∆::HPHMX4 (pHRR25-HA; p3545, HIS3 CEN) | This study |
| DL4528 | MATa W303 hrr25∆::HPHMX4 (phrr25-I82A-HA; p3550, HIS3 CEN) | This study |
| DL4541 | MATa W303 hrr25∆::HPHMX4 (pHRR25-GFP; p3562, HIS3 2µ) | This study |
| DL4542 | MATa W303 hrr25∆::HPHMX4 (phrr25-∆404-GFP; p3567, HIS3 2µ) | This study |
| DL4555 | MATa W303 hrr25∆::HPHMX4 (phrr25-∆404-HA; p3546, HIS3 CEN) | This study |
| DL4556 | MATa W303 hrr25∆::HPHMX4 (phrr25-3A-HA; p3576, HIS3 CEN) | This study |
| JV826 | MATa BY4741 PKC1-HTBeaq::NatMX | This study |
| Plasmid | Description | Source or Reference |
|---|---|---|
| p117 | pRS313 | [39] |
| p118 | pRS314 | [39] |
| p119 | pRS315 | [39] |
| p120 | YEp351 | [40] |
| p813 | YEp351-PKC1-HA | David Levin |
| p1105 | pRS425 | [39] |
| p1202 | pRS425-GFP | David Levin |
| p2062 | pVDG7 PKC1-GFP | [41] |
| p2454 | pRS413 | [39] |
| p2947 | pRNR3-lacZ | Stephen Elledge |
| p3064 | pAG32-RGC2 | [42] |
| p3149 | pRS425-3HA-ADH1T | [42] |
| p3357 | pUG36-HRR25-GFP | Martha Cyert |
| p3358 | pUG36-GFP | Martha Cyert |
| p3484 | pRS425-HRR25-HA | This study |
| p3504 | pRS313-3HA-ADH1T | This study |
| p3517 | YEp351-pkc1-S577A-HA | This study |
| p3521 | YEp351-pkc1-S577A, S626A-HA | This study |
| p3522 | YEp351-pkc1-S577A, T626A, T753A-HA | This study |
| p3523 | YEp351-pkc1-S577A, T626A, T753A, S804A-HA | This study |
| p3525 | pRS315-HRR25-HA | This study |
| p3538 | pRS425-hrr25-∆404-HA | This study |
| p3544 | pRS423-3HA-ADH1T | This study |
| p3545 | pRS313-HRR25-HA | This study |
| p3546 | pRS313-hrr25-∆404-HA | This study |
| p3547 | pRS423-HRR25-HA | This study |
| p3550 | pRS313-hrr25-I82A-HA | This study |
| p3552 | pRS423-hrr25-I82A-HA | This study |
| p3553 | pRS423-hrr25-I82G-HA | This study |
| p3560 | pRS423-GFP | This study |
| p3562 | pRS423-HRR25-GFP | This study |
| p3567 | pRS423-hrr25-∆404-GFP | This study |
| p3570 | pRS313-hrr25-T453A-HA | This study |
| p3572 | pRS313-hrr25-T453A, S405A-HA | This study |
| p3574 | YEp351-pkc1-S577A, T626A, T753A, S761A, S804A-HA | This study |
| p3576 | pRS313-hrr25-T453A, S405A, S438A-HA | This study |
| p3597 | YEp351-pkc1-S577A, T626A, T753A, S761A, S772A, S804A-HA | This study |
| p3603 | YEp351-pkc1-S577A, T626A, S686A, T753A, S761A, S772A, S804A-HA | This study |
| p3604 | YEp351-pkc1-S577A, T626A, S686A, S750A, T753A, S761A, S772A, S804A-HA | This study |
| p3605 | YEp351-pkc1-S152A, S577A, T626A, S686A, S750A, T753A, S761A, S772A, S804A-HA | This study |
| p3606 | YEp351-pkc1-S152A, S577A, T626A, S657A, S686A, S750A, T753A, S761A, S772A, S804A-HA | This study |
| p3608 | YEp351-pkc1-S152A, S577A, T626A, S657A, S686A, S750A, T753A, S761A, S772A, S781A, S804A-HA | This study |
| p3610 | YEp351-pkc1-S75A, S152A, S577A, T626A, S657A, S686A, S750A, T753A, S761A, S772A, S781A, S804A-HA | This study |
| p3612 | YEp351-pkc1-S75A, S152A, S577A, T626A, S657A, S686A, S750A, T753A, S761A, S772A, S781A, S797A, S804A-HA (S/T13A) | This study |
| p3617 | YEp351-pkc1-S2A-HA | This study |
| p3618 | YEp351-pkc1-S2A, S657A-HA | This study |
| p3619 | YEp351-pkc1-S2A, S657A, S577A-HA (S3A) | This study |
| p3623 | pRS314-PKC1-HA | This study |
| p3624 | pRS314-pkc1-S2A, S657A, S577A-HA (S3A) | This study |
| p3625 | pRS314-pkc1-S75A, S152A, S577A, T626A, S657A, S686A, S750A, T753A, S761A, S772A, S781A, S797A, S804A-HA (S/T13A) | This study |
| pWR268 | pFA6a-integrative HTBeaq tag, NatMX | [43] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, L.; Veis, J.; Reiter, W.; Motari, E.; Costello, C.E.; Samuelson, J.C.; Ammerer, G.; Levin, D.E. Regulation of Pkc1 Hyper-Phosphorylation by Genotoxic Stress. J. Fungi 2021, 7, 874. https://doi.org/10.3390/jof7100874
Liu L, Veis J, Reiter W, Motari E, Costello CE, Samuelson JC, Ammerer G, Levin DE. Regulation of Pkc1 Hyper-Phosphorylation by Genotoxic Stress. Journal of Fungi. 2021; 7(10):874. https://doi.org/10.3390/jof7100874
Chicago/Turabian StyleLiu, Li, Jiri Veis, Wolfgang Reiter, Edwin Motari, Catherine E. Costello, John C. Samuelson, Gustav Ammerer, and David E. Levin. 2021. "Regulation of Pkc1 Hyper-Phosphorylation by Genotoxic Stress" Journal of Fungi 7, no. 10: 874. https://doi.org/10.3390/jof7100874
APA StyleLiu, L., Veis, J., Reiter, W., Motari, E., Costello, C. E., Samuelson, J. C., Ammerer, G., & Levin, D. E. (2021). Regulation of Pkc1 Hyper-Phosphorylation by Genotoxic Stress. Journal of Fungi, 7(10), 874. https://doi.org/10.3390/jof7100874