The Human Mucosal Mycobiome and Fungal Community Interactions


{kind=link}
Abstract
1. Introduction
2. The Mycobiome
2.1. Studying the Mycobiome
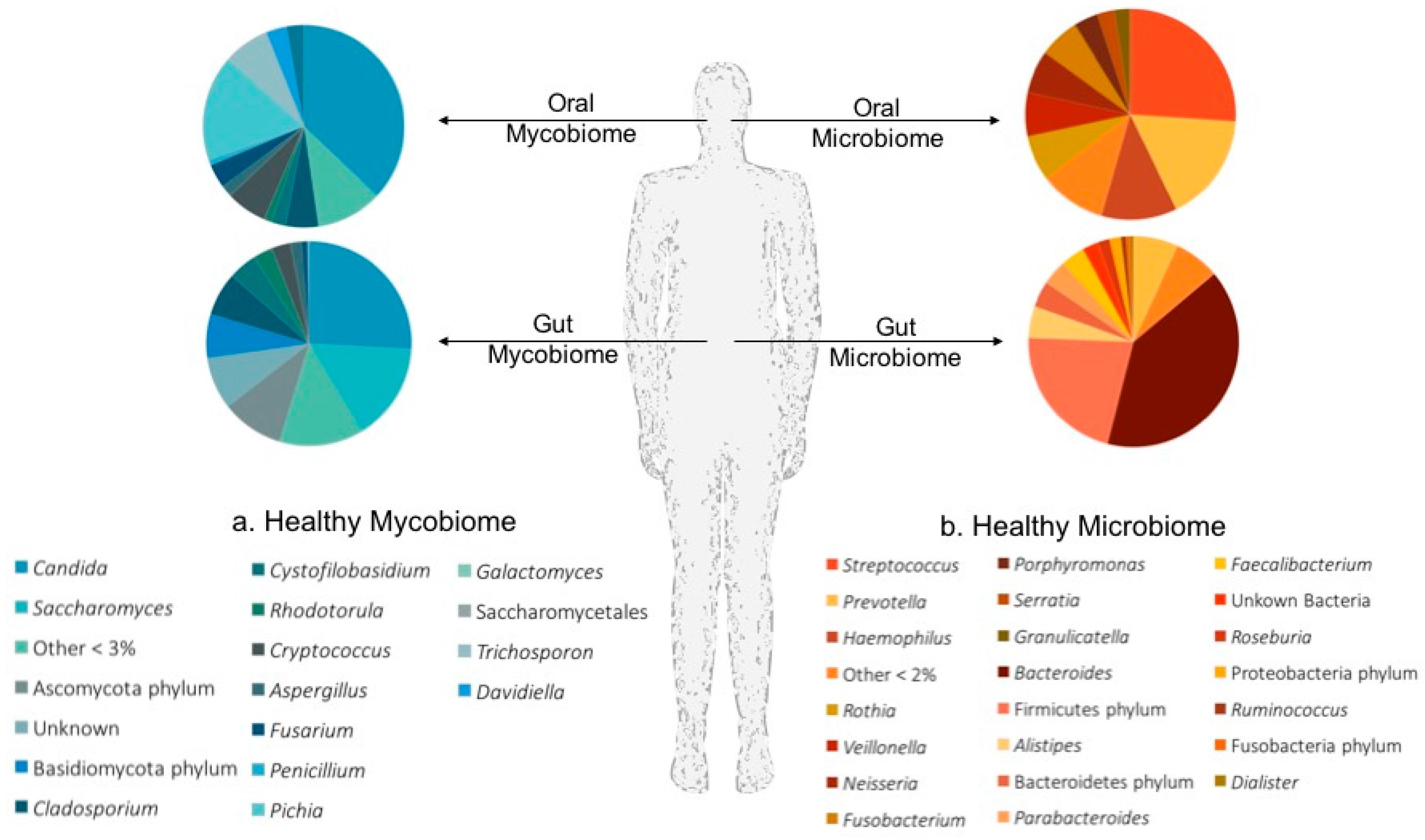
2.2. Mucosal Mycobiomes in Health and Disease
2.2.1. The Oral Mycobiome
2.2.2. The Gut Mycobiome
3. Fungal Interactions
3.1. Polymicrobial Interactions and the Microbiome
3.2. Fungal–Bacterial Interactions
3.3. Fungal–Fungal Interactions
3.4. Host-Fungal Interactions
4. Modeling of the Mycobiome, Microbiome and Host Interactions
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergstrom, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Backhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Krogh Pedersen, H.; et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Zeller, G.; Tap, J.; Voigt, A.Y.; Sunagawa, S.; Kultima, J.R.; Costea, P.I.; Amiot, A.; Bohm, J.; Brunetti, F.; Habermann, N.; et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol. 2014, 10, 766. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y.; et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2017, 66, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Hoarau, G.; Mukherjee, P.K.; Gower-Rousseau, C.; Hager, C.; Chandra, J.; Retuerto, M.A.; Neut, C.; Vermeire, S.; Clemente, J.; Colombel, J.F.; et al. Bacteriome and Mycobiome Interactions Underscore Microbial Dysbiosis in Familial Crohn’s Disease. mBio 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.L.; Bor, B.; Agnello, M.; Shi, W.; He, X. Ecology of the Oral Microbiome: Beyond Bacteria. Trends Microbiol. 2017, 25, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Huffnagle, G.B.; Noverr, M.C. The emerging world of the fungal microbiome. Trends Microbiol. 2013, 21, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Vesty, A.; Biswas, K.; Taylor, M.W.; Gear, K.; Douglas, R.G. Evaluating the Impact of DNA Extraction Method on the Representation of Human Oral Bacterial and Fungal Communities. PLoS ONE 2017, 12, e0169877. [Google Scholar] [CrossRef] [PubMed]
- Dollive, S.; Peterfreund, G.L.; Sherrill-Mix, S.; Bittinger, K.; Sinha, R.; Hoffmann, C.; Nabel, C.S.; Hill, D.A.; Artis, D.; Bachman, M.A.; et al. A tool kit for quantifying eukaryotic rRNA gene sequences from human microbiome samples. Genome Biol. 2012, 13, R60. [Google Scholar] [CrossRef] [PubMed]
- Diaz, P.I.; Hong, B.Y.; Dupuy, A.K.; Strausbaugh, L.D. Mining the oral mycobiome: Methods, components, and meaning. Virulence 2017, 8, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Iliev, I.D.; Leonardi, I. Fungal dysbiosis: Immunity and interactions at mucosal barriers. Nat. Rev. Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Iliev, I.D.; Funari, V.A.; Taylor, K.D.; Nguyen, Q.; Reyes, C.N.; Strom, S.P.; Brown, J.; Becker, C.A.; Fleshner, P.R.; Dubinsky, M.; et al. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 2012, 336, 1314–1317. [Google Scholar] [CrossRef] [PubMed]
- Moyes, D.L.; Naglik, J.R. The mycobiome: Influencing IBD severity. Cell Host Microbe 2012, 11, 551–552. [Google Scholar] [CrossRef] [PubMed]
- Seed, P.C. The human mycobiome. Cold Spring Harb. Perspect. Med. 2015, 5, a019810. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Morris, A.; Ghedin, E. The human mycobiome in health and disease. Genome Med. 2013, 5, 63. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Iliev, I.D.; Brown, J.; Underhill, D.M.; Funari, V.A. Mycobiome: Approaches to analysis of intestinal fungi. J. Immunol. Methods 2015, 421, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Koljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M.; et al. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jia, H.; Cai, X.; Zhong, H.; Feng, Q.; Sunagawa, S.; Arumugam, M.; Kultima, J.R.; Prifti, E.; Nielsen, T.; et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.K.; Chandra, J.; Retuerto, M.; Sikaroodi, M.; Brown, R.E.; Jurevic, R.; Salata, R.A.; Lederman, M.M.; Gillevet, P.M.; Ghannoum, M.A. Oral mycobiome analysis of HIV-infected patients: Identification of Pichia as an antagonist of opportunistic fungi. PLoS Pathog. 2014, 10, e1003996. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.; Dollive, S.; Grunberg, S.; Chen, J.; Li, H.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Archaea and fungi of the human gut microbiome: Correlations with diet and bacterial residents. PLoS ONE 2013, 8, e66019. [Google Scholar] [CrossRef] [PubMed]
- Ghannoum, M.A.; Jurevic, R.J.; Mukherjee, P.K.; Cui, F.; Sikaroodi, M.; Naqvi, A.; Gillevet, P.M. Characterization of the oral fungal microbiome (mycobiome) in healthy individuals. PLoS Pathog. 2010, 6, e1000713. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, A.K.; David, M.S.; Li, L.; Heider, T.N.; Peterson, J.D.; Montano, E.A.; Dongari-Bagtzoglou, A.; Diaz, P.I.; Strausbaugh, L.D. Redefining the human oral mycobiome with improved practices in amplicon-based taxonomy: Discovery of Malassezia as a prominent commensal. PLoS ONE 2014, 9, e90899. [Google Scholar] [CrossRef] [PubMed]
- Zakaria, M.N.; Furuta, M.; Takeshita, T.; Shibata, Y.; Sundari, R.; Eshima, N.; Ninomiya, T.; Yamashita, Y. Oral mycobiome in community-dwelling elderly and its relation to oral and general health conditions. Oral Dis. 2017. [Google Scholar] [CrossRef] [PubMed]
- Findley, K.; Oh, J.; Yang, J.; Conlan, S.; Deming, C.; Meyer, J.A.; Schoenfeld, D.; Nomicos, E.; Park, M.; Kong, H.H.; et al. Topographic diversity of fungal and bacterial communities in human skin. Nature 2013, 498, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Delhaes, L.; Monchy, S.; Frealle, E.; Hubans, C.; Salleron, J.; Leroy, S.; Prevotat, A.; Wallet, F.; Wallaert, B.; Dei-Cas, E.; et al. The airway microbiota in cystic fibrosis: A complex fungal and bacterial community—Implications for therapeutic management. PLoS ONE 2012, 7, e36313. [Google Scholar] [CrossRef] [PubMed]
- Ott, S.J.; Kuhbacher, T.; Musfeldt, M.; Rosenstiel, P.; Hellmig, S.; Rehman, A.; Drews, O.; Weichert, W.; Timmis, K.N.; Schreiber, S. Fungi and inflammatory bowel diseases: Alterations of composition and diversity. Scand. J. Gastroenterol. 2008, 43, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Mar Rodriguez, M.; Perez, D.; Javier Chaves, F.; Esteve, E.; Marin-Garcia, P.; Xifra, G.; Vendrell, J.; Jove, M.; Pamplona, R.; Ricart, W.; et al. Obesity changes the human gut mycobiome. Sci. Rep. 2015, 5, 14600. [Google Scholar] [CrossRef] [PubMed]
- Underhill, D.M.; Iliev, I.D. The mycobiota: Interactions between commensal fungi and the host immune system. Nat. Rev. Immunol. 2014, 14, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, C.; Tang, C.; He, Q.; Li, N.; Li, J. Dysbiosis of gut fungal microbiota is associated with mucosal inflammation in Crohn’s disease. J. Clin. Gastroenterol. 2014, 48, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Sam, Q.H.; Chang, M.W.; Chai, L.Y. The Fungal Mycobiome and Its Interaction with Gut Bacteria in the Host. Int. J. Mol. Sci. 2017, 18, 330. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Pan, C.; Wang, K.; Chen, X.; Wu, X.; Chen, C.A.; Wu, B. Synthetic multispecies microbial communities reveals shifts in secondary metabolism and facilitates cryptic natural product discovery. Environ. Microbiol. 2017, 19, 3606–3618. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.; Opel, V.; Scherlach, K.; Hertweck, C. Biosynthesis of antifungal and antibacterial polyketides by Burkholderia gladioli in coculture with Rhizopus microsporus. Mycoses 2014, 57, 48–55. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Kim, D.; Zhou, X.; Ahn, S.J.; Burne, R.A.; Richards, V.P.; Koo, H. RNA-Seq Reveals Enhanced Sugar Metabolism in Streptococcus mutans Co-cultured with Candida albicans within Mixed-Species Biofilms. Front. Microbiol. 2017, 8, 1036. [Google Scholar] [CrossRef] [PubMed]
- Jakubovics, N.S. Intermicrobial Interactions as a Driver for Community Composition and Stratification of Oral Biofilms. J. Mol. Biol. 2015, 427, 3662–3675. [Google Scholar] [CrossRef] [PubMed]
- Hogan, D.A.; Kolter, R. Pseudomonas-Candida interactions: An ecological role for virulence factors. Science 2002, 296, 2229–2232. [Google Scholar] [CrossRef] [PubMed]
- Kong, E.F.; Tsui, C.; Kucharikova, S.; Andes, D.; Van Dijck, P.; Jabra-Rizk, M.A. Commensal Protection of Staphylococcus aureus against Antimicrobials by Candida albicans Biofilm Matrix. mBio 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Hogan, D.A.; Vik, A.; Kolter, R. A Pseudomonas aeruginosa quorum-sensing molecule influences Candida albicans morphology. Mol. Microbiol. 2004, 54, 1212–1223. [Google Scholar] [CrossRef] [PubMed]
- Nickerson, K.W.; Atkin, A.L.; Hornby, J.M. Quorum Sensing in Dimorphic Fungi: Farnesol and Beyond. Appl. Environ. Microbiol. 2006, 72, 3805–3813. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.R.; Taylor, G.W.; Rutman, A.; Høiby, N.; Cole, P.J.; Wilson, R. Pseudomonas aeruginosa pyocyanin and 1-hydroxyphenazine inhibit fungal growth. J. Clin. Pathol. 1999, 52, 385–387. [Google Scholar] [CrossRef] [PubMed]
- Morales, D.K.; Grahl, N.; Okegbe, C.; Dietrich, L.E.P.; Jacobs, N.J.; Hogan, D.A. Control of Candida albicans Metabolism and Biofilm Formation by Pseudomonas aeruginosa Phenazines. mBio 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.I.; Dolben, E.F.; Okegbe, C.; Harty, C.E.; Golub, Y.; Thao, S.; Ha, D.G.; Willger, S.D.; O’Toole, G.A.; Harwood, C.S.; et al. Candida albicans Ethanol Stimulates Pseudomonas aeruginosa WspR-Controlled Biofilm Formation as Part of a Cyclic Relationship Involving Phenazines. PLOS Pathog. 2014, 10, e1004480. [Google Scholar] [CrossRef] [PubMed]
- Cugini, C.; Calfee, M.W.; Farrow, J.M.; Morales, D.K.; Pesci, E.C.; Hogan, D.A. Farnesol, a common sesquiterpene, inhibits PQS production in Pseudomonas aeruginosa. Mol. Microbiol. 2007, 65, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Holmes, A.R.; McNab, R.; Jenkinson, H.F. Candida albicans binding to the oral bacterium Streptococcus gordonii involves multiple adhesin-receptor interactions. Infect. Immun. 1996, 64, 4680–4685. [Google Scholar] [PubMed]
- Nobbs, A.H.; Vickerman, M.M.; Jenkinson, H.F. Heterologous Expression of Candida albicans Cell Wall-Associated Adhesins in Saccharomyces cerevisiae Reveals Differential Specificities in Adherence and Biofilm Formation and in Binding Oral Streptococcus gordonii. Eukaryot. Cell 2010, 9, 1622–1634. [Google Scholar] [CrossRef] [PubMed]
- Bamford, C.V.; d’Mello, A.; Nobbs, A.H.; Dutton, L.C.; Vickerman, M.M.; Jenkinson, H.F. Streptococcus gordonii Modulates Candida albicans Biofilm Formation through Intergeneric Communication. Infect. Immun. 2009, 77, 3696–3704. [Google Scholar] [CrossRef] [PubMed]
- Vílchez, R.; Lemme, A.; Ballhausen, B.; Thiel, V.; Schulz, S.; Jansen, R.; Sztajer, H.; Wagner-Döbler, I. Streptococcus mutans Inhibits Candida albicans Hyphal Formation by the Fatty Acid Signaling Molecule trans-2-Decenoic Acid (SDSF). ChemBioChem 2010, 11, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Jarosz, L.M.; Deng, D.M.; van der Mei, H.C.; Crielaard, W.; Krom, B.P. Streptococcus mutans Competence-Stimulating Peptide Inhibits Candida albicans Hypha Formation. Eukaryot. Cell 2009, 8, 1658–1664. [Google Scholar] [CrossRef] [PubMed]
- Tati, S.; Davidow, P.; McCall, A.; Hwang-Wong, E.; Rojas, I.G.; Cormack, B.; Edgerton, M. Candida glabrata Binding to Candida albicans Hyphae Enables Its Development in Oropharyngeal Candidiasis. PLoS Pathog. 2016, 12, e1005522. [Google Scholar] [CrossRef] [PubMed]
- Hitchcock, C.A.; Pye, G.W.; Troke, P.F.; Johnson, E.M.; Warnock, D.W. Fluconazole resistance in Candida glabrata. Antimicrob. Agents Chemother. 1993, 37, 1962–1965. [Google Scholar] [CrossRef] [PubMed]
- Lorek, J.; Pöggeler, S.; Weide, M.R.; Breves, R.; Bockmühl, D.P. Influence of farnesol on the morphogenesis of Aspergillus niger. J. Basic Microbiol. 2008, 48, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Semighini, C.P.; Murray, N.; Harris, S.D. Inhibition of Fusarium graminearum growth and development by farnesol. FEMS Microbiol. Lett. 2008, 279, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Luo, L.; Guo, J.; Liu, H.; Wang, B.; Deng, B.; Long, C.-A.; Cheng, Y. Farnesol induces apoptosis and oxidative stress in the fungal pathogen Penicillium expansum. Mycologia 2010, 102, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Tanaka, T.; Fujita, K.-I.; Taniguchi, M. Farnesol-Induced Generation of Reactive Oxygen Species via Indirect Inhibition of the Mitochondrial Electron Transport Chain in the Yeast Saccharomyces cerevisiae. J. Bacteriol. 1998, 180, 4460–4465. [Google Scholar] [PubMed]
- Machida, K.; Tanaka, T. Farnesol-induced generation of reactive oxygen species dependent on mitochondrial transmembrane potential hyperpolarization mediated by F0F1-ATPase in yeast. FEBS Lett. 1999, 462, 108–112. [Google Scholar] [CrossRef]
- Shirtliff, M.E.; Krom, B.P.; Meijering, R.A.M.; Peters, B.M.; Zhu, J.; Scheper, M.A.; Harris, M.L.; Jabra-Rizk, M.A. Farnesol-Induced Apoptosis in Candida albicans. Antimicrob. Agents Chemother. 2009, 53, 2392–2401. [Google Scholar] [CrossRef] [PubMed]
- Quintin, J.; Saeed, S.; Martens, J.H.; Giamarellos-Bourboulis, E.J.; Ifrim, D.C.; Logie, C.; Jacobs, L.; Jansen, T.; Kullberg, B.J.; Wijmenga, C.; et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 2012, 12, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Thaiss, C.A.; Zeevi, D.; Dohnalova, L.; Zilberman-Schapira, G.; Mahdi, J.A.; David, E.; Savidor, A.; Korem, T.; Herzig, Y.; et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 2015, 163, 1428–1443. [Google Scholar] [CrossRef] [PubMed]
- Witherden, E.A.; Moyes, D.L.; Bruce, K.D.; Ehrlich, S.D.; Shoaie, S. Using systems biology approaches to elucidate cause and effect in host-microbiome interactions. Curr. Opin. Syst. Biol. 2017, 3, 141–146. [Google Scholar] [CrossRef]
- Ghaffari, P.; Mardinoglu, A.; Asplund, A.; Shoaie, S.; Kampf, C.; Uhlen, M.; Nielsen, J. Identifying anti-growth factors for human cancer cell lines through genome-scale metabolic modeling. Sci. Rep. 2015, 5, 8183. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Uhlen, M.; Nielsen, J. Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 3083. [Google Scholar] [CrossRef] [PubMed]
- Agren, R.; Liu, L.; Shoaie, S.; Vongsangnak, W.; Nookaew, I.; Nielsen, J. The RAVEN toolbox and its use for generating a genome-scale metabolic model for Penicillium chrysogenum. PLoS Comput. Biol. 2013, 9, e1002980. [Google Scholar] [CrossRef] [PubMed]
- Caspeta, L.; Shoaie, S.; Agren, R.; Nookaew, I.; Nielsen, J. Genome-scale metabolic reconstructions of Pichia stipitis and Pichia pastoris and in silico evaluation of their potentials. BMC Syst. Biol. 2012, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Shoaie, S.; Karlsson, F.; Mardinoglu, A.; Nookaew, I.; Bordel, S.; Nielsen, J. Understanding the interactions between bacteria in the human gut through metabolic modeling. Sci. Rep. 2013, 3, 2532. [Google Scholar] [CrossRef] [PubMed]
- Shoaie, S.; Nielsen, J. Elucidating the interactions between the human gut microbiota and its host through metabolic modeling. Front. Genet. 2014, 5, 86. [Google Scholar] [CrossRef] [PubMed]
- Shoaie, S.; Ghaffari, P.; Kovatcheva-Datchary, P.; Mardinoglu, A.; Sen, P.; Pujos-Guillot, E.; de Wouters, T.; Juste, C.; Rizkalla, S.; Chilloux, J.; et al. Quantifying Diet-Induced Metabolic Changes of the Human Gut Microbiome. Cell Metab. 2015, 22, 320–331. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Witherden, E.A.; Shoaie, S.; Hall, R.A.; Moyes, D.L. The Human Mucosal Mycobiome and Fungal Community Interactions. J. Fungi 2017, 3, 56. https://doi.org/10.3390/jof3040056
Witherden EA, Shoaie S, Hall RA, Moyes DL. The Human Mucosal Mycobiome and Fungal Community Interactions. Journal of Fungi. 2017; 3(4):56. https://doi.org/10.3390/jof3040056
Chicago/Turabian StyleWitherden, Elizabeth A., Saeed Shoaie, Rebecca A. Hall, and David L. Moyes. 2017. "The Human Mucosal Mycobiome and Fungal Community Interactions" Journal of Fungi 3, no. 4: 56. https://doi.org/10.3390/jof3040056
APA StyleWitherden, E. A., Shoaie, S., Hall, R. A., & Moyes, D. L. (2017). The Human Mucosal Mycobiome and Fungal Community Interactions. Journal of Fungi, 3(4), 56. https://doi.org/10.3390/jof3040056