Towards Genomic Criteria for Delineating Fungal Species


Abstract
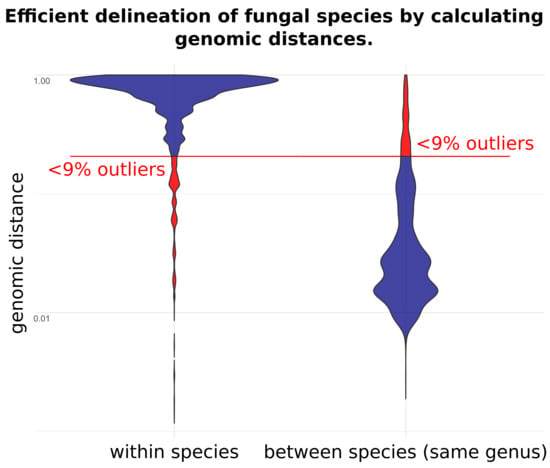
1. Introduction
2. Materials and Methods
2.1. Genome Sequences
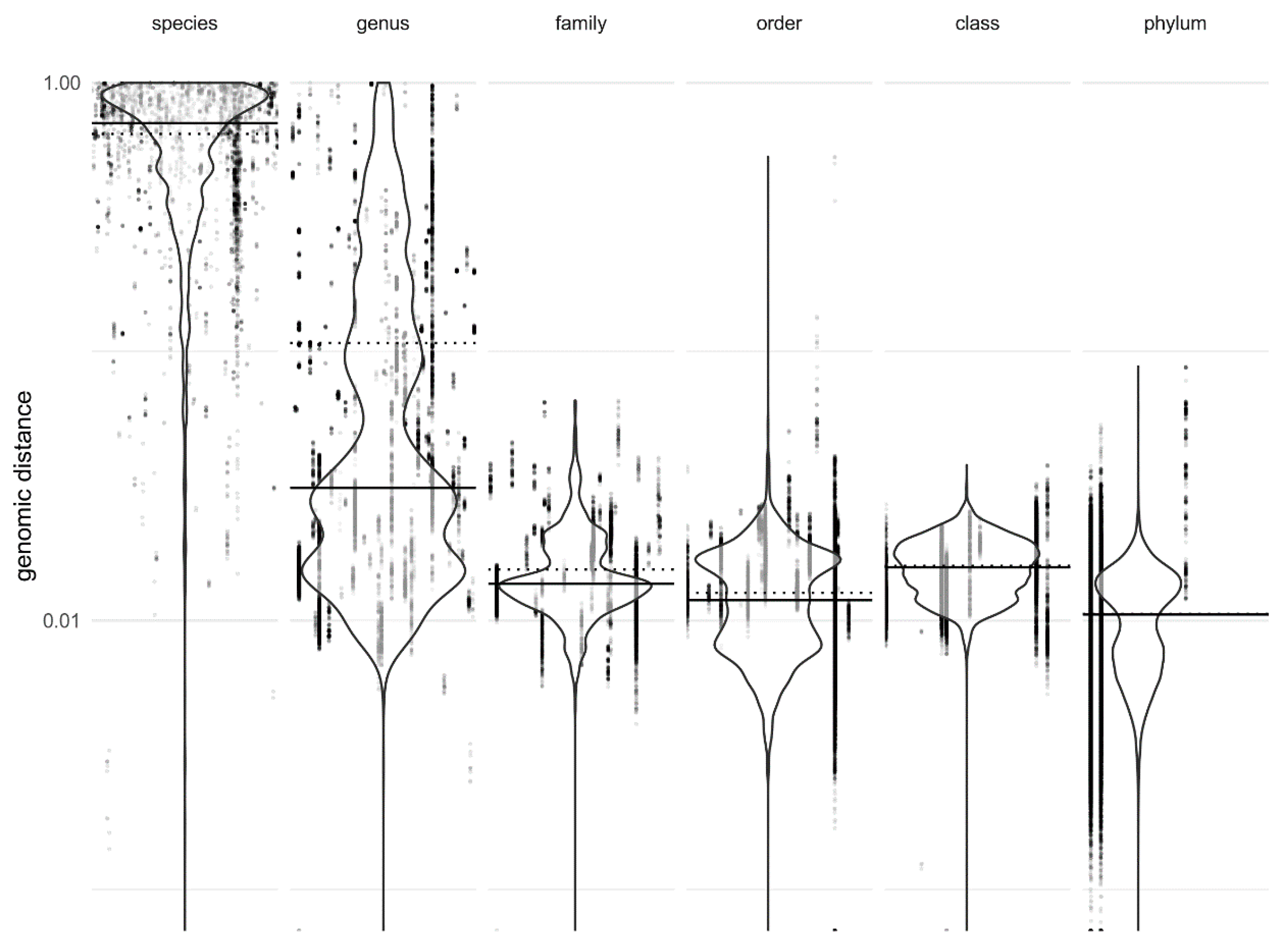
2.2. Calculation of Genomic Distances
2.3. Statistics
2.4. Computational Power
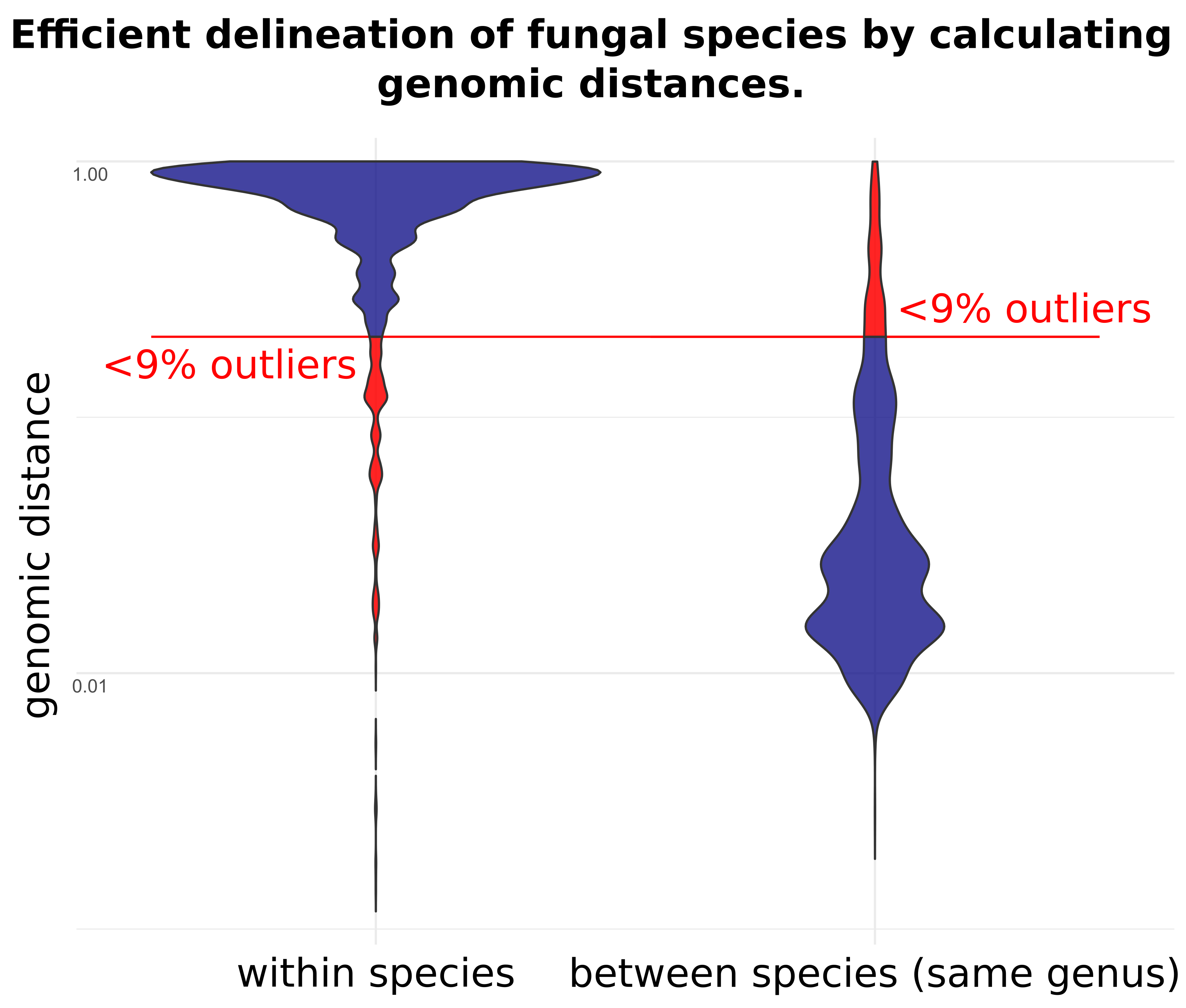
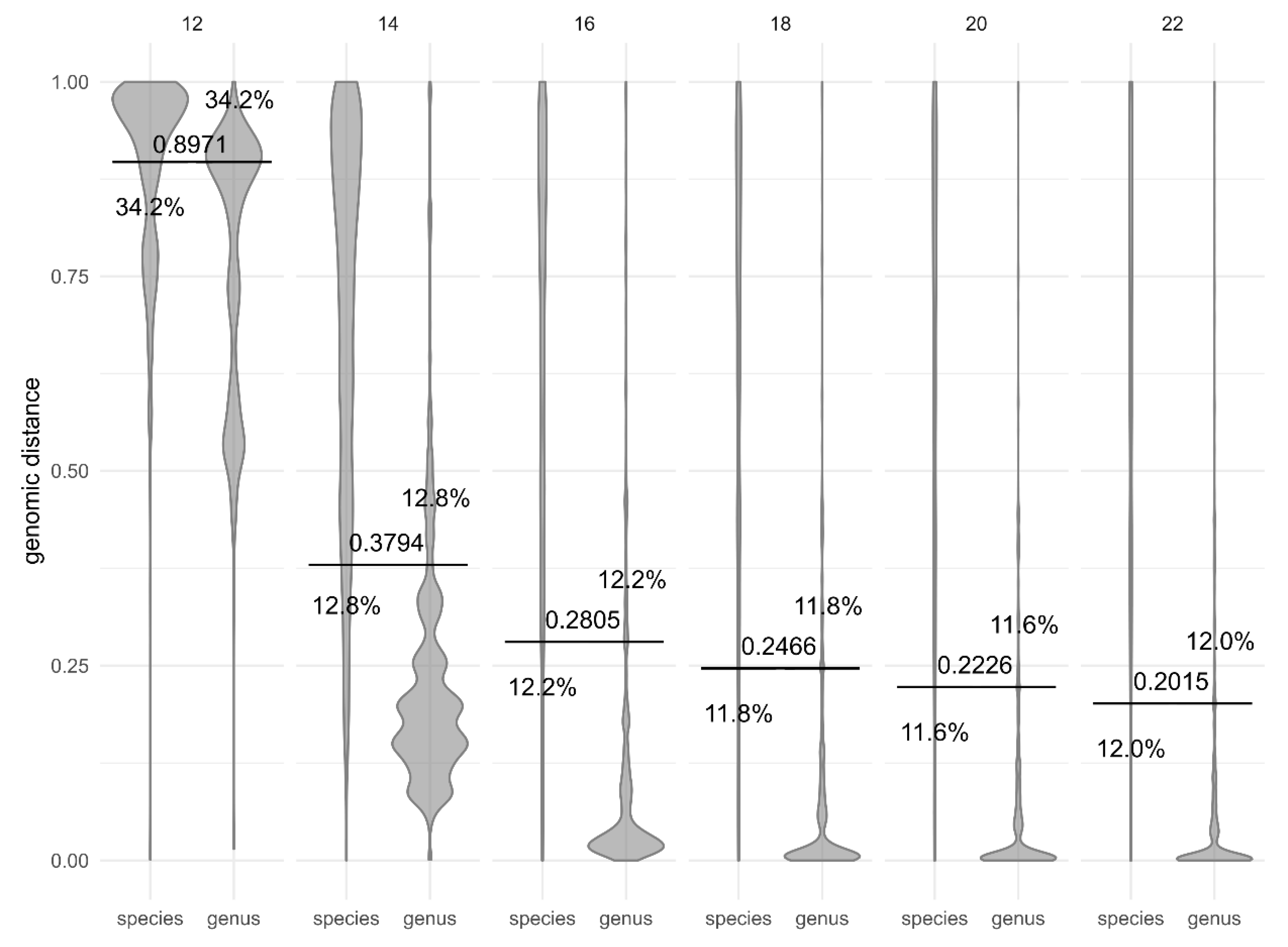
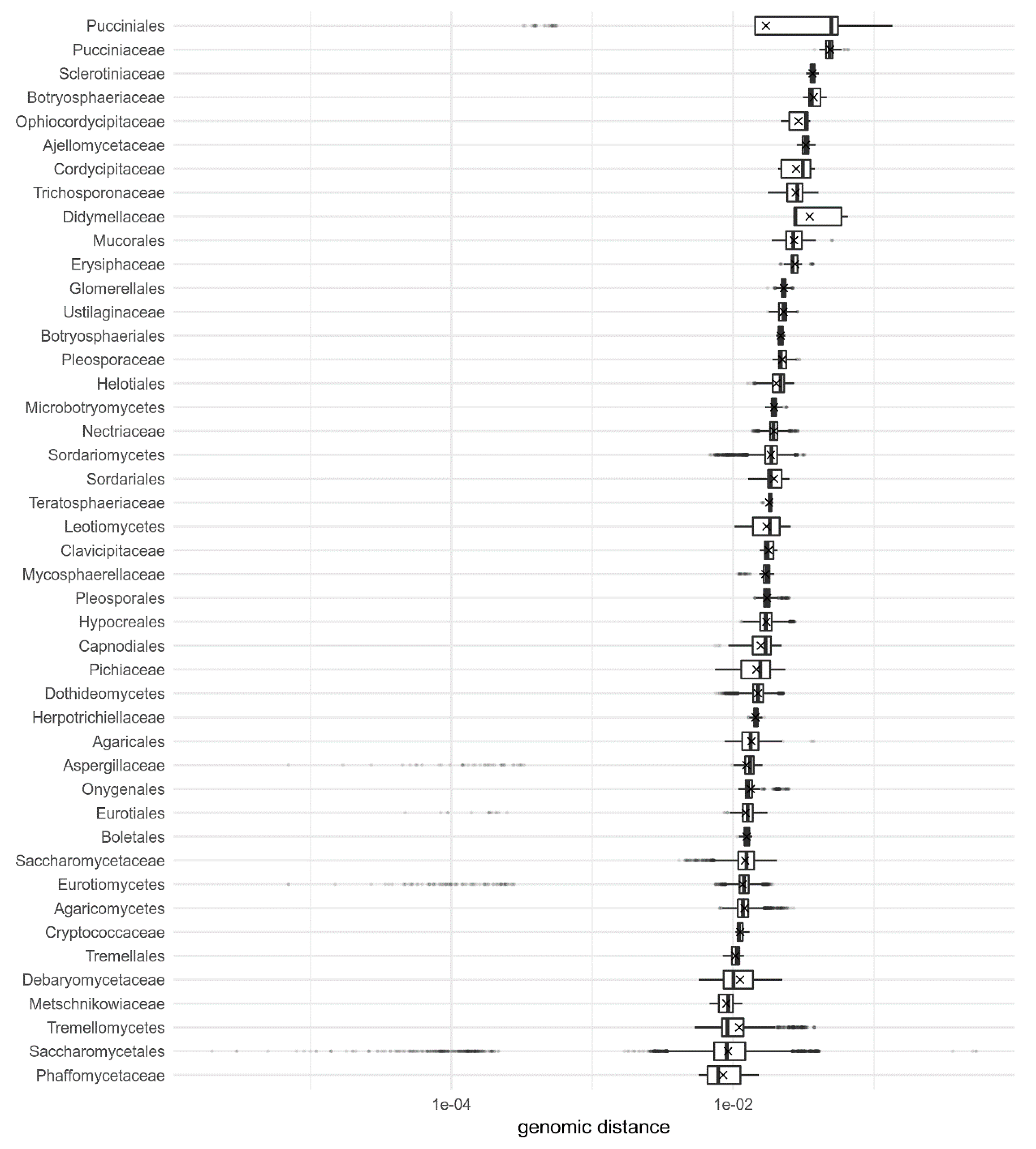
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Funding
Conflicts of Interest
References
- Mayr, E. Systematics and the Origin of Species from the Viewpoint of a Zoologist; Columbia University Press: New York, NY, USA, 1942; p. 334. [Google Scholar]
- Nieuwenhuis, B.P.S.; James, T.Y. The frequency of sex in fungi. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150540. [Google Scholar] [CrossRef] [PubMed]
- Cohan, F.M.; Perry, E.B. A systematics for discovering the fundamental units of bacterial diversity. Curr. Biol. 2007, 17, R373–R386. [Google Scholar] [CrossRef] [PubMed]
- Barbu, E.; Lee, K.Y.; Wahl, R. Contenu en bases puriques et pyrimidiques des acides désoxyribonucléiques des bactéries. Ann. Inst. Pasteur 1956, 91, 212–224. [Google Scholar] [PubMed]
- Schildkraut, C.L.; Marmur, J.; Doty, P. The formation of hybrid DNA molecules and their use in studies of DNA homologies. J. Mol. Biol. 1961, 3, 595–617. [Google Scholar] [CrossRef]
- Moore, W.E.C.; Stackebrandt, E.; Kandler, O.; Colwell, R.R.; Krichevsky, M.I.; Truper, H.G.; Murray, R.G.E.; Wayne, L.G.; Grimont, P.A.D.; Brenner, D.J.; et al. Report of the ad hoc committee on reconciliation of approaches to bacterial systematics. Int. J. Syst. Evol. Microbiol. 1987, 37, 463–464. [Google Scholar] [CrossRef]
- Stackebrandt, E.; Goebel, B.M. Taxonomic note: A place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int. J. Syst. Evol. Microbiol. 1994, 44, 846–849. [Google Scholar] [CrossRef]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef]
- Kim, M.; Oh, H.-S.; Park, S.-C.; Chun, J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 2014, 64, 346–351. [Google Scholar] [CrossRef]
- Ciufo, S.; Kannan, S.; Sharma, S.; Badretdin, A.; Clark, K.; Turner, S.; Brover, S.; Schoch, C.L.; Kimchi, A.; DiCuccio, M. Using average nucleotide identity to improve taxonomic assignments in prokaryotic genomes at the NCBI. Int. J. Syst. Evol. Microbiol. 2018, 68, 2386–2392. [Google Scholar] [CrossRef]
- Chan, J.Z.M.; Halachev, M.R.; Loman, N.J.; Constantinidou, C.; Pallen, M.J. Defining bacterial species in the genomic era: Insights from the genus Acinetobacter. BMC Microbiol. 2012, 12, 302. [Google Scholar] [CrossRef]
- Matute, D.R.; Sepúlveda, V.E. Fungal species boundaries in the genomics era. Fungal Genet. Biol. 2019, 131, 103249. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.W.; Jacobson, D.J.; Kroken, S.; Kasuga, T.; Geiser, D.M.; Hibbett, D.S.; Fisher, M.C. Phylogenetic species recognition and species concepts in fungi. Fungal Genet. Biol. 2000, 31, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Padial, J.M.; De la Riva, I. Taxonomic inflation and the stability of species lists: The perils of ostrich’s behavior. Syst. Biol. 2006, 55, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.W.; Fisher, M.C. Fungal multilocus sequence typing—It’s not just for bacteria. Curr. Opin. Microbiol. 2003, 6, 351–356. [Google Scholar] [CrossRef]
- De Hoog, G.S.; Dukik, K.; Monod, M.; Packeu, A.; Stubbe, D.; Hendrickx, M.; Kupsch, C.; Stielow, J.B.; Freeke, J.; Göker, M.; et al. Toward a novel multilocus phylogenetic taxonomy for the dermatophytes. Mycopathologia 2017, 182, 5–31. [Google Scholar] [CrossRef] [PubMed]
- De Hoog, G.S.; Chaturvedi, V.; Denning, D.W.; Dyer, P.S.; Frisvad, J.C.; Geiser, D.; Gräser, Y.; Guarro, J.; Haase, G.; Kwon-Chung, K.-J.; et al. Name changes in medically important fungi and their implications for clinical practice. J. Clin. Microbiol. 2015, 53, 1056–1062. [Google Scholar] [CrossRef]
- McLaughlin, D.J.; Hibbett, D.S.; Lutzoni, F.; Spatafora, J.W.; Vilgalys, R. The search for the fungal tree of life. Trends Microbiol. 2009, 17, 488–497. [Google Scholar] [CrossRef]
- Naranjo-Ortiz, M.A.; Gabaldón, T. Fungal evolution: Diversity, taxonomy and phylogeny of the Fungi. Biol. Rev. 2019, 94, 2101–2137. [Google Scholar] [CrossRef]
- Nguyen, H.D.T.; Sultana, T.; Kesanakurti, P.; Hambleton, S. Genome sequencing and comparison of five Tilletia species to identify candidate genes for the detection of regulated species infecting wheat. IMA Fungus 2019, 10, 11. [Google Scholar] [CrossRef]
- Gostinčar, C.; Ohm, R.A.; Kogej, T.; Sonjak, S.; Turk, M.; Zajc, J.; Zalar, P.; Grube, M.; Sun, H.; Han, J.; et al. Genome sequencing of four Aureobasidium pullulans varieties: Biotechnological potential, stress tolerance, and description of new species. BMC Genom. 2014, 15, 549. [Google Scholar] [CrossRef]
- Sepúlveda, V.E.; Márquez, R.; Turissini, D.A.; Goldman, W.E.; Matute, D.R. Genome sequences reveal cryptic speciation in the human pathogen Histoplasma capsulatum. MBio 2017, 8. [Google Scholar] [CrossRef]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.N.; Langmead, B. Dashing: Fast and accurate genomic distances with HyperLogLog. Genome Biol. 2019, 20, 265. [Google Scholar] [CrossRef] [PubMed]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef]
- R Development Core Team R: A Language and Environment for Statistical Computing 2020. Available online: https://cran.r-project.org/doc/FAQ/R-FAQ.html#Citing-R (accessed on 20 February 2020).
- Fox, J.; Weisberg, S. An R Companion to Applied Regression, 3rd ed.; Sage: Thousand Oaks, CA, USA, 2019; p. 608. [Google Scholar]
- Searle, S.R.; Speed, F.M.; Milliken, G.A. Population marginal means in the linear model: An alternative to least squares means. Am. Stat. 1980, 34, 216–221. [Google Scholar] [CrossRef]
- Wickham, H.; François, R.; Henry, L.; Müller, K. dplyr: A Grammar of Data Manipulation. 2018. Available online: https://dplyr.tidyverse.org/reference/dplyr-package.html (accessed on 1 January 2014).
- Wickham, H. ggplot2: Elegant Graphics For Data Analysis; Springer-Verlag: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.S.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M.; et al. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Wurzbacher, C.; Larsson, E.; Bengtsson-Palme, J.; Van den Wyngaert, S.; Svantesson, S.; Kristiansson, E.; Kagami, M.; Nilsson, R.H. Introducing ribosomal tandem repeat barcoding for fungi. Mol. Ecol. Resour. 2019, 19, 118–127. [Google Scholar] [CrossRef]
- Fitzpatrick, D.A.; Logue, M.E.; Stajich, J.E.; Butler, G. A fungal phylogeny based on 42 complete genomes derived from supertree and combined gene analysis. BMC Evol. Biol. 2006, 22, 99. [Google Scholar] [CrossRef]
- Spatafora, J.W.; Chang, Y.; Benny, G.L.; Lazarus, K.; Smith, M.E.; Berbee, M.L.; Bonito, G.; Corradi, N.; Grigoriev, I.; Gryganskyi, A.; et al. A phylum-level phylogenetic classification of zygomycete fungi based on genome-scale data. Mycologia 2016, 108, 1028–1046. [Google Scholar] [CrossRef]
- Choi, J.; Kim, S.-H. A genome tree of life for the fungi kingdom. Proc. Natl. Acad. Sci. USA 2017, 114, 9391–9396. [Google Scholar] [CrossRef]
- Robbertse, B.; Strope, P.K.; Chaverri, P.; Gazis, R.; Ciufo, S.; Domrachev, M.; Schoch, C.L. Improving taxonomic accuracy for fungi in public sequence databases: Applying ‘one name one species’ in well-defined genera with Trichoderma/Hypocrea as a test case. Database 2017, 2017, 1–14. [Google Scholar] [CrossRef]
- Magain, N.; Miadlikowska, J.; Mueller, O.; Gajdeczka, M.; Truong, C.; Salamov, A.A.; Dubchak, I.; Grigoriev, I.V.; Goffinet, B.; Sérusiaux, E.; et al. Conserved genomic collinearity as a source of broadly applicable, fast evolving, markers to resolve species complexes: A case study using the lichen-forming genus Peltigera section Polydactylon. Mol. Phylogenet. Evol. 2017, 117, 10–29. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.P.; Gamble, J.A.; Yeomans, T.; Moran, G.P.; Saunders, D.; Harris, D.; Aslett, M.; Barrell, J.F.; Butler, G.; Citiulo, F.; et al. Comparative genomics of the fungal pathogens Candida dubliniensis and Candida albicans. Genome Res. 2009, 19, 2231–2244. [Google Scholar] [CrossRef] [PubMed]
- Leray, M.; Knowlton, N.; Ho, S.-L.; Nguyen, B.N.; Machida, R.J. GenBank is a reliable resource for 21st century biodiversity research. Proc. Natl. Acad. Sci. USA 2019, 116, 22651–22656. [Google Scholar] [CrossRef] [PubMed]
- Kurtzman, C.P. Description of Komagataella phaffii sp. nov. and the transfer of Pichia pseudopastoris to the methylotrophic yeast genus Komagataella. Int. J. Syst. Evol. Microbiol. 2005, 55, 973–976. [Google Scholar] [CrossRef]
- Love, K.R.; Shah, K.A.; Whittaker, C.A.; Wu, J.; Bartlett, M.C.; Ma, D.; Leeson, R.L.; Priest, M.; Borowsky, J.; Young, S.K.; et al. Comparative genomics and transcriptomics of Pichia pastoris. BMC Genom. 2016, 17, 550. [Google Scholar] [CrossRef]
- Taheri, P.; Tarighi, S. Cytomolecular aspects of rice sheath blight caused by Rhizoctonia solani. Eur. J. Plant Pathol. 2011, 129, 511–528. [Google Scholar] [CrossRef]
- Ziegler, R.; Lutz, M.; Piątek, J.; Piątek, M. Dismantling a complex of anther smuts (Microbotryum) on carnivorous plants in the genus Pinguicula. Mycologia 2018, 110, 361–374. [Google Scholar] [CrossRef]
- Woudenberg, J.H.C.; Seidl, M.F.; Groenewald, J.Z.; de Vries, M.; Stielow, J.B.; Thomma, B.P.H.J.; Crous, P.W. Alternaria section Alternaria: Species, formae speciales or pathotypes? Stud. Mycol. 2015, 82, 1–21. [Google Scholar] [CrossRef]
- Zalar, P.; Zupančič, J.; Gostinčar, C.; Zajc, J.; de Hoog, G.S.; De Leo, F.; Azua-Bustos, A.; Gunde-Cimerman, N. The extremely halotolerant black yeast Hortaea werneckii-a model for intraspecific hybridization in clonal fungi. IMA Fungus 2019, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Gostinčar, C.; Stajich, J.E.; Zupančič, J.; Zalar, P.; Gunde-Cimerman, N. Genomic evidence for intraspecific hybridization in a clonal and extremely halotolerant yeast. BMC Genom. 2018, 19, 364. [Google Scholar] [CrossRef]
- Baranova, M.A.; Logacheva, M.D.; Penin, A.A.; Seplyarskiy, V.B.; Safonova, Y.Y.; Naumenko, S.A.; Klepikova, A.V.; Gerasimov, E.S.; Bazykin, G.A.; James, T.Y.; et al. Extraordinary genetic diversity in a wood decay mushroom. Mol. Biol. Evol. 2015, 32, 2775–2783. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Packeu, A.; Ahmed, S.A.; Al-Hatmi, A.M.S.; Blechert, O.; İlkit, M.; Hagen, F.; Gräser, Y.; Liu, W.; Deng, S.; et al. Species distinction in the Trichophyton rubrum complex. J. Clin. Microbiol. 2019, 57, 1–14. [Google Scholar] [CrossRef]
- Gehesquière, B.; Crouch, J.A.; Marra, R.E.; Van Poucke, K.; Rys, F.; Maes, M.; Gobin, B.; Höfte, M.; Heungens, K. Characterization and taxonomic reassessment of the box blight pathogen Calonectria pseudonaviculata, introducing Calonectria henricotiae sp. nov. Plant Pathol. 2016, 65, 37–52. [Google Scholar] [CrossRef]
- Lücking, R.; Hawksworth, D.L. Formal description of sequence-based voucherless Fungi: Promises and pitfalls, and how to resolve them. IMA Fungus 2018, 9, 143–165. [Google Scholar] [CrossRef] [PubMed]
- Zamora, J.C.; Svensson, M.; Kirschner, R.; Olariaga, I.; Ryman, S.; Parra, L.A.; Geml, J.; Rosling, A.; Adamčík, S.; Ahti, T.; et al. Considerations and consequences of allowing DNA sequence data as types of fungal taxa. IMA Fungus 2018, 9, 167–175. [Google Scholar] [CrossRef]
- De Almeida, S.M.; Imano, E.C.M.; Vicente, V.A.; Gomes, R.R.; Trentin, A.P.; Zamarchi, K.; Schneider, G.X.; Pinheiro, R.L.; da Silva, N.M.; de Hoog, G.S. Primary central nervous system infection by Histoplasma in an immunocompetent adult. Mycopathologia 2020, 185, 331–338. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Commands | Notes |
|---|---|---|
| Mash | # with pre-sketching mash sketch -k 16 -s 100000 A.fna mash sketch -k 16 -s 100000 B.fna mash dist A.fna.msh B.fna.msh # alternatively – without pre-sketching mash dist -k 16 -s 100000 A.fna B.fna | sketches of a certain size and k-mer length need to be calculated only once and can then be reused; sketching onto the disk can optionally be skipped and the distance calculated by a single command; only the distance can be extracted by adding “|cut -f 3” to the last command |
| Dashing | # with pre-sketching dashing sketch --sketch-size 20 --kmer-length 16 A.fna dashing sketch --sketch-size 20 --kmer-length 16 B.fna dashing cmp --presketched --full-tsv \ A.fna.w.16.spacing.20.hll B.fna.w.16.spacing.20.hll # alternatively – without pre-sketching dashing cmp --sketch-size 20 --kmer-length 16 --full-tsv A.fna B.fna | sketches of a certain size and k-mer length need to be calculated only once and can then be reused; sketching onto the disk can optionally be skipped and the distance calculated by a single command; only the distance can be extracted by adding “|tail -n1|cut -f 2” to the last command |
| k-mer overlap | jellyfish count -m 16 -s 1000M -t 30 -C A.fna -o A.jelbin jellyfish dump A.jelbin > A.jelly sed ’s/>.*//g’ A.jelly | sed ’/^[[:space:]]*$/d’ > A.edited sort A.edited > A.sorted uniq A.sorted > A.jellykmers # same for genome B KMERSIZE=16 KMERSPACE=$(echo "4^$KMERSIZE" | bc) UKMERA=$(cat A.jellykmers | wc --lines) UKMERB=$(cat B.jellykmers | wc --lines) MINKMER=$(( $UKMERA < $UKMERB ? $UKMERA : $UKMERB )) RANDOVERLAP=$(echo "$UKMERA*$UKMERB/$KMERSPACE" | bc -l) COMKMER=$(comm -12 --nocheck-order A.jellykmers B.jellykmers | wc --lines) RESULT=$(echo "($COMKMER - $RANDOVERLAP) / $MINKMER" | bc -l) echo $RESULT | the method is relatively inefficient and slow—it was used here simply as a proof of concept; calculations could be optimised, e.g., by using multi-threaded sorting and implementation of the method in a faster programming language |
| Method | k-mer Size * | Same Species | Genomic Distance Threshold ** | Different Species |
|---|---|---|---|---|
| Mash | 14 | less than | 0.004129 | greater than |
| Mash | 16 | less than | 0.04317 | greater than |
| Mash | 18 | less than | 0.05781 | greater than |
| Mash | 20 | less than | 0.05915 | greater than |
| Mash | 22 | less than | 0.05833 | greater than |
| Dashing | 14 | greater than | 0.9076 | less than |
| Dashing | 16 | greater than | 0.3736 | less than |
| Dashing | 18 | greater than | 0.2437 | less than |
| Dashing | 20 | greater than | 0.2084 | less than |
| Dashing | 22 | greater than | 0.1863 | less than |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gostinčar, C. Towards Genomic Criteria for Delineating Fungal Species. J. Fungi 2020, 6, 246. https://doi.org/10.3390/jof6040246
Gostinčar C. Towards Genomic Criteria for Delineating Fungal Species. Journal of Fungi. 2020; 6(4):246. https://doi.org/10.3390/jof6040246
Chicago/Turabian StyleGostinčar, Cene. 2020. "Towards Genomic Criteria for Delineating Fungal Species" Journal of Fungi 6, no. 4: 246. https://doi.org/10.3390/jof6040246
APA StyleGostinčar, C. (2020). Towards Genomic Criteria for Delineating Fungal Species. Journal of Fungi, 6(4), 246. https://doi.org/10.3390/jof6040246