
Structure–Activity Relationship of Phytotoxic Natural 10-Membered Lactones and Their Semisynthetic Derivatives

 , ,
, ,  ,
,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. General Experimental Procedures
2.2. Fungal Strain and Toxin Production
2.3. Semisynthetic Derivatives Preparation
2.4. Biological Activity Assays
2.4.1. Leaf Puncture Assay
2.4.2. Seedlings Bioassay
2.4.3. Microalgae Assay
2.4.4. Antimicrobial Assay
2.4.5. Cytotoxicity Assay
2.5. LogP determination
2.6. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Cantrell, C.L.; Dayan, F.E.; Duke, S.O. Natural Products as Sources for New Pesticides. J. Nat. Prod. 2012, 75, 1231–1242. [Google Scholar] [CrossRef]
- Duke, S.O.; Owens, D.K.; Dayan, F.E. Natural Product-Based Chemical Herbicides. In Weed Control: Sustainability, Hazards, and Risks in Cropping Systems Worldwide, 1st ed.; Korres, N.E., Burgos, N.R., Duke, S.O., Eds.; CRC Press: Boca Raton, FL, USA, 2019; Chapter 8; pp. 153–165. [Google Scholar] [CrossRef]
- Duke, S.O.; Dayan, F.E. Discovery of New Herbicide Modes of Action with Natural Phytotoxins. In Discovery and Synthesis of Crop Protection Products; Maienfisch, P., Stevenson, T.M., Eds.; ACS Symposium Series eBooks: Washington, DC, USA, 2015; pp. 79–92. [Google Scholar] [CrossRef]
- Rivero-Cruz, F.J.; García-Aguirre, G.; Cerda-García-Rojas, C.M.; Mata, R. Conformational behavior and absolute stereostructure of two phytotoxic nonenolides from the fungus Phoma herbarum. Tetrahedron 2000, 56, 5337–5344. [Google Scholar] [CrossRef]
- Li, Y.-Y.; Hu, Z.-Y.; Shen, Y.-M. Two New Cyclopeptides and One New Nonenolide from Xylaria sp. 101. Nat. Prod. Commun. 2011, 6, 1843–1846. [Google Scholar] [CrossRef] [PubMed]
- Boonphong, S.; Kittakoop, P.; Isaka, M.; Pittayakhajonwut, D.; Tanticharoen, M.; Thebtaranonth, Y. Multiplolides A and B, New Antifungal 10-Membered Lactones from Xylaria multiplex. J. Nat. Prod. 2001, 64, 965–967. [Google Scholar] [CrossRef]
- Nukina, M.; Ikeda, M.; Sassa, T. Two New Pyrenolides, Fungal Morphogenic Substances Produced by Pyrenophora teres (Diedicke) Drechsler. Agric. Biol. Chem. 1980, 44, 2761–2762. [Google Scholar] [CrossRef][Green Version]
- Ishida, T.; Wada, K. A steroid hydroxylase inhibitor, diplodialide A from Diplodia pinea. J. Chem. Soc. Chem. Comm. 1975, 6, 209–210. [Google Scholar] [CrossRef]
- Wada, K.; Ishida, T. A new pentaketide, diplodialide-D, from Diplodia pinea. J. Chem. Soc. Chem. Comm. 1976, 10, 340. [Google Scholar] [CrossRef]
- Dalinova, A.; Dubovik, V.; Chisty, L.; Kochura, D.; Ivanov, A.; Smirnov, S.; Petrova, M.; Zolatarev, A.; Evidente, A.; Berestetskiy, A. Stagonolides J and K and Stagochromene A, two new natural substituted nonenolides and a new disubstituted chromene-4,5-dione isolated from Stagonospora cirsii S-47 proposed for the biocontrol of Sonchus arvensis. J. Agric. Food Chem. 2019, 67, 13040–13050. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Ree, T.V.; Krohn, K.; Li, L.; Zhang, W. Nonanolides of Natural Origin: Structure, Synthesis, and Biological Activity. Curr. Med. Chem. 2012, 19, 3417–3455. [Google Scholar] [CrossRef] [PubMed]
- Berestetskiy, A.; Dmitriev, A.; Mitina, G.; Lisker, I.; Andolfi, A.; Evidente, A. Nonenolides and cytochalasins with phytotoxic activity against Cirsium arvense and Sonchus arvensis: A structure–activity relationships study. Phytochemistry 2008, 69, 953–960. [Google Scholar] [CrossRef]
- Evidente, A.; Andolfi, A.; Cimmino, A. Relationships between the stereochemistry and biological activity of fungal phytotoxins. Chirality 2011, 23, 674–693. [Google Scholar] [CrossRef] [PubMed]
- Evidente, A.; Lanzetta, R.; Capasso, R.; Andolfi, A.; Bottalico, A.; Vurro, M.; Zonno, M.C. Putaminoxin, A Phytotoxic Nonenolide from Phoma putaminum. Phytochemistry 1995, 40, 1637–1641. [Google Scholar] [CrossRef]
- Yuzikhin, O.; Mitina, G.; Berestetskiy, A. Herbicidal potential of stagonolide, a new phytotoxic nonenolide from Stagonospora cirsii. J. Agric. Food Chem. 2007, 55, 7707–7711. [Google Scholar] [CrossRef]
- Evidente, A.; Cimmino, A.; Berestetskiy, A.; Mitina, G.; Andolfi, A.; Motta, A. Stagonolides B-F, nonenolides produced by Stagonospora cirsii, a potential mycoherbicide of Cirsium arvense. J. Nat. Prod. 2008, 71, 31–34. [Google Scholar] [CrossRef]
- Evidente, A.; Cimmino, A.; Berestetskiy, A.; Andolfi, A.; Motta, A. Stagonolides G-I and Modiolide A, nonenolides produced by Stagonospora cirsii, a potential mycoherbicide for Cirsium arvense. J. Nat. Prod. 2008, 71, 1897–1901. [Google Scholar] [CrossRef] [PubMed]
- Berestetskiy, A.; Dalinova, A.; Dubovik, V. Strain Stagonospora cirsii G-51 VIZR - Producer of Herbarumin I and Stagonolide A. RU Patent 2701817 C1; filed 28 December 2018, and issued 1 October 2019,
- Dubovik, V.; Dalinova, A.; Berestetskiy, A. Effect of Adjuvants on Herbicidal Activity and Selectivity of Three Phytotoxins Produced by the Fungus, Stagonospora cirsii. Plants 2020, 9, 1621. [Google Scholar] [CrossRef]
- Evidente, A.; Capasso, R.; Andolfi, A.; Vurro, M.; Zonno, M.C. Putaminoxins D and E from Phoma putaminum. Phytochemistry 1998, 48, 941–945. [Google Scholar] [CrossRef]
- Evidente, A.; Capasso, R.; Andolfi, A.; Vurro, M.; Zonno, M.C. Structure-activity relationship studies of putaminoxins and pinolidoxins: Phytotoxic nonenolides produced by phytopathogenic Phoma and Ascochyta species. Nat. Toxins 1999, 6, 183–188. [Google Scholar] [CrossRef]
- Firouzabadi, H.; Mostafavipoor, Z. Barium Manganate. A Versatile Oxidant in Organic Synthesis. Bull. Chem. Soc. Jpn. 1983, 56, 914–917. [Google Scholar] [CrossRef]
- Berestetskiy, A.O.; Yuzikhin, O.S.; Katkova, A.S.; Dobrodumov, A.V.; Sivogrivov, D.E.; Kolombet, L.V. Isolation, identification, and characteristics of the phytotoxin produced by the fungus Alternaria cirsinoxia. Appl. Biochem. Microbiol. 2010, 46, 75–79. [Google Scholar] [CrossRef]
- Dornbos, D.L.; Spencer, G.F. Natural products phytotoxicity A bioassay suitable for small quantities of slightly water-soluble compounds. J. Chem. Ecol. 1990, 16, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Chelebieva, E.S.; Dantsyuk, N.V.; Chekanov, K.A.; Chubchikova, I.N.; Drobetskaya, I.V.; Minyuk, G.S.; Lobakova, E.S.; Solovchenko, A.E. Identification and morphological-physiological characterization of Astaxanthin producer strains of Haematococcus pluvialis from the Black Sea Region. Appl. Biochem. Microbiol. 2018, 54, 639–648. [Google Scholar] [CrossRef]
- Fabregas, J.; Dominguez, A.; Regueiro, M.; Maseda, A.; Otero, A. Optimization of culture medium for the continuous cultivation of the microalga Haematococcus pluvialis. Appl. Microbiol. Biotechnol. 2000, 53, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhu, Y.; Tao, Y.; Zhang, Y.; Li, A.; Li, T.; Sang, M.; Zhang, C. Freshwater microalgae harvested via flocculation induced by pH decrease. Biotechnol. Biofuels 2013, 6, 98. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.W.; Kirby, W.M.; Sherris, J.C.; Turck, M. Antibiotic Susceptibility Testing by a Standardized Single Disk Method. Am. J. Clin. Pathol. 1966, 45, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Strober, W. Trypan Blue Exclusion Test of Cell Viability. Curr. Protoc. Immunol. 2015, 111, A3.B.1–A3.B.3. [Google Scholar] [CrossRef]
- Paschke, A.; Neitzel, P.L.; Walther, W.; Schüürmann, G. Octanol/Water Partition Coefficient of Selected Herbicides: Determination Using Shake-Flask Method and Reversed-Phase High-Performance Liquid Chromatography. J. Chem. Eng. Data 2004, 49, 1639–1642. [Google Scholar] [CrossRef]
- Organisation for Economic Co-Operation and Development (OECD). OECD Guideline for Testing of Chemicals: Partition Coefficient (n-Octanol/Water), High Performance Liquid Chromatography (HPLC) Method; Section 1, No. 117; OECD: Paris, France, 1989. [Google Scholar]
- Tomlin, C.D.S. (Ed.) The Pesticide Manual: A World Compendium of Pesticides, 13th ed.; British Crop Protection Council: Farnham, UK, 2003; p. 1344. [Google Scholar]
- Sparapano, L.; Bruno, G.; Fierro, O.; Evidente, A. Studies on Structure-Activity Relationship of Sphaeropsidins A—F, Phytotoxins Produced by Sphaeropsis sapinea f. sp. cupressi. Phytochemistry 2004, 65, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Masi, M.; Freda, F.; Clement, S.; Cimmino, A.; Cristofaro, M.; Meyer, S.; Evidente, A. Phytotoxic Activity and Structure-Activity Relationships of Radicinin Derivatives against the Invasive Weed Buffelgrass (Cenchrus ciliaris). Molecules 2019, 24, 2793. [Google Scholar] [CrossRef]
- Renaud, J.B.; DesRochers, N.; Hoogstra, S.; Garnham, C.P.; Sumarah, M.W. Structure Activity Relationship for Fumonisin Phytotoxicity. Chem. Res. Toxicol. 2021, 34, 1604–1611. [Google Scholar] [CrossRef]
- Evidente, A.; Lanzetta, R.; Capasso, R.; Vurro, M.; Botralico, A. Pinolidoxin, a phytotoxic nonenolide from Ascochyta pinodes. Phytochemistry 1993, 34, 999–1003. [Google Scholar] [CrossRef]
- Fortino, J.; Splittstoesser, W.E. The use of Metribuzin for Weed Control in Tomato. Weed Sci. 1974, 22, 615–619. [Google Scholar] [CrossRef]
- Adu-Tutu, K.O.; Drennan, D.S.H. Studies on the effects of Metsulfuron methyl on the parasitism of sorghum by Striga hermonthica (Del.) Benth. Trop. Pest Manag. 1991, 37, 252–255. [Google Scholar] [CrossRef]
- Bhullar, M.S.; Kaur, S.; Kaur, T.; Singh, T.; Singh, M.; Jhala, A.J. Control of broadleaf weeds with post-emergence herbicides in four barley (Hordeum spp.) cultivars. Crop Prot. 2013, 43, 216–222. [Google Scholar] [CrossRef]
- Avram, S.; Funar-Timofei, S.; Borota, A.; Chennamaneni, S.R.; Manchala, A.K.; Muresan, S. Quantitative estimation of pesticide-likeness for agrochemical discovery. J. Cheminform. 2014, 6, 42. [Google Scholar] [CrossRef] [PubMed]
- Rivero-Cruz, F.J.; Macias, M.; Cerda-Garcia-Rojas, C.M.; Mata, R. A New phytotoxic nonenolide from Phoma herbarum. J. Nat. Prod. 2003, 66, 511–514. [Google Scholar] [CrossRef]
- Grossmann, K. What it takes to get a herbicide’s mode of action. Physionomics, a classical approach in a new complexion. Pest Manag. Sci. 2005, 61, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, J.; Tang, H.; Su, L.; Chou, Y.; Soong, K.; Li, J.; Zhuang, C.-L.; Luo, Y.-P.; Xu, C.-C. Osteoclastogenesis Inhibitory Polyketides from the Sponge-associated fungus Xylaria feejeenisis. Chem. Biodivers. 2018, 15, e1800358. [Google Scholar] [CrossRef]
- Liu, Y.; Cheng, L.; Shen, Y. Two New Nonenolides from Diaporthe sp. SXZ-19, an Endophytic Fungus of Camptotheca Acuminata. Chem. Biodivers. 2021, 18, e2001055. [Google Scholar] [CrossRef]
- Wang, W.-X.; Zheng, M.-J.; Li, J.; Feng, T.; Li, Z.-H.; Huang, R.; Zheng, Y.-S.; Sun, H.; Ai, H.-L.; Liu, J.-K. Cytotoxic polyketides from endophytic fungus Phoma bellidis harbored in Tricyrtis maculate. Phytochem. Lett. 2019, 29, 41–46. [Google Scholar] [CrossRef]
- Hu, M.; Yang, X.-Q.; Wan, C.-P.; Wang, B.-Y.; Yin, H.-Y.; Shi, L.-J.; Wu, Y.-M.; Yang, Y.-B.; Zhou, H.; Ding, Z.-T. Potential antihyperlipidemic polyketones from endophytic Diaporthe sp. JC-J7 in Dendrobium nobile. RSC Adv. 2018, 8, 41810–41817. [Google Scholar] [CrossRef]
- Cimmino, A.; Andolfi, A.; Fondevilla, S.; Abouzeid, M.A.; Rubiales, D.; Evidente, A. Pinolide, a New Nonenolide Produced by Didymella pinodes, the Causal Agent of Ascochyta Blight on Pisum sativum. J. Agric. Food Chem. 2012, 60, 5273–5278. [Google Scholar] [CrossRef] [PubMed]





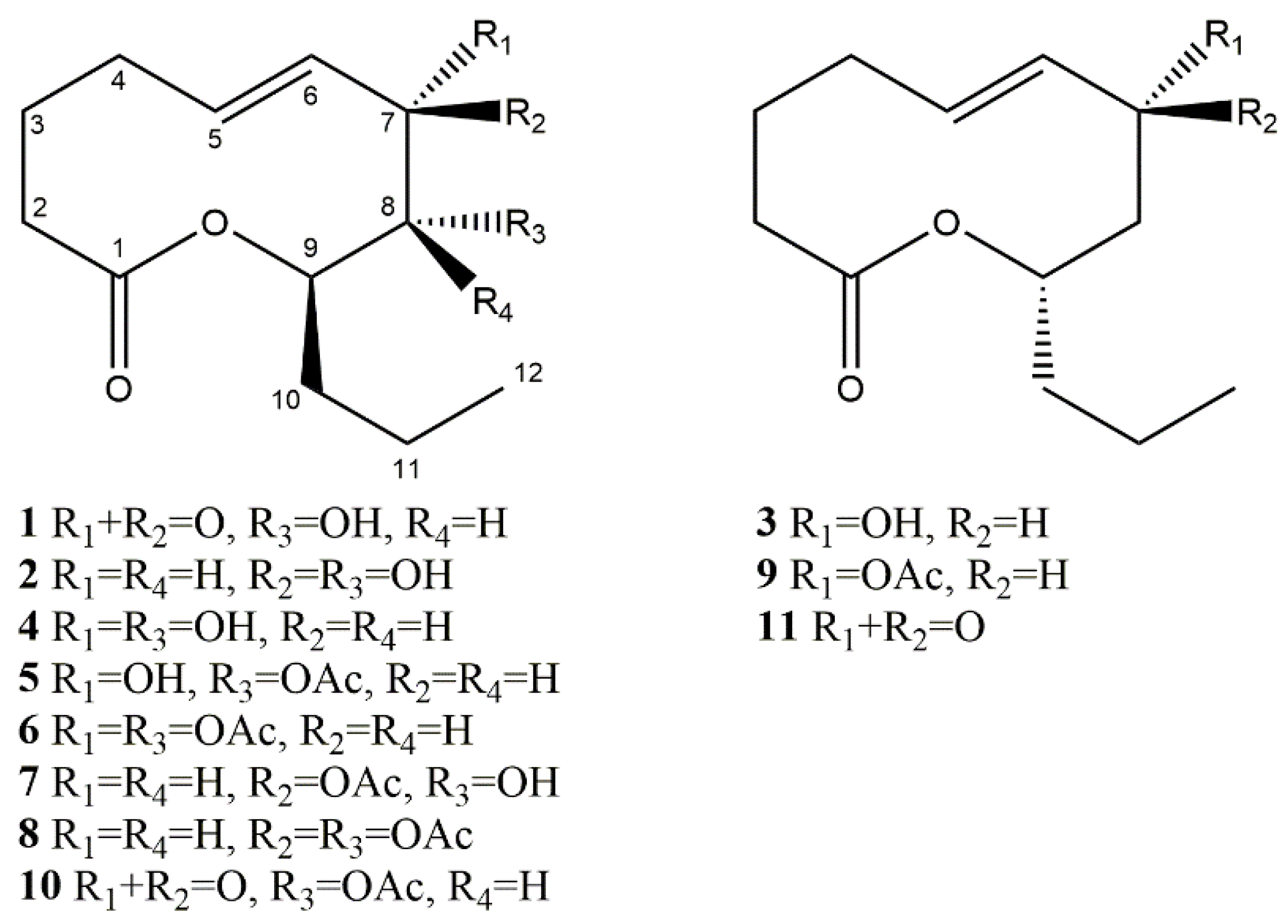{kind=link}
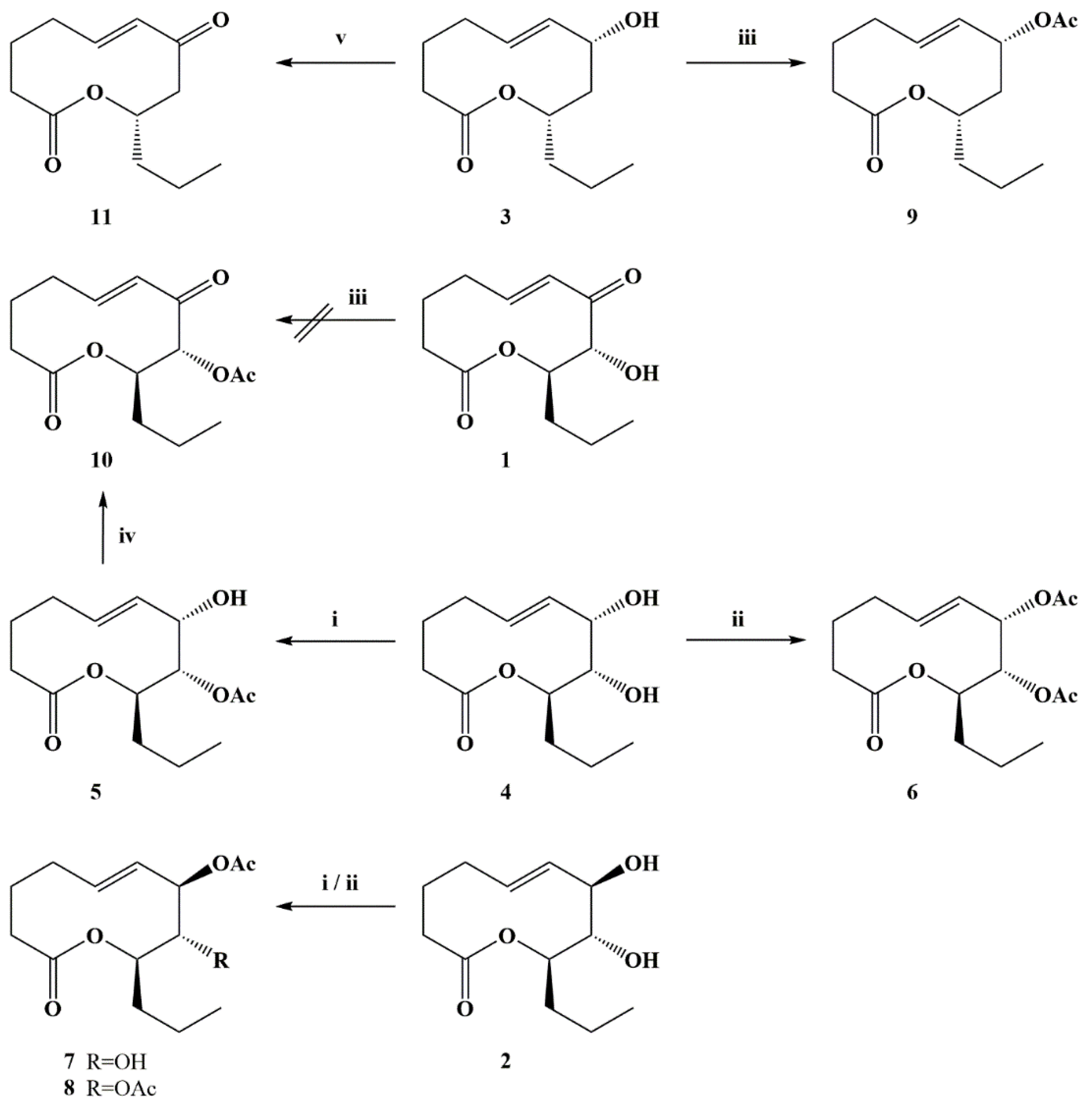{kind=link}
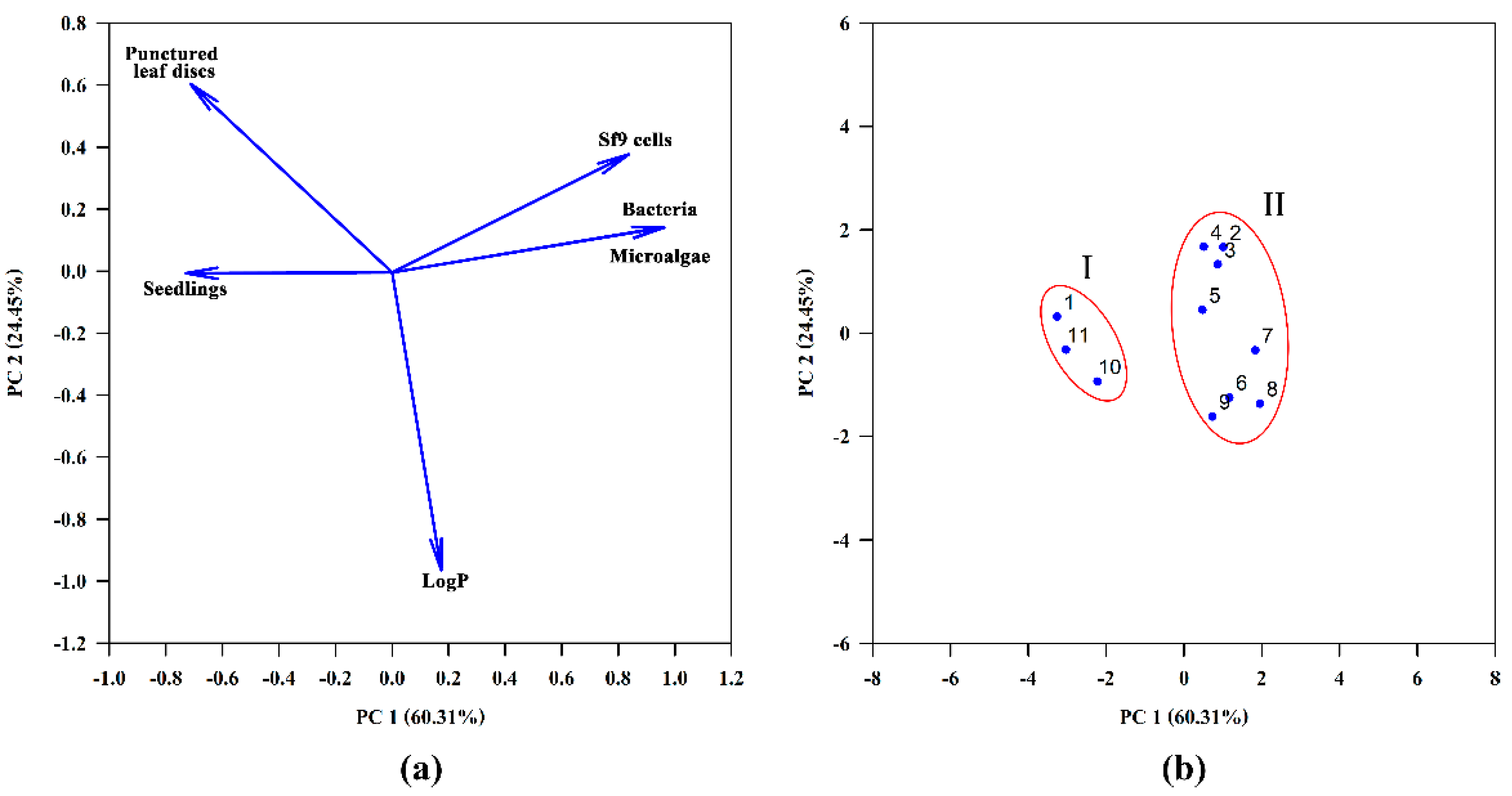{kind=link}
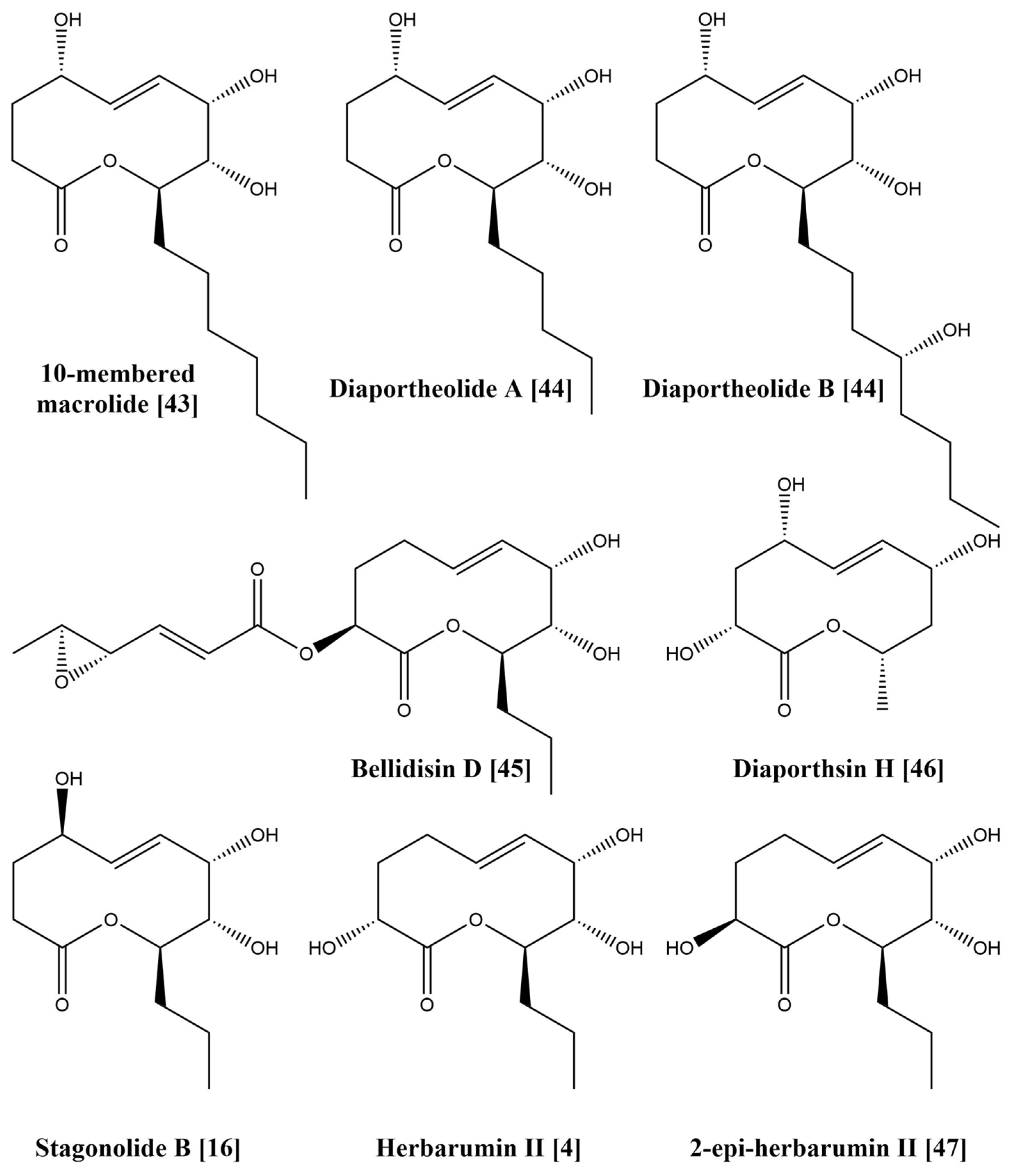{kind=link}
| Compound | tR, min | LogP [32] |
|---|---|---|
| Imidacloprid (Sigma-Aldrich, St. Louis, MO, USA) | 2.59 | 0.57 |
| Acetamiprid (Sigma-Aldrich) | 2.77 | 0.80 |
| Metribuzin (Sigma-Aldrich) | 5.36 | 1.70 |
| Metsulfuron-methyl (Dr. Ehrenstorfer, Augsburg, Germany) | 7.42 | 2.20 |
| Propiconazole (Sigma-Aldrich) | 42.59 | 3.72 |
| Pyraclostrobin (Sigma-Aldrich) | 104.11 | 3.99 |
| Compound | Phytotoxic Activity | Antimicrobial Activity, Bacillus subtilis, Radius of Growth Inhibition Zone, mm | Cytotoxic Activity, Sf9 Cells, LC50, μg/mL (μM) | LogP (HPLC-RP Method) | |||
|---|---|---|---|---|---|---|---|
| Sowthistle Leaves, Necrosis Diameter, mm | Lettuce Seedlings, ID50, 10−8 mole/seed | Haematococcus lacustris, LC50, μg/mL (μM) | |||||
| 48 h | 120 h | ||||||
| 1 | 6.3 ± 0.4 f * | 6.4 ± 0.3 g | 3 | 1 (4.4) | 16.0 ± 1.0 | 3 (13) | 1.87 |
| 2 | 1.6 ± 0.5 d | 4.2 ± 0.4 bcd | n/a | n/a | 0 | n/a | 1.31 |
| 3 | 4.3 ± 0.2 c | 4.9 ± 0.3 de | 94 | n/a | 0 | n/a | 1.73 |
| 4 | 2.8 ± 0.2 b | 4.8 ± 0.3 cde | 35 | n/a | 0 | n/a | 1.39 |
| 5 | 3.7 ± 0.3 c | 4.0 ± 0.3 bc | 4 | n/a | 0 | n/a | 2.32 |
| 6 | 0.3 ± 0.2 a | 0.5 ± 0.2 a | 7 | n/a | 0 | n/a | 3.18 |
| 7 | 0 a | 0 a | n/a | n/a | 0 | n/a | 2.22 |
| 8 | 0 a | 0 a | n/a | n/a | 0 | n/a | 3.20 |
| 9 | 2.6 ± 0.4 b | 2.6 ± 0.4 f | n/a | n/a | 0 | 88 (346.4) | 3.31 |
| 10 | 3.8 ± 0.3 c | 3.9 ± 0.3 b | 30 | 3 (11.2) | 8.3 ± 0.6 | 6 (22.4) | 2.51 |
| 11 | 5.3 ± 0.3 e | 5.3 ± 0.3 e | 4 | 2 (9.5) | 14.0 ± 1.0 | 5 (23.7) | 2.23 |
| metribuzin | 4.4 ± 0.5 c | 5.4 ± 0.7 e | 18 | 2 (9.3) | n/t | n/t | 1.70 [32] |
| metsulfuron-methyl | 0 a | 0 a | 3 | >50 (>131.0) | n/t | n/t | 2.20 [32] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dalinova, A.; Fedorov, A.; Dubovik, V.; Voitsekhovskaja, O.; Tyutereva, E.; Smirnov, S.; Kochura, D.; Chisty, L.; Senderskiy, I.; Berestetskiy, A. Structure–Activity Relationship of Phytotoxic Natural 10-Membered Lactones and Their Semisynthetic Derivatives. J. Fungi 2021, 7, 829. https://doi.org/10.3390/jof7100829
Dalinova A, Fedorov A, Dubovik V, Voitsekhovskaja O, Tyutereva E, Smirnov S, Kochura D, Chisty L, Senderskiy I, Berestetskiy A. Structure–Activity Relationship of Phytotoxic Natural 10-Membered Lactones and Their Semisynthetic Derivatives. Journal of Fungi. 2021; 7(10):829. https://doi.org/10.3390/jof7100829
Chicago/Turabian StyleDalinova, Anna, Anatoly Fedorov, Vsevolod Dubovik, Olga Voitsekhovskaja, Elena Tyutereva, Sergey Smirnov, Dmitry Kochura, Leonid Chisty, Igor Senderskiy, and Alexander Berestetskiy. 2021. "Structure–Activity Relationship of Phytotoxic Natural 10-Membered Lactones and Their Semisynthetic Derivatives" Journal of Fungi 7, no. 10: 829. https://doi.org/10.3390/jof7100829
APA StyleDalinova, A., Fedorov, A., Dubovik, V., Voitsekhovskaja, O., Tyutereva, E., Smirnov, S., Kochura, D., Chisty, L., Senderskiy, I., & Berestetskiy, A. (2021). Structure–Activity Relationship of Phytotoxic Natural 10-Membered Lactones and Their Semisynthetic Derivatives. Journal of Fungi, 7(10), 829. https://doi.org/10.3390/jof7100829