Vulvovaginal Candidiasis: A Current Understanding and Burning Questions


 and
and
Abstract
1. Pathology and Epidemiology of Vulvovaginal Candidiasis
2. Fungal Pathogenicity Mechanisms and Host Response
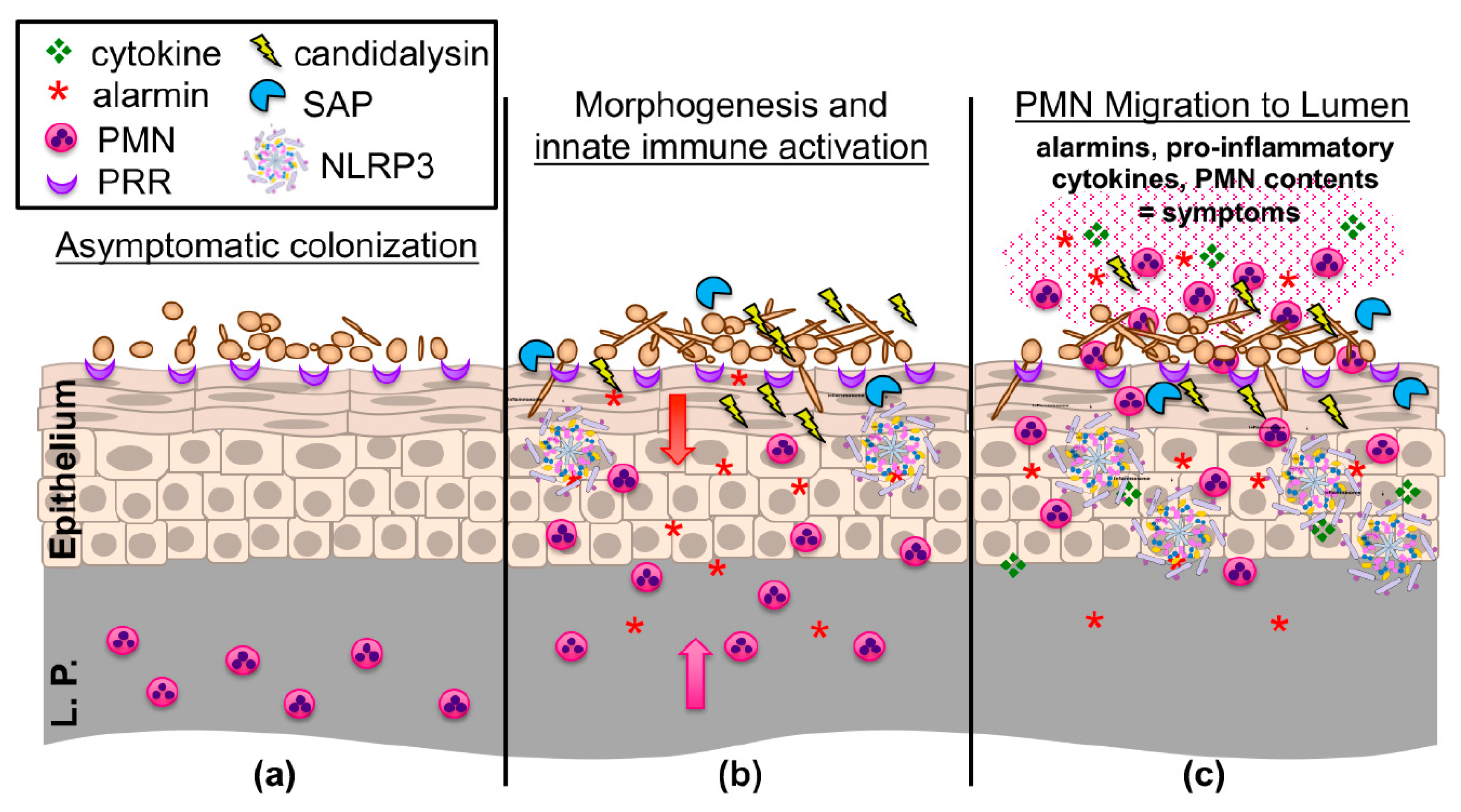
2.1. VVC as an Immunopathology
2.2. Sensing Fungi at the Vaginal Mucosa
2.3. Invasion and Secreted Virulence Effectors
2.4. An Indispensable Role for Candidalysin
2.5. A Controversial Role for IL-17 Signaling?
2.6. The NLRP3 Inflammasome: A Key Role in VVC Pathogenesis
2.7. Estrogen-Dependent Immunomodulation
3. A Role for the Microbiome?
4. Established and Novel Treatment Modalities
5. Conclusions and Burning Questions
Author Contributions
Funding
Conflicts of Interest
References
- Achkar, J.M.; Fries, B.C. Candida infections of the genitourinary tract. Clin. Microbiol. Rev. 2010, 23, 253–273. [Google Scholar] [CrossRef]
- Sobel, J.D. Vaginitis. N. Engl. J. Med. 1997, 337, 1896–1903. [Google Scholar] [CrossRef] [PubMed]
- Denning, D.W.; Kneale, M.; Sobel, J.D.; Rautemaa-Richardson, R. Global burden of recurrent vulvovaginal candidiasis: A systematic review. Lancet Infect. Dis. 2018, 18, e339–e347. [Google Scholar] [CrossRef]
- Sobel, J.D.; Faro, S.; Force, R.W.; Foxman, B.; Ledger, W.J.; Nyirjesy, P.R.; Reed, B.D.; Summers, P.R. Vulvovaginal candidiasis: Epidemiologic, diagnostic, and therapeutic considerations. Am. J. Obstet. Gynecol. 1998, 178, 203–211. [Google Scholar] [CrossRef]
- Nyirjesy, P.; Zhao, Y.; Ways, K.; Usiskin, K. Evaluation of vulvovaginal symptoms and Candida colonization in women with type 2 diabetes mellitus treated with canagliflozin, a sodium glucose co-transporter 2 inhibitor. Curr. Med. Res. Opin. 2012, 28, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Fidel, P.L., Jr. Distinct protective host defenses against oral and vaginal candidiasis. Med. Mycol. 2002, 40, 359–375. [Google Scholar] [CrossRef]
- Foxman, B.; Barlow, R.; D‘Arcy, H.; Gillespie, B.; Sobel, J.D. Candida vaginitis: Self-reported incidence and associated costs. Sex. Transm. Dis. 2000, 27, 230–235. [Google Scholar] [CrossRef]
- Bongomin, F.; Gago, S.; Oladele, R.O.; Denning, D.W. Global and multi-national prevalence of fungal diseases-estimate precision. J. Fungi 2017, 3, 57. [Google Scholar] [CrossRef]
- Dave, S.; Peter, T.; Fogarty, C.; Karatzas, N.; Belinsky, N.; Pant Pai, N. Which community-based HIV initiatives are effective in achieving UNAIDS 90-90-90 targets? A systematic review and meta-analysis of evidence (2007–2018). PLoS ONE 2019, 14, e0219826. [Google Scholar] [CrossRef]
- Rabeneck, L.; Crane, M.M.; Risser, J.M.; Lacke, C.E.; Wray, N.P. A simple clinical staging system that predicts progression to AIDS using CD4 count, oral thrush, and night sweats. J. Gen. Intern. Med. 1993, 8, 5–9. [Google Scholar] [CrossRef]
- Redding, S.W.; Zellars, R.C.; Kirkpatrick, W.R.; McAtee, R.K.; Caceres, M.A.; Fothergill, A.W.; Lopez-Ribot, J.L.; Bailey, C.W.; Rinaldi, M.G.; Patterson, T.F. Epidemiology of oropharyngeal Candida colonization and infection in patients receiving radiation for head and neck cancer. J. Clin. Microbiol. 1999, 37, 3896–3900. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.K.; Capoor, M.R.; Nair, D.; Bhowmik, K.T. Spectrum of fungal infection in head and neck cancer patients on chemoradiotherapy. J. Egypt. Natl. Canc. Inst. 2017, 29, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Paya, C.V. Infections in solid-organ transplant recipients. Clin. Microbiol. Rev. 1997, 10, 86–124. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.; Rao, R.S.; Majumdar, B.; Anil, S. Clinical appearance of oral Candida infection and therapeutic strategies. Front. Microbiol. 2015, 6, 1391. [Google Scholar] [CrossRef] [PubMed]
- WorldBank. Birth Rate, Crude (Per 1000 People); The World Bank Group: Washington, DC, USA, 2017; Available online: https://data.worldbank.org/indicator/SP.DYN.CBRT.IN (accessed on 22 January 2020).
- WHO. Infant Mortality. In Global Health Observatory; World Heatlh Organization: Geneva, Switzerland, 2017; Available online: https://www.who.int/gho/child_health/mortality/neonatal_infant_text/en/ (accessed on 22 January 2020).
- Sobel, J.D.; Sobel, R. Current treatment options for vulvovaginal candidiasis caused by azole-resistant Candida species. Expert Opin. Pharmacother. 2018, 19, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.A.; Sobel, J.D. Vulvovaginal candidiasis caused by non-albicans Candida Species: New insights. Curr. Infect. Dis. Rep. 2010, 12, 465–470. [Google Scholar] [CrossRef]
- Parazzini, F.; Di Cintio, E.; Chiantera, V.; Guaschino, S. Determinants of different Candida species infections of the genital tract in women. Sporachrom Study Geoup. Eur. J. Obstet. Gynecol. Reprod. Biol. 2000, 93, 141–145. [Google Scholar] [CrossRef]
- Richter, S.S.; Galask, R.P.; Messer, S.A.; Hollis, R.J.; Diekema, D.J.; Pfaller, M.A. Antifungal susceptibilities of Candida species causing vulvovaginitis and epidemiology of recurrent cases. J. Clin. Microbiol. 2005, 43, 2155–2162. [Google Scholar] [CrossRef]
- Dan, M.; Poch, F.; Levin, D. High rate of vaginal infections caused by non-C. albicans Candida species among asymptomatic women. Med. Mycol. 2002, 40, 383–386. [Google Scholar] [CrossRef]
- Bitew, A.; Abebaw, Y. Vulvovaginal candidiasis: Species distribution of Candida and their antifungal susceptibility pattern. BMC Womens Health 2018, 18, 94–104. [Google Scholar] [CrossRef]
- Choukri, F.; Benderdouche, M.; Sednaoui, P. In vitro susceptibility profile of 200 recent clinical isolates of Candida spp. to topical antifungal treatments of vulvovaginal candidiasis, the imidazoles and nystatin agents. J. Mycol. Med. 2014, 24, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Powell, A.M.; Gracely, E.; Nyirjesy, P. Non-albicans Candida Vulvovaginitis: Treatment experience at a tertiary care vaginitis center. J. Low. Genit.Tract Dis. 2016, 20, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.; Goswami, R.; Banerjee, U.; Dadhwal, V.; Goswami, D.; Mandal, P.; Sreenivas, V.; Kochupillai, N. Prevalence of Candida glabrata and its response to boric acid vaginal suppositories in comparison with oral fluconazole in patients with diabetes and vulvovaginal candidiasis. Diabetes Care 2007, 30, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Hamad, M.; Kazandji, N.; Awadallah, S.; Allam, H. Prevalence and epidemiological characteristics of vaginal candidiasis in the UAE. Mycoses 2014, 57, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Spinillo, A.; Capuzzo, E.; Gulminetti, R.; Marone, P.; Colonna, L.; Piazzi, G. Prevalence of and risk factors for fungal vaginitis caused by non-albicans species. Am. J. Obstet. Gynecol. 1997, 176, 138–141. [Google Scholar] [CrossRef]
- Kalaiarasan, K.; Singh, R.; Chaturvedula, L. Fungal profile of vulvovaginal candidiasis in a tertiary care hospital. J. Clin. Diagn. Res. 2017, 11, DC06–DC09. [Google Scholar] [CrossRef] [PubMed]
- Spinillo, A.; Nicola, S.; Colonna, L.; Marangoni, E.; Cavanna, C.; Michelone, G. Frequency and significance of drug resistance in vulvovaginal candidiasis. Gynecol. Obstet. Invest. 1994, 38, 130–133. [Google Scholar] [CrossRef]
- Moyes, D.L.; Murciano, C.; Runglall, M.; Kohli, A.; Islam, A.; Naglik, J.R. Activation of MAPK/c-Fos induced responses in oral epithelial cells is specific to Candida albicans and Candida dubliniensis hyphae. Med. Microbiol. Immunol. 2012, 201, 93–101. [Google Scholar] [CrossRef]
- Willems, H.M.E.; Lowes, D.J.; Barker, K.S.; Palmer, G.E.; Peters, B.M. Comparative analysis of the capacity of the candida species to elicit vaginal immunopathology. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef]
- Hong, E.; Dixit, S.; Fidel, P.L.; Bradford, J.; Fischer, G. Vulvovaginal candidiasis as a chronic disease: Diagnostic criteria and definition. J. Low. Genit. Tract Dis. 2013. [Google Scholar] [CrossRef]
- Hamad, M.; Abu-Elteen, K.H.; Ghaleb, M. Estrogen-dependent induction of persistent vaginal candidosis in naive mice. Mycoses 2004, 47, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Yeater, K.M.; Hoyer, L.L. Cellular and molecular biology of Candida albicans estrogen response. Eukaryot. Cell 2006, 5, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Wira, C.R.; Fahey, J.V.; Sentman, C.L.; Pioli, P.A.; Shen, L. Innate and adaptive immunity in female genital tract: Cellular responses and interactions. Immunol. Rev. 2005, 206, 306–335. [Google Scholar] [CrossRef] [PubMed]
- Fidel, P.L., Jr.; Lynch, M.E.; Redondo-Lopez, V.; Sobel, J.D.; Robinson, R. Systemic cell-mediated immune reactivity in women with recurrent vulvovaginal candidiasis. J. Infect. Dis. 1993, 168, 1458–1465. [Google Scholar] [CrossRef]
- Fong, I.W.; McCleary, P.; Read, S. Cellular immunity of patients with recurrent or refractory vulvovaginal moniliasis. Am. J. Obstet. Gynecol. 1992, 166, 887–890. [Google Scholar] [CrossRef]
- Mendling, W.; Koldovsky, U. Investigations by cell-mediated immunologic tests and therapeutic trials with thymopentin in vaginal mycoses. Infect. Dis. Obstet. Gynecol. 1996, 4, 225–231. [Google Scholar] [CrossRef]
- Fidel, P.L., Jr.; Lynch, M.E.; Sobel, J.D. Circulating CD4 and CD8 T cells have little impact on host defense against experimental vaginal candidiasis. Infect. Immun. 1995, 63, 2403–2408. [Google Scholar] [CrossRef]
- Fidel, P.L., Jr.; Barousse, M.; Espinosa, T.; Ficarra, M.; Sturtevant, J.; Martin, D.H.; Quayle, A.J.; Dunlap, K. An intravaginal live Candida challenge in humans leads to new hypotheses for the immunopathogenesis of vulvovaginal candidiasis. Infect. Immun. 2004, 72, 2939–2946. [Google Scholar] [CrossRef]
- Sudbery, P.E. Growth of Candida albicans hyphae. Nat. Rev. Microbiol. 2011, 9, 737–748. [Google Scholar] [CrossRef]
- Lo, H.J.; Kohler, J.R.; DiDomenico, B.; Loebenberg, D.; Cacciapuoti, A.; Fink, G.R. Nonfilamentous, C. Albicans mutants are avirulent. Cell 1997, 90, 939–949. [Google Scholar] [CrossRef]
- Peters, B.M.; Palmer, G.E.; Nash, A.K.; Lilly, E.A.; Fidel, P.L., Jr.; Noverr, M.C. Fungal morphogenetic pathways are required for the hallmark inflammatory response during Candida albicans vaginitis. Infect. Immun. 2014, 82, 532–543. [Google Scholar] [CrossRef] [PubMed]
- Black, C.A.; Eyers, F.M.; Russell, A.; Dunkley, M.L.; Clancy, R.L.; Beagley, K.W. Acute neutropenia decreases inflammation associated with murine vaginal candidiasis but has no effect on the course of infection. Infect. Immun. 1998, 66, 1273–1275. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.C.; Joosten, L.A.; Kullberg, B.J.; Netea, M.G. Interplay between Candida albicans and the mammalian innate host defense. Infect. Immun. 2012, 80, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Moyes, D.L.; Murciano, C.; Runglall, M.; Islam, A.; Thavaraj, S.; Naglik, J.R. Candida albicans yeast and hyphae are discriminated by MAPK signaling in vaginal epithelial cells. PLoS ONE 2011, 6, e26580. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.M.; Yano, J.; Noverr, M.C.; Fidel, P.L., Jr. Candida vaginitis: When opportunism knocks, the host responds. PLoS Pathog. 2014, 10, e1003965. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D.; Taylor, P.R.; Reid, D.M.; Willment, J.A.; Williams, D.L.; Martinez-Pomares, L.; Wong, S.Y.; Gordon, S. Dectin-1 is a major beta-glucan receptor on macrophages. J. Exp. Med. 2002, 196, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Ferwerda, B.; Ferwerda, G.; Plantinga, T.S.; Willment, J.A.; van Spriel, A.B.; Venselaar, H.; Elbers, C.C.; Johnson, M.D.; Cambi, A.; Huysamen, C.; et al. Human dectin-1 deficiency and mucocutaneous fungal infections. N. Engl. J. Med. 2009, 361, 1760–1767. [Google Scholar] [CrossRef]
- Usluogullari, B.; Gumus, I.; Gunduz, E.; Kaygusuz, I.; Simavli, S.; Acar, M.; Oznur, M.; Gunduz, M.; Kafali, H. The role of human dectin-1 Y238X gene polymorphism in recurrent vulvovaginal candidiasis infections. Mol. Biol. Rep. 2014, 41, 6763–6768. [Google Scholar] [CrossRef]
- Carvalho, A.; Giovannini, G.; De Luca, A.; D’Angelo, C.; Casagrande, A.; Iannitti, R.G.; Ricci, G.; Cunha, C.; Romani, L. Dectin-1 isoforms contribute to distinct Th1/Th17 cell activation in mucosal candidiasis. Cell. Mol. Immunol. 2012, 9, 276–286. [Google Scholar] [CrossRef]
- Yano, J.; Palmer, G.E.; Eberle, K.E.; Peters, B.M.; Vogl, T.; McKenzie, A.N.; Fidel, P.L., Jr. Vaginal epithelial cell-derived S100 alarmins induced by Candida albicans via pattern recognition receptor interactions are sufficient but not necessary for the acute neutrophil response during experimental vaginal candidiasis. Infect. Immun. 2014, 82, 783–792. [Google Scholar] [CrossRef]
- Rosentul, D.C.; Plantinga, T.S.; Oosting, M.; Scott, W.K.; Velez Edwards, D.R.; Smith, P.B.; Alexander, B.D.; Yang, J.C.; Laird, G.M.; Joosten, L.A.; et al. Genetic variation in the Dectin-1/CARD9 recognition pathway and susceptibility to candidemia. J. Infect. Dis. 2011, 204, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- Babula, O.; Lazdane, G.; Kroica, J.; Ledger, W.J.; Witkin, S.S. Relation between recurrent vulvovaginal candidiasis, vaginal concentrations of mannose-binding lectin, and a mannose-binding lectin gene polymorphism in Latvian women. Clin. Infect. Dis. 2003, 37, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Donders, G.G.; Babula, O.; Bellen, G.; Linhares, I.M.; Witkin, S.S. Mannose-binding lectin gene polymorphism and resistance to therapy in women with recurrent vulvovaginal candidiasis. BJOG 2008, 115, 1225–1231. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, M.; Pinelli, M.; Borghi, M.; Constantini, C.; Dindo, M.; van Emst, L.; Puccetti, M.; Pariano, M.; Ricano-Ponce, I.; Bull, C.; et al. A systems genomics approach identifies SIGLEC15 as a susceptibility factor in recurrent vulvovaginal candidiasis. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Jabra-Rizk, M.A.; Kong, E.; Tsui, C.; Nguyen, M.; Clancy, C.J.; Fidel, P.L., Jr.; Noverr, M. Candida albicans pathogenesis: Fitting within the “host-microbe damage response framework”. Infect. Immun. 2016, 84, 2724–2739. [Google Scholar] [CrossRef] [PubMed]
- Lew, R.R. How does a hypha grow? The biophysics of pressurized growth in fungi. Nat. Rev. Microbiol. 2011, 9, 509–518. [Google Scholar] [CrossRef]
- Wachtler, B.; Wilson, D.; Hube, B. Candida albicans adhesion to and invasion and damage of vaginal epithelial cells: Stage-specific inhibition by clotrimazole and bifonazole. Antimicrob. Agents Chemother. 2011, 55, 4436–4439. [Google Scholar] [CrossRef]
- Harriott, M.M.; Lilly, E.A.; Rodriguez, T.E.; Fidel, P.L.; Noverr, M.C. Candida albicans forms biofilms on the vaginal mucosa. Microbiology 2010, 156, 3635–3644. [Google Scholar] [CrossRef]
- Phan, Q.T.; Myers, C.L.; Fu, Y.; Sheppard, D.C.; Yeaman, M.R.; Welch, W.H.; Ibrahim, A.S.; Edwards, J.E., Jr.; Filler, S.G. Als3 is a Candida albicans invasin that binds to cadherins and induces endocytosis by host cells. PLoS Biol. 2007, 5, e64. [Google Scholar] [CrossRef]
- Roudbarmohammadi, S.; Roudbary, M.; Bakhshi, B.; Katiraee, F.; Mohammadi, R.; Falahati, M. ALS1 and ALS3 gene expression and biofilm formation in Candida albicans isolated from vulvovaginal candidiasis. Adv. Biomed. Res. 2016, 5, 105. [Google Scholar] [CrossRef]
- Schaller, M.; Korting, H.C.; Borelli, C.; Hamm, G.; Hube, B. Candida albicans-secreted aspartic proteinases modify the epithelial cytokine response in an in vitro model of vaginal candidiasis. Infect. Immun. 2005, 73, 2758–2765. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.; Schackert, C.; Korting, H.C.; Januschke, E.; Hube, B. Invasion of Candida albicans correlates with expression of secreted aspartic proteinases during experimental infection of human epidermis. J. Investig. Dermatol. 2000, 114, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Borg-von Zepelin, M.; Beggah, S.; Boggian, K.; Sanglard, D.; Monod, M. The expression of the secreted aspartyl proteinases Sap4 to Sap6 from Candida albicans in murine macrophages. Mol. Microbiol. 1998, 28, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Bruno, V.M.; Shetty, A.C.; Yano, J.; Fidel, P.L., Jr.; Noverr, M.C.; Peters, B.M. Transcriptomic analysis of vulvovaginal candidiasis identifies a role for the NLRP3 inflammasome. MBio 2015, 6. [Google Scholar] [CrossRef]
- De Bernardis, F.; Arancia, S.; Morelli, L.; Hube, B.; Sanglard, D.; Schafer, W.; Cassone, A. Evidence that members of the secretory aspartyl proteinase gene family, in particular SAP2, are virulence factors for Candida vaginitis. J. Infect. Dis. 1999, 179, 201–208. [Google Scholar] [CrossRef]
- Pericolini, E.; Gabrielli, E.; Amacker, M.; Kasper, L.; Roselletti, E.; Luciano, E.; Sabbatini, S.; Kaeser, M.; Moser, C.; Hube, B.; et al. Secretory Aspartyl Proteinases cause vaginitis and can mediate vaginitis caused by Candida albicans in mice. MBio 2015, 6, e00724. [Google Scholar] [CrossRef]
- Willems, H.M.E.; Bruner, W.S.; Barker, K.S.; Liu, J.; Palmer, G.E.; Peters, B.M. Overexpression of Candida albicans Secreted Aspartyl Proteinases 2 or 5 is not sufficient for exacerbation of immunopathology in a murine model of vaginitis. Infect. Immun. 2017. [Google Scholar] [CrossRef]
- Schaller, M.; Borelli, C.; Korting, H.C.; Hube, B. Hydrolytic enzymes as virulence factors of Candida albicans. Mycoses 2005, 48, 365–377. [Google Scholar] [CrossRef]
- Schofield, D.A.; Westwater, C.; Warner, T.; Balish, E. Differential Candida albicans lipase gene expression during alimentary tract colonization and infection. FEMS Microbiol. Lett. 2005, 244, 359–365. [Google Scholar] [CrossRef]
- Schofield, D.A.; Westwater, C.; Warner, T.; Nicholas, P.J.; Paulling, E.E.; Balish, E. Hydrolytic gene expression during oroesophageal and gastric candidiasis in immunocompetent and immunodeficient gnotobiotic mice. J. Infect. Dis. 2003, 188, 591–599. [Google Scholar] [CrossRef]
- Roustan, J.L.; Chu, A.R.; Moulin, G.; Bigey, F. A novel lipase/acyltransferase from the yeast Candida albicans: Expression and characterisation of the recombinant enzyme. Appl. Microbiol. Biotechnol. 2005, 68, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Gacser, A.; Stehr, F.; Kroger, C.; Kredics, L.; Schafer, W.; Nosanchuk, J.D. Lipase 8 affects the pathogenesis of Candida albicans. Infect. Immun. 2007, 75, 4710–4718. [Google Scholar] [CrossRef] [PubMed]
- Trofa, D.; Soghier, L.; Long, C.; Nosanchuk, J.D.; Gacser, A.; Goldman, D.L. A rat model of neonatal candidiasis demonstrates the importance of lipases as virulence factors for Candida albicans and Candida parapsilosis. Mycopathologia 2011, 172, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Birse, C.E.; Irwin, M.Y.; Fonzi, W.A.; Sypherd, P.S. Cloning and characterization of ECE1, a gene expressed in association with cell elongation of the dimorphic pathogen Candida albicans. Infect. Immun. 1993, 61, 3648–3655. [Google Scholar] [CrossRef]
- Moyes, D.L.; Wilson, D.; Richardson, J.P.; Mogavero, S.; Tang, S.X.; Wernecke, J.; Hofs, S.; Gratacap, R.L.; Robbins, J.; Runglall, M.; et al. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature 2016, 532, 64–68. [Google Scholar] [CrossRef]
- Bader, O.; Krauke, Y.; Hube, B. Processing of predicted substrates of fungal Kex2 proteinases from Candida albicans, C. glabrata, Saccharomyces cerevisiae and Pichia pastoris. BMC Microbiol. 2008, 8, 116. [Google Scholar] [CrossRef]
- Richardson, J.P.; Mogavero, S.; Moyes, D.L.; Blagojevic, M.; Kruger, T.; Verma, A.H.; Coleman, B.M.; De La Cruz Diaz, J.; Schulz, D.; Ponde, N.O.; et al. Processing of Candida albicans ece1p is critical for candidalysin maturation and fungal virulence. MBio 2018, 9. [Google Scholar] [CrossRef]
- Kasper, L.; Konig, A.; Koenig, P.A.; Gresnigt, M.S.; Westman, J.; Drummond, R.A.; Lionakis, M.S.; Gross, O.; Ruland, J.; Naglik, J.R.; et al. The fungal peptide toxin Candidalysin activates the NLRP3 inflammasome and causes cytolysis in mononuclear phagocytes. Nat. Commun. 2018, 9, 4260. [Google Scholar] [CrossRef]
- Richardson, J.P.; Willems, H.M.E.; Moyes, D.L.; Shoaie, S.; Barker, K.S.; Tan, S.L.; Palmer, G.E.; Hube, B.; Naglik, J.R.; Peters, B.M. Candidalysin drives epithelial signaling, neutrophil recruitment, and immunopathology at the vaginal mucosa. Infect. Immun. 2017. [Google Scholar] [CrossRef]
- Rogiers, O.; Frising, U.C.; Kucharikova, S.; Jabra-Rizk, M.A.; van Loo, G.; Van Dijck, P.; Wullaert, A. Candidalysin crucially contributes to Nlrp3 inflammasome activation by Candida albicans hyphae. MBio 2019, 10. [Google Scholar] [CrossRef]
- Swidergall, M.; Khalaji, M.; Solis, N.V.; Moyes, D.L.; Drummond, R.A.; Hube, B.; Lionakis, M.S.; Murdoch, C.; Filler, S.G.; Naglik, J.R. Candidalysin is required for neutrophil recruitment and virulence during systemic candida albicans infection. J. Infect. Dis. 2019. [Google Scholar] [CrossRef] [PubMed]
- Roselletti, E.; Monari, C.; Sabbatini, S.; Perito, S.; Vecchiarelli, A.; Sobel, J.D.; Cassone, A. A role for yeast/pseudohyphal cells of Candida albicans in the correlated expression of NLRP3 inflammasome inducers in women with acute vulvovaginal Candidiasis. Front. Microbiol. 2019, 10, 2669. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; Yang, X.; Nikou, S.A.; Kichik, N.; Donkin, A.; Ponde, N.O.; Richardson, J.P.; Gratacap, R.L.; Archambault, L.S.; Zwirner, C.P.; et al. Candidalysin activates innate epithelial immune responses via epidermal growth factor receptor. Nat. Commun. 2019, 10, 2297. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.H.; Richardson, J.P.; Zhou, C.; Coleman, B.M.; Moyes, D.L.; Ho, J.; Huppler, A.R.; Ramani, K.; McGeachy, M.J.; Mufazalov, I.A.; et al. Oral epithelial cells orchestrate innate type 17 responses to Candida albicans through the virulence factor candidalysin. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.M.; Coleman, B.M.; Willems, H.M.E.; Barker, K.S.; Aggor, F.E.Y.; Cipolla, E.; Verma, A.H.; Bishu, S.; Huppler, A.H.; Bruno, V.M.; et al. The IL-17R/IL-22R signaling axis is dispensable for vulvovaginal candidiasis regardless of estrogen status. J. Infect. Dis. 2019. [Google Scholar] [CrossRef]
- Pietrella, D.; Rachini, A.; Pines, M.; Pandey, N.; Mosci, P.; Bistoni, F.; D‘Enfert, C.; Vecchiarelli, A. Th17 cells and IL-17 in protective immunity to vaginal candidiasis. PLoS ONE 2011, 6, e22770. [Google Scholar] [CrossRef]
- Yano, J.; Kolls, J.K.; Happel, K.I.; Wormley, F.; Wozniak, K.L.; Fidel, P.L., Jr. The acute neutrophil response mediated by S100 alarmins during vaginal Candida infections is independent of the Th17-pathway. PLoS ONE 2012, 7, e46311. [Google Scholar] [CrossRef]
- Boisson, B.; Wang, C.; Pedergnana, V.; Wu, L.; Cypowyj, S.; Rybojad, M.; Belkadi, A.; Picard, C.; Abel, L.; Fieschi, C.; et al. A biallelic ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity 2013, 39, 676–686. [Google Scholar] [CrossRef]
- Li, J.; Casanova, J.L.; Puel, A. Mucocutaneous IL-17 immunity in mice and humans: Host defense vs. excessive inflammation. Mucosal. Immunol. 2017. [Google Scholar] [CrossRef]
- Matzinger, P. The danger model: A renewed sense of self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef]
- Altmeier, S.; Toska, A.; Sparber, F.; Teijeira, A.; Halin, C.; LeibundGut-Landmann, S. IL-1 coordinates the neutrophil response to C. albicans in the oral mucosa. PLoS Pathog. 2016, 12, e1005882. [Google Scholar] [CrossRef] [PubMed]
- Hise, A.G.; Tomalka, J.; Ganesan, S.; Patel, K.; Hall, B.A.; Brown, G.D.; Fitzgerald, K.A. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe 2009, 5, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Borghi, M.; De Luca, A.; Puccetti, M.; Jaeger, M.; Mencacci, A.; Oikonomou, V.; Pariano, M.; Garlanda, C.; Moretti, S.; Bartoli, A.; et al. Pathogenic NLRP3 inflammasome activity during Candida infection is negatively regulated by IL-22 via activation of NLRC4 and IL-1Ra. Cell Host Microbe 2015, 18, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Joly, S.; Ma, N.; Sadler, J.J.; Soll, D.R.; Cassel, S.L.; Sutterwala, F.S. Cutting edge: Candida albicans hyphae formation triggers activation of the Nlrp3 inflammasome. J. Immunol. 2009, 183, 3578–3581. [Google Scholar] [CrossRef] [PubMed]
- Lowes, D.J.; Hevener, K.E.; Peters, B.M. The second-generation anti-diabetic sulfonylureas inhibit Candida albicans and Candidalysin mediated activation of the NLRP3 inflammasome. Antimicrob. Agents Chemother. 2019. [Google Scholar] [CrossRef] [PubMed]
- Toth, A.; Zajta, E.; Csonka, K.; Vagvolgyi, C.; Netea, M.G.; Gacser, A. Specific pathways mediating inflammasome activation by Candida parapsilosis. Sci. Rep. 2017, 7, 43129. [Google Scholar] [CrossRef] [PubMed]
- Pietrella, D.; Pandey, N.; Gabrielli, E.; Pericolini, E.; Perito, S.; Kasper, L.; Bistoni, F.; Cassone, A.; Hube, B.; Vecchiarelli, A. Secreted aspartic proteases of Candida albicans activate the NLRP3 inflammasome. Eur. J. Immunol. 2013, 43, 679–692. [Google Scholar] [CrossRef]
- Roselletti, E.; Perito, S.; Gabrielli, E.; Mencacci, A.; Pericolini, E.; Sabbatini, S.; Cassone, A.; Vecchiarelli, A. NLRP3 inflammasome is a key player in human vulvovaginal disease caused by Candida albicans. Sci. Rep. 2017, 7, 17877. [Google Scholar] [CrossRef]
- Jaeger, M.; Carvalho, A.; Cunha, C.; Plantinga, T.S.; van de Veerdonk, F.; Puccetti, M.; Galosi, C.; Joosten, L.A.; Dupont, B.; Kullberg, B.J.; et al. Association of a variable number tandem repeat in the NLRP3 gene in women with susceptibility to RVVC. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 797–801. [Google Scholar] [CrossRef]
- Byers, S.L.; Wiles, M.V.; Dunn, S.L.; Taft, R.A. Mouse estrous cycle identification tool and images. PLoS ONE 2012, 7, e35538. [Google Scholar] [CrossRef]
- Lasarte, S.; Samaniego, R.; Salinas-Munoz, L.; Guia-Gonzalez, M.A.; Weiss, L.A.; Mercader, E.; Ceballos-Garcia, E.; Navarro-Gonzalez, T.; Moreno-Ochoa, L.; Perez-Millan, F.; et al. Sex hormones coordinate neutrophil immunity in the vagina by controlling chemokine gradients. J. Infect. Dis. 2016, 213, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Relloso, M.; Aragoneses-Fenoll, L.; Lasarte, S.; Bourgeois, C.; Romera, G.; Kuchler, K.; Corbi, A.L.; Munoz-Fernandez, M.A.; Nombela, C.; Rodriguez-Fernandez, J.L.; et al. Estradiol impairs the Th17 immune response against Candida albicans. J. Leukoc. Biol. 2012, 91, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.; Peters, B.M.; Noverr, M.C.; Fidel, P.L., Jr. Novel mechanism behind the immunopathogenesis of vulvovaginal candidiasis: “Neutrophil Anergy”. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef] [PubMed]
- Clemons, K.V.; Spearow, J.L.; Parmar, R.; Espiritu, M.; Stevens, D.A. Genetic susceptibility of mice to Candida albicans vaginitis correlates with host estrogen sensitivity. Infect. Immun. 2004, 72, 4878–4880. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.; Noverr, M.C.; Fidel, P.L., Jr. Vaginal heparan sulfate linked to neutrophil dysfunction in the acute inflammatory response associated with experimental vulvovaginal candidiasis. MBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Forney, L.J.; Ravel, J. Vaginal microbiome: Rethinking health and disease. Annu. Rev. Microbiol. 2012, 66, 371–389. [Google Scholar] [CrossRef]
- Ceccarani, C.; Foschi, C.; Parolin, C.; D‘Antuono, A.; Gaspari, V.; Consolandi, C.; Laghi, L.; Camboni, T.; Vitali, B.; Severgnini, M.; et al. Diversity of vaginal microbiome and metabolome during genital infections. Sci. Rep. 2019, 9, 14095. [Google Scholar] [CrossRef]
- Chen, C.; Song, X.; Wei, W.; Zhong, H.; Dai, J.; Lan, Z.; Li, F.; Yu, X.; Feng, Q.; Wang, Z.; et al. The microbiota continuum along the female reproductive tract and its relation to uterine-related diseases. Nat. Commun. 2017, 8, 875. [Google Scholar] [CrossRef]
- Hong, K.H.; Hong, S.K.; Cho, S.I.; Ra, E.; Han, K.H.; Kang, S.B.; Kim, E.C.; Park, S.S.; Seong, M.W. Analysis of the vaginal microbiome by next-generation sequencing and evaluation of its performance as a clinical diagnostic tool in vaginitis. Ann. Lab. Med. 2016, 36, 441–449. [Google Scholar] [CrossRef]
- Rampersaud, R.; Randis, T.M.; Ratner, A.J. Microbiota of the upper and lower genital tract. Semin. Fetal Neonatal Med. 2012, 17, 51–57. [Google Scholar] [CrossRef]
- Ravel, J.; Gajer, P.; Abdo, Z.; Schneider, G.M.; Koenig, S.S.; McCulle, S.L.; Karlebach, S.; Gorle, R.; Russell, J.; Tacket, C.O.; et al. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. USA 2011, 108, 4680–4687. [Google Scholar] [CrossRef]
- Miller, E.A.; Beasley, D.E.; Dunn, R.R.; Archie, E.A. Lactobacilli dominance and vaginal pH: Why is the human vaginal microbiome unique? Front. Microbiol. 2016, 7, 1936. [Google Scholar] [CrossRef]
- Auger, P.; Joly, J. Microbial flora associated with Candida albicans vulvovaginitis. Obstet. Gynecol. 1980, 55, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.B.; Xu, S.R.; He, Y.; Deng, G.H.; Sheng, H.F.; Huang, X.M.; Ouyang, C.Y.; Zhou, H.W. Diverse vaginal microbiomes in reproductive-age women with vulvovaginal candidiasis. PLoS ONE 2013, 8, e79812. [Google Scholar] [CrossRef]
- McClelland, R.S.; Richardson, B.A.; Hassan, W.M.; Graham, S.M.; Kiarie, J.; Baeten, J.M.; Mandaliya, K.; Jaoko, W.; Ndinya-Achola, J.O.; Holmes, K.K. Prospective study of vaginal bacterial flora and other risk factors for vulvovaginal candidiasis. J. Infect. Dis. 2009, 199, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Swidsinski, A.; Guschin, A.; Tang, Q.; Dorffel, Y.; Verstraelen, H.; Tertychnyy, A.; Khayrullina, G.; Luo, X.; Sobel, J.D.; Jiang, X. Vulvovaginal candidiasis: Histologic lesions are primarily polymicrobial and invasive and do not contain biofilms. Am. J. Obstet. Gynecol. 2019, 220. [Google Scholar] [CrossRef]
- Pramanick, R.; Mayadeo, N.; Warke, H.; Begum, S.; Aich, P.; Aranha, C. Vaginal microbiota of asymptomatic bacterial vaginosis and vulvovaginal candidiasis: Are they different from normal microbiota? Microb. Pathog. 2019, 134, 103599. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Westman, R.; Hickey, R.; Hansmann, M.A.; Kennedy, C.; Osborn, T.W.; Forney, L.J. Vaginal microbiota of women with frequent vulvovaginal candidiasis. Infect. Immun. 2009, 77, 4130–4135. [Google Scholar] [CrossRef]
- De Gregorio, P.R.; Silva, J.A.; Marchesi, A.; Nader-Macias, M.E.F. Anti-Candida activity of beneficial vaginal lactobacilli in in vitro assays and in a murine experimental model. FEMS Yeast Res. 2019, 19. [Google Scholar] [CrossRef]
- Dos Santos, C.I.; Franca, Y.R.; Lima Campos, C.D.; Quaresma Bomfim, M.R.; Melo, B.O.; Assuncao Holanda, R.; Santos, V.L.; Gomes Monteiro, S.; Buozzi Moffa, E.; Souza Monteiro, A.; et al. Antifungal and antivirulence activity of vaginal Lactobacillus Spp. Products against Candida vaginal isolates. Pathogens 2019, 8, 150. [Google Scholar] [CrossRef]
- Jang, S.J.; Lee, K.; Kwon, B.; You, H.J.; Ko, G. Vaginal lactobacilli inhibit growth and hyphae formation of Candida albicans. Sci. Rep. 2019, 9, 8121. [Google Scholar] [CrossRef] [PubMed]
- Fuochi, V.; Cardile, V.; Petronio Petronio, G.; Furneri, P.M. Biological properties and production of bacteriocins-like-inhibitory substances by Lactobacillus sp. strains from human vagina. J. Appl. Microbiol. 2019, 126, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, Q.; Yang, E.; Yan, L.; Li, T.; Zhuang, H. Antimicrobial compounds produced by vaginal Lactobacillus crispatus are able to strongly inhibit Candida albicans growth, hyphal formation and regulate virulence-related gene expressions. Front. Microbiol. 2017, 8, 564. [Google Scholar] [CrossRef] [PubMed]
- Buffo, J.; Herman, M.A.; Soll, D.R. A characterization of pH-regulated dimorphism in Candida albicans. Mycopathologia 1984, 85, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, F.C.; de Barros, P.P.; Rossoni, R.D.; Junqueira, J.C.; Jorge, A.O. Lactobacillus rhamnosus inhibits Candida albicans virulence factors in vitro and modulates immune system in Galleria mellonella. J. Appl. Microbiol. 2017, 122, 201–211. [Google Scholar] [CrossRef]
- Coudeyras, S.; Jugie, G.; Vermerie, M.; Forestier, C. Adhesion of human probiotic Lactobacillus rhamnosus to cervical and vaginal cells and interaction with vaginosis-associated pathogens. Infect. Dis. Obstet. Gynecol. 2008, 2008, 549640. [Google Scholar] [CrossRef]
- Martinez, R.C.; Seney, S.L.; Summers, K.L.; Nomizo, A.; De Martinis, E.C.; Reid, G. Effect of Lactobacillus rhamnosus GR-1 and Lactobacillus reuteri RC-14 on the ability of Candida albicans to infect cells and induce inflammation. Microbiol. Immunol. 2009, 53, 487–495. [Google Scholar] [CrossRef]
- Wagner, R.D.; Johnson, S.J. Probiotic lactobacillus and estrogen effects on vaginal epithelial gene expression responses to Candida albicans. J. Biomed. Sci. 2012, 19, 58. [Google Scholar] [CrossRef]
- De Seta, F.; Parazzini, F.; De Leo, R.; Banco, R.; Maso, G.P.; De Santo, D.; Sartore, A.; Stabile, G.; Inglese, S.; Tonon, M.; et al. Lactobacillus plantarum P17630 for preventing Candida vaginitis recurrence: A retrospective comparative study. Eur. J. Obstet. Gynecol. Reprod. Biol. 2014, 182, 136–139. [Google Scholar] [CrossRef]
- Murina, F.; Graziottin, A.; Vicariotto, F.; De Seta, F. Can Lactobacillus fermentum LF10 and Lactobacillus acidophilus LA02 in a slow-release vaginal product be useful for prevention of recurrent vulvovaginal candidiasis?: A clinical study. J. Clin. Gastroenterol. 2014, 48, S102–S105. [Google Scholar] [CrossRef]
- Pappas, P.G.; Kauffman, C.A.; Andes, D.R.; Clancy, C.J.; Marr, K.A.; Ostrosky-Zeichner, L.; Reboli, A.C.; Schuster, M.G.; Vazquez, J.A.; Walsh, T.J.; et al. Clinical practice guideline for the management of candidiasis: 2016 update by the infectious diseases society of America. Clin. Infect. Dis. 2016, 62, e1–e50. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.K.; Clenney, T.L. Management of vaginitis. Am. Fam. Physician. 2004, 70, 2125–2132. [Google Scholar] [PubMed]
- Sobel, J.D.; Wiesenfeld, H.C.; Martens, M.; Danna, P.; Hooton, T.M.; Rompalo, A.; Sperling, M.; Livengood, C., III; Horowitz, B.; Von Thron, J.; et al. Maintenance fluconazole therapy for recurrent vulvovaginal candidiasis. N. Engl. J. Med. 2004, 351, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Marchaim, D.; Lemanek, L.; Bheemreddy, S.; Kaye, K.S.; Sobel, J.D. Fluconazole-resistant Candida albicans vulvovaginitis. Obstet. Gynecol. 2012, 120, 1407–1414. [Google Scholar] [CrossRef]
- Sobel, J.D. Factors involved in patient choice of oral or vaginal treatment for vulvovaginal candidiasis. Patient Prefer. Adherence 2013, 8, 31–34. [Google Scholar] [CrossRef]
- Pericolini, E.; Gabrielli, E.; Ballet, N.; Sabbatini, S.; Roselletti, E.; Cayzeele Decherf, A.; Pelerin, F.; Luciano, E.; Perito, S.; Justen, P.; et al. Therapeutic activity of a Saccharomyces cerevisiae-based probiotic and inactivated whole yeast on vaginal candidiasis. Virulence 2017, 8, 74–90. [Google Scholar] [CrossRef]
- Sobel, J.D.; Vazquez, J.; Lynch, M.; Meriwether, C.; Zervos, M.J. Vaginitis due to Saccharomyces cerevisiae: Epidemiology, clinical aspects, and therapy. Clin. Infect. Dis. 1993, 16, 93–99. [Google Scholar] [CrossRef]
- Schmidt, C.S.; White, C.J.; Ibrahim, A.S.; Filler, S.G.; Fu, Y.; Yeaman, M.R.; Edwards, J.E., Jr.; Hennessey, J.P., Jr. NDV-3, a recombinant alum-adjuvanted vaccine for Candida and Staphylococcus aureus, is safe and immunogenic in healthy adults. Vaccine 2012, 30, 7594–7600. [Google Scholar] [CrossRef]
- Ibrahim, A.S.; Luo, G.; Gebremariam, T.; Lee, H.; Schmidt, C.S.; Hennessey, J.P., Jr.; French, S.W.; Yeaman, M.R.; Filler, S.G.; Edwards, J.E., Jr. NDV-3 protects mice from vulvovaginal candidiasis through T- and B-cell immune response. Vaccine 2013, 31, 5549–5556. [Google Scholar] [CrossRef]
- Uppuluri, P.; Singh, S.; Alqarihi, A.; Schmidt, C.S.; Hennessey, J.P., Jr.; Yeaman, M.R.; Filler, S.G.; Edwards, J.E.; Ibrahim, A.S. Human anti-Als3p antibodies are surrogate markers of NDV-3A vaccine efficacy against recurrent vulvovaginal candidiasis. Front. Immunol. 2018, 9, 1349. [Google Scholar] [CrossRef]
- De Bernardis, F.; Amacker, M.; Arancia, S.; Sandini, S.; Gremion, C.; Zurbriggen, R.; Moser, C.; Cassone, A. A virosomal vaccine against candidal vaginitis: Immunogenicity, efficacy and safety profile in animal models. Vaccine 2012, 30, 4490–4498. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, D.; Di Paola, M.; Rizzetto, L.; Tocci, N.; De Filippo, C.; Lionetti, P.; Ardizzoni, A.; Colombari, B.; Paulone, S.; Gut, I.G.; et al. Genomic and phenotypic variation in morphogenetic networks of two Candida albicans isolates subtends their different pathogenic potential. Front. Immunol. 2017, 8, 1997. [Google Scholar] [CrossRef] [PubMed]
- Geiger, A.M.; Foxman, B. Risk factors for vulvovaginal candidiasis: A case-control study among university students. Epidemiology 1996, 7, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Foxman, B.; Marsh, J.V.; Gillespie, B.; Sobel, J.D. Frequency and response to vaginal symptoms among white and African American women: Results of a random digit dialing survey. J. Womens Health 1998, 7, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Cassone, A.; Sobel, J.D. Experimental models of vaginal candidiasis and their relevance to human candidiasis. Infect. Immun. 2016, 84, 1255–1261. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Candidiasis Route | Avg. Incidence | Approx. Target Population 1 | Approx. Individuals Impacted | Refs. |
|---|---|---|---|---|
| Invasive | 0.009% | 7,800,000,000 | 702,000 | [8] |
| Oropharyngeal | - | - | 15,337,200 | |
| HIV/AIDS 2 | 72.5% | 15,200,000 | 11,020,000 | [9,10] |
| Head/neck cancer | 40% | 650,000 | 260,000 | [11,12] |
| Is Organ transplant | 11.6% | 100,800 | 11,690 | [13] |
| Infants (< 6 mos.) | 6% | 67,425,000 3 | 4,045,500 | [14,15,16] |
| Recurrent vulvovaginal | 8% | 1,682,200,000 | 134,560,000 | [17] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Willems, H.M.E.; Ahmed, S.S.; Liu, J.; Xu, Z.; Peters, B.M. Vulvovaginal Candidiasis: A Current Understanding and Burning Questions. J. Fungi 2020, 6, 27. https://doi.org/10.3390/jof6010027
Willems HME, Ahmed SS, Liu J, Xu Z, Peters BM. Vulvovaginal Candidiasis: A Current Understanding and Burning Questions. Journal of Fungi. 2020; 6(1):27. https://doi.org/10.3390/jof6010027
Chicago/Turabian StyleWillems, Hubertine M. E., Salman S. Ahmed, Junyan Liu, Zhenbo Xu, and Brian M. Peters. 2020. "Vulvovaginal Candidiasis: A Current Understanding and Burning Questions" Journal of Fungi 6, no. 1: 27. https://doi.org/10.3390/jof6010027
APA StyleWillems, H. M. E., Ahmed, S. S., Liu, J., Xu, Z., & Peters, B. M. (2020). Vulvovaginal Candidiasis: A Current Understanding and Burning Questions. Journal of Fungi, 6(1), 27. https://doi.org/10.3390/jof6010027