Aspiring Antifungals: Review of Current Antifungal Pipeline Developments



Abstract
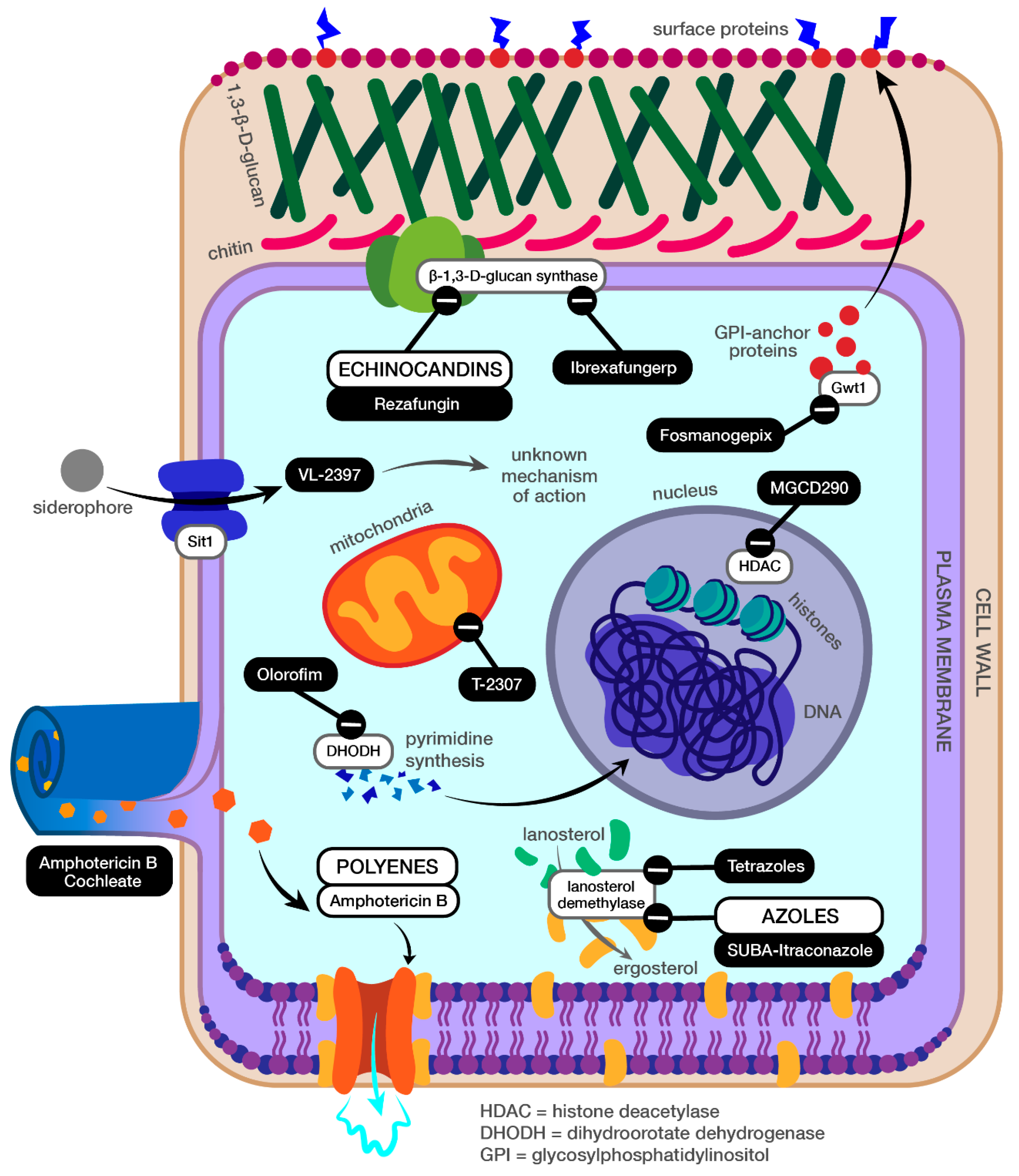
1. Introduction
2. SUBA-itraconazole
3. Rezafungin
4. Ibrexafungerp
5. Olorofim (F901318)
6. MGCD290
7. Amphotericin B Cochleate
8. Tetrazoles (VT-1129, VT-1161, and VT-1598)
9. Fosmanogepix (APX001)
10. VL-2397
11. T-2307
12. Miscellaneous
13. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- McCarthy, M.W.; Kontoyiannis, D.P.; Cornely, O.A.; Perfect, J.R.; Walsh, T.J. Novel agents and drug targets to meet the challenges of resistant fungi. J. Infect. Dis. 2017, 216, S474–S483. [Google Scholar] [CrossRef]
- Van Daele, R.; Spriet, I.; Wauters, J.; Maertens, J.; Mercier, T.; van Hecke, S.; Brüggemann, R. Antifungal drugs: What brings the future? Med. Mycol. 2019, 57, S328–S343. [Google Scholar] [CrossRef]
- Perfect, J.R. The antifungal pipeline: A reality check. Nat. Rev. Drug Discov. 2017, 16, 603–616. [Google Scholar] [CrossRef]
- Nguyen, M.-V.H.; Davis, M.R.; Wittenberg, R.; Mchardy, I.; Baddley, J.W.; Young, Y.; Odermatt, A.; Thompson, G.R., III. Posaconazole Serum Drug Levels Associated with Pseudohyperaldosteronism. Clin. Infect. Dis. 2019. [Google Scholar] [CrossRef]
- Thompson, G.R.; Chang, D.; Wittenberg, R.R.; McHardy, I.; Semrad, A. In vivo 11β-hydroxysteroid dehydrogenase inhibition in posaconazole-induced hypertension and hypokalemia. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef]
- Thompson, G.R.; Krois, C.R.; Affolter, V.K.; Everett, A.D.; Varjonen, E.K.; Sharon, V.R.; Singapuri, A.; Dennis, M.; McHardy, I.; Yoo, H.S.; et al. Examination of fluconazole-induced alopecia in an animal model and human cohort. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- Qu, Y.; Fang, M.; Gao, B.X. Itraconazole decreases left ventricular contractility in isolated rabbit heart: Mechanism of action. Toxicol. Appl. Pharmacol. 2013, 268, 113–122. [Google Scholar] [CrossRef]
- Thompson, G.R.; Cadena, J.; Patterson, T.F. Overview of Antifungal Agents. Clin. Chest. Med. 2009, 30, 203–215. [Google Scholar] [CrossRef]
- Balfour, J.A.; Faulds, D. Drug Evaluation Terbinafine a Review of Its Pharmacodynamic and Pharmacokinetic Properties, and Therapeutic Potential in Superficial Mycoses. Drugs 1992, 43, 259–284. [Google Scholar] [CrossRef]
- Nield, B.; Larsen, S.R.; van Hal, S.J. Clinical experience with new formulation SUBA®-itraconazole for prophylaxis in patients undergoing stem cell transplantation or treatment for haematological malignancies. J. Antimicrob. Chemother. 2019, 74, 3049–3055. [Google Scholar] [CrossRef]
- Abuhelwa, A.Y.; Foster, D.J.R.; Mudge, S.; Hayes, D.; Upton, R.N. Population pharmacokinetic modeling of itraconazole and hydroxyitraconazole for oral SUBA-itraconazole and sporanox capsule formulations in healthy subjects in fed and fasted states. Antimicrob. Agents Chemother. 2015, 59, 5681–5696. [Google Scholar] [CrossRef]
- Lindsay, J.; Mudge, S.; Thompson, G.R. Effects of food and omeprazole on a novel formulation of super bioavailability itraconazole in healthy subjects. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef]
- Lindsay, J.; Sandaradura, I.; Wong, K.; Arthur, C.; Stevenson, W.; Kerridge, I.; Fay, K.; Coyle, L.; Greenwood, M. Serum levels, safety and tolerability of new formulation SUBA-itraconazole prophylaxis in patients with haematological malignancy or undergoing allogeneic stem cell transplantation. J. Antimicrob. Chemother. 2017, 72, 3414–3419. [Google Scholar] [CrossRef]
- Sandison, T.; Ong, V.; Lee, J.; Thye, D. Safety and pharmacokinetics of CD101 IV, a novel echinocandin, in healthy adults. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef]
- Gangneux, J.-P.; Lortholary, O.; Cornely, O.; Pagano, L. 9th Trends in Medical Mycology Held on 11–14 October 2019, Nice, France, Organized under the Auspices of EORTC-IDG and ECMM. J. Fungi 2019, 5, 95. [Google Scholar] [CrossRef]
- Zhao, Y.; Prideaux, B.; Nagasaki, Y. Unraveling Drug Penetration of Echinocandin Antifungals at the Site of Infection in an Intra-abdominal Abscess Model. Antimicrob. Agents Chemother. 2017. [Google Scholar] [CrossRef]
- Lepak, A.J.; Zhao, M.; Andesa, D.R. Pharmacodynamic evaluation of rezafungin (CD101) against Candida auris in the neutropenic mouse invasive candidiasis model. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef]
- Davis, M.R.; Donnelley, M.A.; Thompson, G.R. Ibrexafungerp: A novel oral glucan synthase inhibitor. Med. Mycol. 2019. [Google Scholar] [CrossRef]
- Aruanno, M.; Glampedakis, E.; Lamoth, F. Echinocandins for the Treatment of Invasive Aspergillosis: From Laboratory to Bedside. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- Spec, A.; Pullman, J.; Thompson, G.R. MSG-10: A Phase 2 study of oral ibrexafungerp (SCY-078) following initial echinocandin therapy in non-neutropenic patients with invasive candidiasis. J. Antimicrob. Chemother. 2019, 74, 3056–3062. [Google Scholar] [CrossRef]
- Nunnally, N.S.; Etienne, K.A.; Angulo, D.; Lockhart, S.R.; Berkow, E.L. In Vitro Activity of Ibrexafungerp, a Novel Glucan Synthase Inhibitor against Candida glabrata Isolates with FKS Mutations. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- Hope, W.; Mcentee, L.; Johnson, A.; Farrington, N.; Whalley, S.; Santoyo-Castelazo, A.; Birch, M.; Law, D.; Kennedy, T.; Heep, M.; et al. Session: OS173 Challenges in Antifungal Treatment against Aspergillus Fumigatus in a Rabbit Model of Invasive Pulmonary Aspergillosis (IPA); ECCMID: Viena, Austria, 2017. [Google Scholar]
- Kennedy, T.; Allen, G.; Steiner, J.; Heep, M.; Birch, M. Assessment of the Duration of Infusion on the Tolerability and Repeat Dose Pharmacokinetics of F901318 in Healthy Volunteers; ECCMID: Viena, Austria, 2017. [Google Scholar]
- Rivero-Menendez, O.; Cuenca-Estrella, M.; Alastruey-Izquierdo, A. In vitro activity of olorofim (F901318) against clinical isolates of cryptic species of Aspergillus by EUCAST and CLSI methodologies. J. Antimicrob. Chemother. 2019, 74, 1586–1590. [Google Scholar] [CrossRef]
- Wiederhold, N.P.; Najvar, L.K.; Jaramillo, R.; Olivo, M.; Birch, M.; Law, D.; Rex, J.H.; Catano, G.; Patterson, T.F. The orotomide olorofim is efficacious in an experimental model of central nervous system coccidioidomycosis. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef]
- Pfaller, M.A.; Messer, S.A.; Georgopapadakou, N.; Martell, L.A.; Besterman, J.M.; Diekema, D.J. Activity of MGCD290, a Hos2 histone deacetylase inhibitor, in combination with azole antifungals against opportunistic fungal pathogens. J. Clin. Microbiol. 2009, 47, 3797–3804. [Google Scholar] [CrossRef]
- Pfaller, M.A.; Rhomberg, P.R.; Messer, S.A.; Castanheira, M. In vitro activity of a Hos2 deacetylase inhibitor, MGCD290, in combination with echinocandins against echinocandin-resistant Candida species. Diagn. Microbiol. Infect. Dis. 2015. [Google Scholar] [CrossRef]
- Augenbraun, M.; Livingston, J.; Parker, R.; Lederman, R. Fluconazole and MGCD290 in Vulvo Vaginal Candidiasis (VVC): Results from a Randomized Phase II Study; ID Week: San Francisco, CA, USA, 2013. [Google Scholar]
- Gallis, H.A.; Drew, R.H.; Pickard, W.W. (Eds.) Amphotericin B: 30 Years of Clinical Experience. Rev. Infect. Disesases 1990, 12, 308–329. [Google Scholar]
- Hamill, R.J. Amphotericin B formulations: A comparative review of efficacy and toxicity. Drugs 2013, 73, 919–934. [Google Scholar] [CrossRef]
- Santangelo, R.; Paderu, P.; Delmas, G.; Chen, Z.-W.; Mannino, R.; Zarif, L.; Perlin, D.S. Efficacy of oral cochleate-amphotericin B in a mouse model of systemic candidiasis. Antimicrob. Agents Chemother. 2000, 44, 2356–2360. [Google Scholar] [CrossRef]
- Shende, P.; Khair, R.; Gaud, R.S. Nanostructured cochleates: A multi-layered platform for cellular transportation of therapeutics. Drug Dev. Ind. Pharm. 2019, 45, 869–881. [Google Scholar] [CrossRef]
- Wiederhold, N.P.; Xu, X.; Wang, A.; Najvar, L.K.; Garvey, E.P.; Ottinger, E.A.; Alimardanov, A.; Cradock, J.; Behnke, M.; Hoekstra, W.J.; et al. In vivo efficacy of VT-1129 against experimental cryptococcal meningitis with the use of a loading dose-maintenance dose administration strategy. Antimicrob. Agents Chemother. 2018, 62, 1–10. [Google Scholar] [CrossRef]
- Hoekstra, W.J.; Garvey, E.P.; Moore, W.R.; Rafferty, S.W.; Yates, C.M.; Schotzinger, R.J. Design and optimization of highly-selective fungal CYP51 inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 3455–3458. [Google Scholar] [CrossRef] [PubMed]
- Wiederhold, N.P.; Shubitz, L.F.; Najvar, L.K.; Jaramillo, R.; Olivo, M.; Catano, G.; Trinh, H.T.; Yates, C.M.; Schotzinger, R.J.; Garvey, E.P.; et al. The novel fungal Cyp51 inhibitor VT-1598 is efficacious in experimental models of central nervous system coccidioidomycosis caused by Coccidioides posadasii and Coccidioides immitis. Antimicrob. Agents Chemother. 2018, 62, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, A.T.; Wiederhold, N.P.; Flowers, S.A.; Zhang, Q.; Kelly, S.L.; Morschhäuser, J.; Yates, C.M.; Hoekstra, W.J.; Schotzinger, R.J.; Garvey, E.P.; et al. In vitro activities of the novel investigational tetrazoles vt-1161 and vt-1598 compared to the triazole antifungals against azole-resistant strains and clinical isolates of Candida albicans. Antimicrob. Agents Chemother. 2019, 63, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hata, K.; Horii, T.; Miyazaki, M.; Watanabe, N.; Okubo, M.; Sonoda, J.; Nakamoto, K.; Tanaka, K.; Shirotori, S.; Murai, N.; et al. Efficacy of oral E1210, a new broad-spectrum antifungal with a novel mechanism of action, in murine models of candidiasis, aspergillosis, and fusariosis. Antimicrob. Agents Chemother. 2011, 55, 4543–4551. [Google Scholar] [CrossRef] [PubMed]
- Alkhazraji, S.; Gebremariam, T.; Alqarihi, A. Fosmanogepix (APX001) is Effective in the Treatment of Immunocompromised Mice Infected with Invasive Pulmonary Scedosporiosis or Disseminated Fusariosis. Antimicrob. Agents Chemother. 2019. [Google Scholar] [CrossRef] [PubMed]
- Viriyakosol, S.; Kapoor, M.; Okamoto, S. APX001 and Other Gwt1 Inhibitor Prodrugs Are Effective in Experimental Coccidioides immitis Pneumonia. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- Kovanda, L.L.; Sullivan, S.M.; Smith, L.R.; Desai, A.V.; Bonate, P.L.; Hope, W.W. Population pharmacokinetic modeling of VL-2397, a novel systemic antifungal agent: Analysis of a single- And multiple-ascending-dose study in healthy subjects. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- Abe, M.; Nakamura, S.; Kinjo, Y. Efficacy of T-2307, a novel arylamidine, against ocular complications of disseminated candidiasis in mice. J. Antimicrob. Chemother. 2019, 74, 1327–1332. [Google Scholar] [CrossRef]
- Shubitz, L.F.; Trinh, H.T.; Perrill, R.H. Modeling Nikkomycin Z dosing and pharmacology in murine pulmonary coccidioidomycosis preparatory to phase 2 clinical trials. J. Infect. Dis. 2014, 209, 1949–1954. [Google Scholar] [CrossRef]
- Rhein, J.; Huppler Hullsiek, K.; Tugume, L.; Nuwagira, E.; Mpoza, E.; Evans, E.E.; Kiggundu, R.; Pastick, K.A.; Ssebambulidde, K.; Akampurira, A.; et al. Adjunctive sertraline for HIV-associated cryptococcal meningitis: A randomised, placebo-controlled, double-blind phase 3 trial. Lancet Infect. Dis. 2019, 8, 843–851. [Google Scholar] [CrossRef]
- Wiederhold, N.P.; Patterson, T.F.; Srinivasan, A. Repurposing auranofin as an antifungal: In vitro activity against a variety of medically important fungi. Virulence 2017, 8, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Dolan, K.; Montgomery, S.; Buchheit, B.; DiDone, L.; Wellington, M.; Krysan, D.J. Antifungal activity of tamoxifen: In vitro and in vivo activities and mechanistic characterization. Antimicrob. Agents Chemother. 2009, 53, 3337–3346. [Google Scholar] [CrossRef] [PubMed]
- Villanueva-Lozano, H.; Treviño-Rangel, R.J.; Téllez-Marroquín, R.; Bonifaz, A.; Rojas, O.C. In vitro inhibitory activity of sertraline against clinical isolates of Sporothrix schenckii. Rev. Iberoam Micol. 2019, 36, 139–141. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Class | Antifungal Agent | Mechanism of Action | Spectrum of Activity | Clinical Phase and Company | Clinical Advantages |
|---|---|---|---|---|---|
| Azole | SUBA-itraconazole | Interferes with cytochrome P450 activity, decreasing ergosterol synthesis, inhibiting cell membrane formation | blastomycosis, histoplasmosis, and aspergillosis | FDA approved Mayne Pharma Ltd. | Increased bioavailability compared to itraconazole |
| Echinocandin | Rezafungin | Inhibition of 1,3-β-D-glucan synthesis | Candida albicans, Candida auris Candida krusei, Candida tropicalis, Aspergillus spp. Pneumocystis spp. | Phase III Cidara Therapeutics, Inc. | Once-weekly dosing regimen. Treatment and potential role for prophylaxis |
| Terpenoid | Ibrexafungerp | Triterpenoid enfumafungin derivative that inhibits 1,3-β-D-glucan synthesis | Candida spp. including Candida glabrata and Candida auris Aspergillus spp. | Phase III SCYNEXIS Inc. | Oral and IV formulation Maintains activity against echinocandin-resistant Candida spp. and Aspergillus spp. |
| Orotomides | Olorofim | Inhibition of dihydroorotate dehydrogenase, thereby inhibiting pyrimidine production which negatively affects fungal nucleic acid, cell wall, and phospholipid synthesis, as well as cell regulation and protein production | Aspergillus fumigatus, Aspergillus nidulans, Aspergillus terreus, and Aspergillus niger and multidrug resistant strains of Aspergillus spp. Uncommon moulds such as Lomentospora prolificans and Scedosporium spp. Endemic Fungi | Phase II F2G Ltd. | Oral and IV formulation Activity against Aspergillus spp. including multidrug resistant and uncommon moulds |
| HDAC Inhibitor | MGCD290 | Fungal histone deacetylase (HDAC) inhibitor | Candida spp. Aspergillus spp. | Phase II Mirati Therapeutics, Inc. | Possible role as an adjunctive antifungal in combination with an azole or echinocandin |
| Polyene | Amphotericin B Cochleate | Cochleate are a multilayered structure that forms a solid, lipid bilayer, configured into a spiral. Following oral administration, the cochleate is absorbed from the GI tract, enters circulation, and once calcium concentrations in cochleates are decreased, the spiral formation opens and releases the encapsulated drug into the cell. | Candida spp. | Phase II Matinas BioPharma | Oral formulation of amphotericin delivered via cochleate |
| Tetrazole | VT-1129 | Interferes with cytochrome P450 activity, decreasing ergosterol synthesis, inhibiting cell membrane formation | Cryptococus spp. Candida spp. | Pre-clinical Viamet Pharmaceuticals, Inc. | May have reduced P450 drug interactions |
| VT-1161 | Interferes with cytochrome P450 activity, decreasing ergosterol synthesis, inhibiting cell membrane formation | Candida spp. Coccidioides spp. Rhizopus spp. | Phase III Mycovia Pharmaceuticals | May have reduced P450 drug interactions | |
| VT-1598 | Interferes with cytochrome P450 activity, decreasing ergosterol synthesis, inhibiting cell membrane formation | Candida spp. including C. auris Aspergillus spp. Cryptococcus spp. | Phase I Mycovia Pharmaceuticals | May have reduced P450 drug interactions | |
| Glycosylphosphatidylinositol inhibitor | Fosmanogepix (APX001) | Inhibits fungal Gwt1 GPI anchor protein. Low affinity for human GPI anchor proteins | Candida spp. including C. auris Cryptococcus Coccidioides Aspergillus and hyaline moulds Mucorales Not active against C. krusei | Phase II Amplyx Pharmaceuticals | Broad spectrum, oral formulation little toxicity in human studies thus far |
| Siderophore | VL-2397 | Uptake via siderophore iron transporter | Aspergillus Some Candida spp. and Aspergillus spp. Mucorales | No current development plans – phase II trial terminated early | Activity against triazole resistant Aspergillus isolates |
| Arylamidine | T-2307 | Thought to inhibit fungal mitochondrial synthesis | Candida spp. Aspergillus and some hyaline moulds | Phase I Toyama Chemical Company Ltd. | Structurally similar to pentamidine |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gintjee, T.J.; Donnelley, M.A.; Thompson, G.R. Aspiring Antifungals: Review of Current Antifungal Pipeline Developments. J. Fungi 2020, 6, 28. https://doi.org/10.3390/jof6010028
Gintjee TJ, Donnelley MA, Thompson GR. Aspiring Antifungals: Review of Current Antifungal Pipeline Developments. Journal of Fungi. 2020; 6(1):28. https://doi.org/10.3390/jof6010028
Chicago/Turabian StyleGintjee, Thomas J., Monica A. Donnelley, and George R. Thompson. 2020. "Aspiring Antifungals: Review of Current Antifungal Pipeline Developments" Journal of Fungi 6, no. 1: 28. https://doi.org/10.3390/jof6010028
APA StyleGintjee, T. J., Donnelley, M. A., & Thompson, G. R. (2020). Aspiring Antifungals: Review of Current Antifungal Pipeline Developments. Journal of Fungi, 6(1), 28. https://doi.org/10.3390/jof6010028