Immune Recognition of Fungal Polysaccharides


Abstract

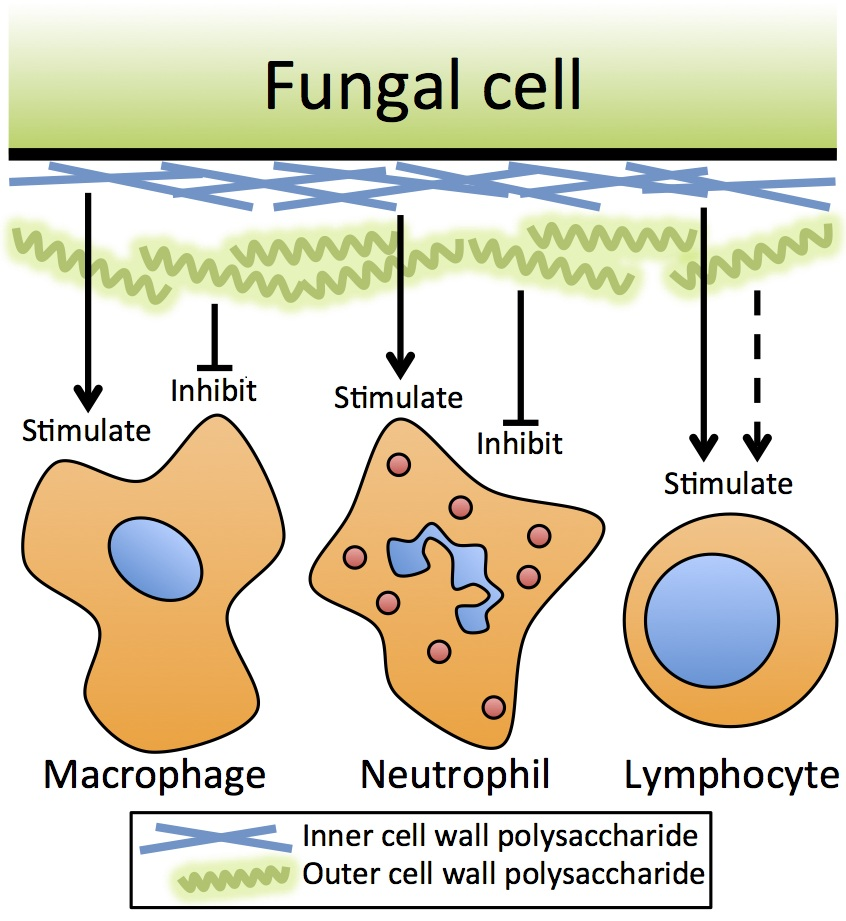{kind=link}
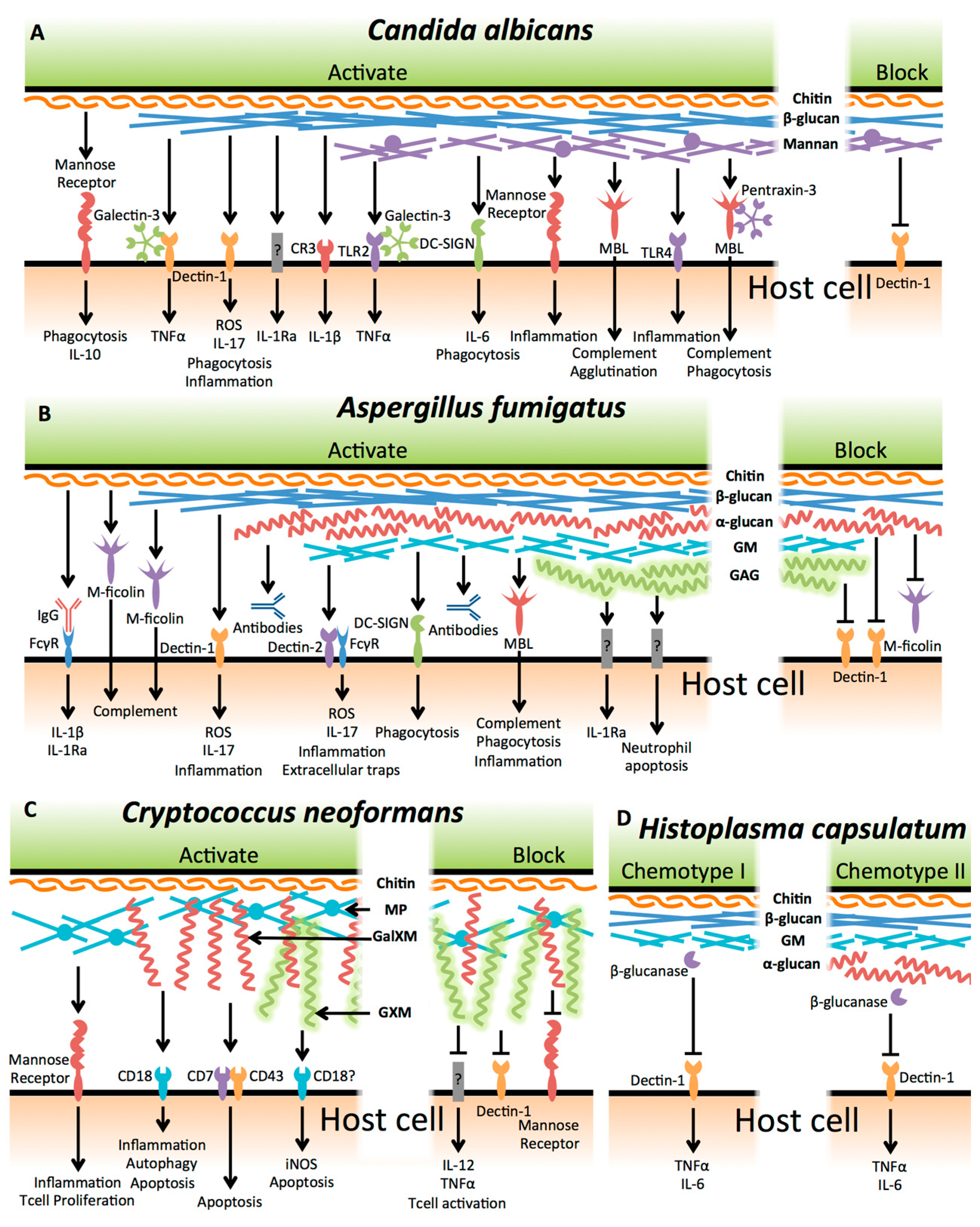{kind=link}
1. Introduction
2. Candida albicans
2.1. Beta-Glucan
2.2. Mannan
2.3. Chitin
2.4. Candida albicans Biofilms
2.5. Non-albicans Candida Species
3. Aspergillus fumigatus
3.1. Beta-Glucan
3.2. Galactomannan
3.3. Alpha-Glucan
3.4. Chitin
3.5. Galactosaminogalactan
3.6. Non-fumigatus Aspergillus Species
4. Cryptococcus neoformans
4.1. Glucuronoxylomannan
4.2. Galactoxylomannan
4.3. Mannoproteins
4.4. Cryptococcus gattii
5. Histoplasma capsulatum
Alpha-(1,3)-Glucan
6. Thoughts and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vallabhaneni, S.; Mody, R.K.; Walker, T.; Chiller, T. The Global Burden of Fungal Diseases. Infect. Dis. Clin. N. Am. 2016, 30, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Shoham, S.; Levitz, S.M. The immune response to fungal infections. Br. J. Haematol. 2005, 129, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Ramage, G.; Robertson, S.N.; Williams, C. Strength in numbers: Antifungal strategies against fungal biofilms. Int. J. Antimicrob. Agents 2014, 43, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Tsui, C.; Kong, E.F.; Jabra-Rizk, M.A. Pathogenesis of Candida albicans biofilm. Pathog. Dis. 2016, 74, ftw018. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D.; Gordon, S. Immune recognition of fungal β-glucans. Cell. Microbiol. 2005, 7, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D.; Taylor, P.R.; Reid, D.M.; Willment, J.A.; Williams, D.L.; Martinez-Pomares, L.; Wong, S.Y.C.; Gordon, S. Dectin-1 is a major β-glucan receptor on macrophages. J. Exp. Med. 2002, 196, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; O’Callaghan, C.A.; Marshall, A.S.J.; Gilbert, R.J.C.; Siebold, C.; Gordon, S.; Brown, G.D.; Jones, E.Y. Structure of the fungal β-glucan-binding immune receptor dectin-1: Implications for function. Protein Sci. 2007, 16, 1042–1052. [Google Scholar] [CrossRef]
- Adams, E.L.; Rice, P.J.; Graves, B.; Ensley, H.E.; Yu, H.; Brown, G.D.; Gordon, S.; Monteiro, M.A.; Papp-Szabo, E.; Lowman, D.W.; et al. Differential High-Affinity Interaction of Dectin-1 with Natural or Synthetic Glucans Is Dependent upon Primary Structure and Is Influenced by Polymer Chain Length and Side-Chain Branching. J. Pharmacol. Exp. Ther. 2008, 325, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Latgé, J.-P. Tasting the fungal cell wall. Cell. Microbiol. 2010, 12, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Leal, S.M.; Cowden, S.; Hsia, Y.C.; Ghannoum, M.A.; Momany, M.; Pearlman, E. Distinct roles for Dectin-1 and TLR4 in the pathogenesis of Aspergillus fumigatus keratitis. PLoS Pathog. 2010, 6, e1000976. [Google Scholar] [CrossRef] [PubMed]
- Dennehy, K.M.; Ferwerda, G.; Faro-Trindade, I.; Pyż, E.; Willment, J.A.; Taylor, P.R.; Kerrigan, A.; Tsoni, S.V.; Gordon, S.; Meyer-Wentrup, F.; et al. Syk kinase is required for collaborative cytokine production induced through Dectin-1 and Toll-like receptors. Eur. J. Immunol. 2008, 38, 500–506. [Google Scholar] [CrossRef]
- Gringhuis, S.I.; Dunnen, D.J.; Litjens, M. Dectin-1 directs T helper cell differentiation by controlling noncanonical NF-κB activation through Raf-1 and Syk. Nature 2009, 10, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.S.; Kasperkovitz, P.V.; Timmons, A.K.; Mansour, M.K.; Tam, J.M.; Seward, M.W.; Reedy, J.L.; Puranam, S.; Feliu, M.; Vyas, J.M. Dectin-1 Controls TLR9 Trafficking to Phagosomes Containing β-1,3 Glucan. J. Immunol. 2016, 196, 2249–2261. [Google Scholar] [CrossRef]
- Tam, J.M.; Mansour, M.K.; Khan, N.S.; Seward, M.; Puranam, S.; Tanne, A.; Sokolovska, A.; Becker, C.E.; Acharya, M.; Baird, M.A.; et al. Dectin-1-Dependent LC3 Recruitment to Phagosomes Enhances Fungicidal Activity in Macrophages. J. Infect. Dis. 2014, 210, 1844–1854. [Google Scholar] [CrossRef] [PubMed]
- Drummond, R.A.; Brown, G.D. The role of Dectin-1 in the host defence against fungal infections. Curr. Opin. Microbiol. 2011, 14, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Esteban, A.; Popp, M.W.; Vyas, V.K.; Strijbis, K.; Ploegh, H.L.; Fink, G.R. Fungal recognition is mediated by the association of dectin-1 and galectin-3 in macrophages. Proc. Natl. Acad. Sci. USA 2011, 108, 14270–14275. [Google Scholar] [CrossRef] [PubMed]
- Dumic, J.; Dabelic, S.; Flögel, M. Galectin-3: An open-ended story. Biochim. Biophys. Acta (BBA) Gen. Subj. 2006, 1760, 616–635. [Google Scholar] [CrossRef] [PubMed]
- Jouault, T.; El Abed-El Behi, M.; Martínez-Esparza, M.; Breuilh, L.; Trinel, P.-A.; Chamaillard, M.; Trottein, F.; Poulain, D. Specific recognition of Candida albicans by macrophages requires galectin-3 to discriminate Saccharomyces cerevisiae and needs association with TLR2 for signaling. J. Immunol. 2006, 177, 4679–4687. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-Y.; Huang, J.-H.; Chen, W.-Y.; Chan, Y.-C.; Lin, C.-H.; Chen, Y.-C.; Liu, F.-T.; Wu-Hsieh, B.A. Cell Intrinsic Galectin-3 Attenuates Neutrophil ROS-Dependent Killing of Candida by Modulating CR3 Downstream Syk Activation. Front. Immunol. 2017, 8, 616. [Google Scholar] [CrossRef]
- Linden, J.R.; De Paepe, M.E.; Laforce-Nesbitt, S.S.; Bliss, J.M. Galectin-3 plays an important role in protection against disseminated candidiasis. Med. Mycol. 2013, 51, 641–651. [Google Scholar] [CrossRef]
- Rubin-Bejerano, I.; Abeijon, C.; Magnelli, P.; Grisafi, P.; Fink, G.R. Phagocytosis by human neutrophils is stimulated by a unique fungal cell wall component. Cell Host Microbe 2007, 2, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Utomo, A.; Cullere, X.; Choi, M.M.; Milner, D.A.; Venkatesh, D.; Yun, S.-H.; Mayadas, T.N. The β-glucan receptor Dectin-1 activates the integrin Mac-1 in neutrophils via Vav protein signaling to promote Candida albicans clearance. Cell Host Microbe 2011, 10, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, S.; Rathinam, V.A.K.; Bossaller, L.; Army, K.; Kaiser, W.J.; Mocarski, E.S.; Dillon, C.P.; Green, D.R.; Mayadas, T.N.; Levitz, S.M.; et al. Caspase-8 Modulates Dectin-1 and Complement Receptor 3–Driven IL-1β Production in Response to β-Glucans and the Fungal Pathogen, Candida albicans. J. Immunol. 2014, 193, 2519–2530. [Google Scholar] [CrossRef] [PubMed]
- Smeekens, S.P.; Gresnigt, M.S.; Becker, K.L.; Cheng, S.-C.; Netea, S.A.; Jacobs, L.; Jansen, T.; van de Veerdonk, F.L.; Williams, D.L.; Joosten, L.A.B.; et al. An anti-inflammatory property of Candida albicans β-glucan: Induction of high levels of interleukin-1 receptor antagonist via a Dectin-1/CR3 independent mechanism. Cytokine 2015, 71, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Gow, N.A.R.; Joosten, L.A.B.; Verschueren, I.; Van der Meer, J.W.M.; Kullberg, B.J. Variable recognition of Candida albicans strains by TLR4 and lectin recognition receptors. Med. Mycol. 2010, 48, 897–903. [Google Scholar] [CrossRef] [PubMed]
- MacCallum, D.M.; Castillo, L.; Nather, K.; Munro, C.A.; Brown, A.J.P.; Gow, N.A.R.; Odds, F.C. Property Differences among the Four Major Candida albicans Strain Clades. Eukaryot. Cell 2009, 8, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Szabo, E.K.; MacCallum, D.M. A novel renal epithelial cell in vitro assay to assess Candida albicans virulence. Virulence 2014, 5, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Torosantucci, A.; Bromuro, C.; Chiani, P.; De Bernardis, F.; Berti, F.; Galli, C.; Norelli, F.; Bellucci, C.; Polonelli, L.; Costantino, P.; et al. A novel glyco-conjugate vaccine against fungal pathogens. J. Exp. Med. 2005, 202, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Torosantucci, A.; Chiani, P.; Bromuro, C.; De Bernardis, F. Protection by anti-β-glucan antibodies is associated with restricted β-1, 3 glucan binding specificity and inhibition of fungal growth and adherence. PLoS ONE 2009, 4, e5392. [Google Scholar] [CrossRef] [PubMed]
- Pietrella, D.; Rachini, A.; Torosantucci, A.; Chiani, P.; Brown, A.J.P.; Bistoni, F.; Costantino, P.; Mosci, P.; d’Enfert, C.; Rappuoli, R.; et al. A β-glucan-conjugate vaccine and anti-β-glucan antibodies are effective against murine vaginal candidiasis as assessed by a novel in vivo imaging technique. Vaccine 2010, 28, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Rachini, A.; Pietrella, D.; Lupo, P.; Torosantucci, A.; Chiani, P.; Bromuro, C.; Proietti, C.; Bistoni, F.; Cassone, A.; Vecchiarelli, A. An anti-β-glucan monoclonal antibody inhibits growth and capsule formation of Cryptococcus neoformans in vitro and exerts therapeutic, anticryptococcal activity in vivo. Infect. Immun. 2007, 75, 5085–5094. [Google Scholar] [CrossRef] [PubMed]
- De Groot, P.W.J.; Kraneveld, E.A.; Yin, Q.Y.; Dekker, H.L.; Gross, U.; Crielaard, W.; de Koster, C.G.; Bader, O.; Klis, F.M.; Weig, M. The cell wall of the human pathogen Candida glabrata: Differential incorporation of novel adhesin-like wall proteins. Eukaryot. Cell 2008, 7, 1951–1964. [Google Scholar] [CrossRef] [PubMed]
- Gow, N.A.; Hube, B. Importance of the Candida albicans cell wall during commensalism and infection. Curr. Opin. Microbiol. 2012, 15, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Shibata, N.; Suzuki, A.; Kobayashi, H.; Okawa, Y. Chemical structure of the cell-wall mannan of Candida albicans serotype A and its difference in yeast and hyphal forms. Biochem. J. 2007, 404, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Courjol, F.; Jouault, T.; Mille, C.; Hall, R.; Maes, E.; Sendid, B.; Mallet, J.M.; Guerardel, Y.; Gow, N.A.R.; Poulain, D.; et al. β-1,2-Mannosyltransferases 1 and 3 Participate in Yeast and Hyphae O- and N-Linked Mannosylation and Alter Candida albicans Fitness During Infection. Open Forum Infect. Dis. 2015, 2, ofv116. [Google Scholar] [CrossRef] [PubMed]
- Ueno, K.; Okawara, A.; Yamagoe, S.; Naka, T.; Umeyama, T.; Utena-Abe, Y.; Tarumoto, N.; Niimi, M.; Ohno, H.; Doe, M.; et al. The mannan of Candida albicans lacking β-1,2-linked oligomannosides increases the production of inflammatory cytokines by dendritic cells. Med. Mycol. 2013, 51, 385–395. [Google Scholar] [CrossRef]
- Mora-Montes, H.M.; Bates, S.; Netea, M.G.; Castillo, L.; Brand, A.; Buurman, E.T.; Diaz-Jimenez, D.F.; Jan Kullberg, B.; Brown, A.J.P.; Odds, F.C.; et al. A Multifunctional Mannosyltransferase Family in Candida albicans Determines Cell Wall Mannan Structure and Host-Fungus Interactions. J. Biol. Chem. 2010, 285, 12087–12095. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Q.; Zou, Z.; Shen, H.; Shen, S.S.; Miao, Q.; Huang, X.; Liu, W.; Li, L.P.; Chen, S.M.; Yan, L.; et al. Mnn10 Maintains Pathogenicity in Candida albicans by Extending α-1,6-Mannose Backbone to Evade Host Dectin-1 Mediated Antifungal Immunity. PLoS Pathog. 2016, 12, e1005617. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.M.; Louw, J.; Lewis, L.E.; Okai, B.; Walls, C.A.; Ballou, E.R.; Walker, L.A.; Reid, D.; Munro, C.A.; Brown, A.J.P.; et al. Candida albicans hypha formation and mannan masking of β-glucan inhibit macrophage phagosome maturation. mBio 2014, 5, e01874. [Google Scholar] [CrossRef] [PubMed]
- Boxx, G.M.; Kozel, T.R.; Nishiya, C.T.; Zhang, M.X. Influence of Mannan and Glucan on Complement Activation and C3 Binding by Candida albicans. Infect. Immun. 2010, 78, 1250–1259. [Google Scholar] [CrossRef] [PubMed]
- Cambi, A.; Netea, M.G.; Mora-Montes, H.M.; Gow, N.A.R.; Hato, S.V.; Lowman, D.W.; Kullberg, B.J.; Torensma, R.; Williams, D.L.; Figdor, C.G. Dendritic Cell Interaction with Candida albicans Critically Depends on N-Linked Mannan. J. Biol. Chem. 2008, 283, 20590–20599. [Google Scholar] [CrossRef]
- Barreto-Bergter, E.; Figueiredo, R.T. Fungal glycans and the innate immune recognition. Front. Cell Infect. Microbiol. 2014, 4, 145. [Google Scholar] [CrossRef] [PubMed]
- Appelmelk, B.J.; van Die, I.; van Vliet, S.J.; Vandenbroucke-Grauls, C.M.J.E.; Geijtenbeek, T.B.H.; van Kooyk, Y. Cutting Edge: Carbohydrate Profiling Identifies New Pathogens that Interact with Dendritic Cell-Specific ICAM-3-Grabbing Nonintegrin on Dendritic Cells. J. Immunol. 2003, 170, 1635–1639. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Gómez, D.; Sierra-Filardi, E.; Martínez-Nuñez, R.T.; Caparrós, E.; Delgado, R.; Muñoz-Fernández, M.A.; Abad, M.A.; Jiménez-Barbero, J.; Leal, M.; Corbí, A.L. Structural requirements for multimerization of the pathogen receptor dendritic cell-specific ICAM3-grabbing non-integrin (CD209) on the cell surface. J. Biol. Chem. 2008, 283, 3889–3903. [Google Scholar] [CrossRef] [PubMed]
- Cambi, A.; Gijzen, K.; de Vries, L.J.M.; Torensma, R.; Joosten, B.; Adema, G.J.; Netea, M.G.; Kullberg, B.J.; Romani, L.; Figdor, C.G. The C-type lectin DC-SIGN (CD209) is an antigen-uptake receptor for Candida albicans on dendritic cells. Eur. J. Immunol. 2003, 33, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Gow, N.A.R.; Munro, C.A.; Bates, S.; Collins, C.; Ferwerda, G.; Hobson, R.P.; Bertram, G.; Hughes, H.B.; Jansen, T.; et al. Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and Toll-like receptors. J. Clin. Investig. 2006, 116, 1642–1650. [Google Scholar] [CrossRef] [PubMed]
- Vautier, S.; MacCallum, D.M.; Brown, G.D. C-type lectin receptors and cytokines in fungal immunity. Cytokine 2012, 58, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Graham, L.M.; Brown, G.D. The Dectin-2 family of C-type lectins in immunity and homeostasis. Cytokine 2009, 48, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Ifrim, D.C.; Quintin, J.; Courjol, F.; Verschueren, I.; van Krieken, J.H.; Koentgen, F.; Fradin, C.; Gow, N.A.R.; Joosten, L.A.B.; Van der Meer, J.W.M.; et al. The Role of Dectin-2 for Host Defense against Disseminated Candidiasis. J. Interferon Cytokine Res. 2016, 36, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.-L.; Zhao, X.-Q.; Jiang, C.; You, Y.; Chen, X.-P.; Jiang, Y.-Y.; Jia, X.-M.; Lin, X. C-Type Lectin Receptors Dectin-3 and Dectin-2 Form a Heterodimeric Pattern-Recognition Receptor for Host Defense against Fungal Infection. Immunity 2013, 39, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Toyonaga, K.; Mori, D.; Kakuta, S.; Hoshino, Y.; Oyamada, A.; Yamada, H.; Ono, K.-I.; Suyama, M.; Iwakura, Y.; et al. C-type Lectin MCL Is an FcRg-Coupled Receptor that Mediates the Adjuvanticityof Mycobacterial Cord Factor. Immunity 2013, 38, 1050–1062. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Yang, X.-L.; Yudate, T.; Chung, J.-S.; Wu, J.; Luby-Phelps, K.; Kimberly, R.P.; Underhill, D.; Cruz, P.D.; Ariizumi, K. Dectin-2 is a pattern recognition receptor for fungi that couples with the Fc receptor gamma chain to induce innate immune responses. J. Biol. Chem. 2006, 281, 38854–38866. [Google Scholar] [CrossRef]
- Ifrim, D.C.; Bain, J.M.; Reid, D.M.; Oosting, M.; Verschueren, I.; Gow, N.A.R.; van Krieken, J.H.; Brown, G.D.; Kullberg, B.J.; Joosten, L.A.B.; et al. Role of Dectin-2 for Host Defense against Systemic Infection with Candida glabrata. Infect. Immun. 2014, 82, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Kohatsu, L.; Hsu, D.K.; Jegalian, A.G.; Liu, F.-T.; Baum, L.G. Galectin-3 induces death of Candida species expressing specific β-1,2-linked mannans. J. Immunol. 2006, 177, 4718–4726. [Google Scholar] [CrossRef] [PubMed]
- Holmskov, U.; Malhotra, R.; Sim, R.B.; Jensenius, J.C. Collectins: Collagenous C-type lectins of the innate immune defense system. Immunol. Today 1994, 15, 67–74. [Google Scholar] [CrossRef]
- Matsushita, M.; Fujita, T. Activation of the classical complement pathway by mannose-binding protein in association with a novel C1s-like serine protease. J. Exp. Med. 1992, 176, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- Lillegard, J.B.; Sim, R.B.; Thorkildson, P.; Gates, M.A.; Kozel, T.R. Recognition of Candida albicans by mannan-binding lectin in vitro and in vivo. J. Infect. Dis. 2006, 193, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Ip, W.-K.; Lau, Y.-L. Role of mannose-binding lectin in the innate defense against Candida albicans: Enhancement of complement activation, but lack of opsonic function, in phagocytosis by human dendritic cells. J. Infect. Dis. 2004, 190, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Van Asbeck, E.C.; Hoepelman, A.I.; Scharringa, J.; Herpers, B.L.; Verhoef, J. Mannose binding lectin plays a crucial role in innate immunity against yeast by enhanced complement activation and enhanced uptake of polymorphonuclear cells. BMC Microbiol. 2008, 8, 229. [Google Scholar] [CrossRef]
- Ma, Y.J.; Doni, A.; Skjoedt, M.O.; Honore, C.; Arendrup, M.; Mantovani, A.; Garred, P. Heterocomplexes of Mannose-binding Lectin and the Pentraxins PTX3 or Serum Amyloid P Component Trigger Cross-activation of the Complement System. J. Biol. Chem. 2011, 286, 3405–3417. [Google Scholar] [CrossRef] [PubMed]
- Nedovic, B.; Posteraro, B.; Leoncini, E.; Ruggeri, A.; Amore, R.; Sanguinetti, M.; Ricciardi, W.; Boccia, S. Mannose-Binding Lectin Codon 54 Gene Polymorphism and Vulvovaginal Candidiasis: A Systematic Review and Meta-Analysis. BioMed Res. Int. 2014, 2014, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Timpel, C.; Strahl-Bolsinger, S.; Ziegelbauer, K.; Ernst, J.F. Multiple functions of Pmt1p-mediated protein O-mannosylation in the fungal pathogen Candida albicans. J. Biol. Chem. 1998, 273, 20837–20846. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.M.; Paes, H.C.; Nanjappa, S.G.; Sorkness, R.; Gasper, D.; Sterkel, A.; Wuthrich, M.; Klein, B.S. Complement Component 3C3 and C3a Receptor Are Required in Chitin-Dependent Allergic Sensitization to Aspergillus fumigatus but Dispensable in Chitin-Induced Innate Allergic Inflammation. mBio 2013, 4, e00162-13. [Google Scholar] [CrossRef] [PubMed]
- Mora-Montes, H.M.; Netea, M.G.; Ferwerda, G.; Lenardon, M.D.; Brown, G.D.; Mistry, A.R.; Kullberg, B.J.; O’Callaghan, C.A.; Sheth, C.C.; Odds, F.C.; et al. Recognition and Blocking of Innate Immunity Cells by Candida albicans Chitin. Infect. Immun. 2011, 79, 1961–1970. [Google Scholar] [CrossRef]
- Hoang, A. Caspofungin acetate: An antifungal agent. Am. J. Health Syst. Pharm. 2001, 58, 1206–1214. [Google Scholar] [PubMed]
- Van Dyken, S.J.; Mohapatra, A.; Nussbaum, J.C.; Molofsky, A.B.; Thornton, E.E.; Ziegler, S.F.; McKenzie, A.N.J.; Krummel, M.F.; Liang, H.-E.; Locksley, R.M. Chitin Activates Parallel Immune Modules that Direct Distinct Inflammatory Responses via Innate Lymphoid Type 2 and gd T Cells. Immunity 2014, 40, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Wagener, J.; Malireddi, R.K.S.; Lenardon, M.D.; Köberle, M.; Vautier, S.; MacCallum, D.M.; Biedermann, T.; Schaller, M.; Netea, M.G.; Kanneganti, T.-D.; et al. Fungal Chitin Dampens Inflammation through IL-10 Induction Mediated by NOD2 and TLR9 Activation. PLoS Pathog. 2014, 10, e1004050. [Google Scholar] [CrossRef] [PubMed]
- Douglas, L.J. Candida biofilms and their role in infection. Trends Microbiol. 2003, 11, 30–36. [Google Scholar] [CrossRef]
- Kumamoto, C.A.; Vinces, M.D. Alternative Candida albicans lifestyles: Growth on surfaces. Annu. Rev. Microbiol. 2005, 59, 113–133. [Google Scholar] [CrossRef] [PubMed]
- Nett, J.E. The host’s reply to Candida biofilm. Pathogens 2016, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Chandra, J.; McCormick, T.S.; Imamura, Y.; Mukherjee, P.K.; Ghannoum, M.A. Interaction of Candida albicans with adherent human peripheral blood mononuclear cells increases C. albicans biofilm formation and results in differential expression of pro- and anti-inflammatory cytokines. Infect. Immun. 2007, 75, 2612–2620. [Google Scholar] [CrossRef] [PubMed]
- Katragkou, A.; Kruhlak, M.J.; Simitsopoulou, M.; Chatzimoschou, A.; Taparkou, A.; Cotten, C.J.; Paliogianni, F.; Diza Mataftsi, E.; Tsantali, C.; Walsh, T.J.; et al. Interactions between human phagocytes and Candida albicans biofilms alone and in combination with antifungal agents. J. Infect. Dis. 2010, 201, 1941–1949. [Google Scholar] [CrossRef] [PubMed]
- Katragkou, A.; Simitsopoulou, M.; Chatzimoschou, A.; Georgiadou, E.; Walsh, T.J.; Roilides, E. Effects of interferon-γ and granulocyte colony-stimulating factor on antifungal activity of human polymorphonuclear neutrophils against Candida albicans grown as biofilms or planktonic cells. Cytokine 2011, 55, 330–334. [Google Scholar] [CrossRef]
- Xie, Z.; Thompson, A.; Sobue, T.; Kashleva, H.; Xu, H.; Vasilakos, J.; Dongari-Bagtzoglou, A. Candida albicans biofilms do not trigger reactive oxygen species and evade neutrophil killing. J. Infect. Dis. 2012, 206, 1936–1945. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, P.; Thompson, A.; Xie, Z.; Kashleva, H.; Ganguly, S.; Mitchell, A.P.; Dongari-Bagtzoglou, A. Role of Bcr1-activated genes Hwp1 and Hyr1 in Candida albicans oral mucosal biofilms and neutrophil evasion. PLoS ONE 2011, 6, e16218. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.; Palmer, G.E.; Eberle, K.E.; Peters, B.M.; Vogl, T.; McKenzie, A.N.; Fidel, P.L. Vaginal epithelial cell-derived S100 alarmins induced by Candida albicans via pattern recognition receptor interactions are sufficient but not necessary for the acute neutrophil response during experimental vaginal candidiasis. Infect. Immun. 2014, 82, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.; Lilly, E.; Barousse, M.; Fidel, P.L. Epithelial cell-derived S100 calcium-binding proteins as key mediators in the hallmark acute neutrophil response during Candida vaginitis. Infect. Immun. 2010, 78, 5126–5137. [Google Scholar] [CrossRef] [PubMed]
- Nett, J.E.; Marchillo, K.; Spiegel, C.A.; Andes, D.R. Development and validation of an in vivo Candida albicans biofilm denture model. Infect. Immun. 2010, 78, 3650–3659. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Fries, B.C. A murine model for catheter-associated candiduria. J. Med. Microbiol. 2011, 60, 1523–1529. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, M.T.; Hernandez, M.; Novak-Frazer, L.; Kuula, H.; Ramage, G.; Bowyer, P.; Warn, P.; Sorsa, T.; Rautemaa, R. DL-2-hydroxyisocaproic acid attenuates inflammatory responses in a murine Candida albicans biofilm model. Clin. Vaccine Immunol. 2014, 21, 1240–1245. [Google Scholar] [CrossRef] [PubMed]
- Conti, H.R.; Gaffen, S.L. IL-17–Mediated immunity to the opportunistic fungal pathogen Candida albicans. J. Immunol. 2015, 195, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Hebecker, B.; Naglik, J.R.; Hube, B.; Jacobsen, I.D. Pathogenicity mechanisms and host response during oral Candida albicans infections. Expert Rev. Anti Infect. Ther. 2014, 12, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.; Noverr, M.C.; Fidel, P.L. Cytokines in the host response to Candida vaginitis: Identifying a role for non-classical immune mediators, S100 alarmins. Cytokine 2012, 58, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Montagna, M.T.; Lovero, G.; Borghi, E.; Amato, G.; Andreoni, S.; Campion, L.; Lo Cascio, G.; Lombardi, G.; Luzzaro, F.; Manso, E.; et al. Candidemia in intensive care unit: A nationwide prospective observational survey (GISIA-3 study) and review of the European literature from 2000 through 2013. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 661–674. [Google Scholar] [PubMed]
- Kobayashi, H.; Mitobe, H.; Takahashi, K.; Yamamoto, T.; Shibata, N.; Suzuki, S. Structural study of a cell wall mannan-protein complex of the pathogenic yeast Candida glabrata IFO 0622 strain. Arch. Biochem. Biophys. 1992, 294, 662–669. [Google Scholar] [CrossRef]
- Kobayashi, H.; Oyamada, H.; Iwadate, N.; Suzuki, H.; Mitobe, H.; Takahashi, K.; Shibata, N.; Suzuki, S.; Okawa, Y. Structural and immunochemical characterization of β-1,2-linked mannobiosyl phosphate residue in the cell wall mannan of Candida glabrata. Arch. Microbiol. 1998, 169, 188–194. [Google Scholar] [CrossRef] [PubMed]
- West, L.; Lowman, D.W.; Mora-Montes, H.M.; Grubb, S.; Murdoch, C.; Thornhill, M.H.; Gow, N.A.R.; Williams, D.; Haynes, K. Differential virulence of Candida glabrata glycosylation mutants. J. Biol. Chem. 2013, 288, 22006–22018. [Google Scholar] [CrossRef] [PubMed]
- Keppler-Ross, S.; Douglas, L.; Konopka, J.B.; Dean, N. Recognition of yeast by murine macrophages requires mannan but not glucan. Eukaryot. Cell 2010, 9, 1776–1787. [Google Scholar] [CrossRef] [PubMed]
- Seider, K.; Gerwien, F.; Kasper, L.; Allert, S.; Brunke, S.; Jablonowski, N.; Schwarzmüller, T.; Barz, D.; Rupp, S.; Kuchler, K.; et al. Immune evasion, stress resistance, and efficient nutrient acquisition are crucial for intracellular survival of Candida glabrata within macrophages. Eukaryot. Cell 2014, 13, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Lima-Neto, R.G.; Beltrão, E.I.C.; Oliveira, P.C.; Neves, R.P. Adherence of Candida albicans and Candida parapsilosis to epithelial cells correlates with fungal cell surface carbohydrates. Mycoses 2011, 54, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Pérez-García, L.A.; Csonka, K.; Flores-Carreón, A.; Estrada-Mata, E.; Mellado-Mojica, E.; Németh, T.; López-Ramírez, L.A.; Toth, R.; López, M.G.; Vizler, C.; et al. Role of Protein Glycosylation in Candida parapsilosis Cell Wall Integrity and Host Interaction. Front. Microbiol. 2016, 7, 306. [Google Scholar] [CrossRef] [PubMed]
- Latge, J.P. Aspergillus fumigatus and aspergillosis. Clin. Microbiol. Rev. 1999, 12, 310–350. [Google Scholar] [PubMed]
- Lee, M.J.; Sheppard, D.C. Recent advances in the understanding of the Aspergillus fumigatus cell wall. J. Microbiol. 2016, 54, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Filler, S.G.; Sheppard, D.C. Fungal Invasion of Normally Non-Phagocytic Host Cells. PLoS Pathog. 2006, 2, e129. [Google Scholar] [CrossRef] [PubMed]
- Balloy, V.; Chignard, M. The innate immune response to Aspergillus fumigatus. Microbes Infect. 2009, 11, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Dichtl, K.; Samantaray, S.; Aimanianda, V.; Zhu, Z.; Prevost, M.-C.; Latgé, J.-P.; Ebel, F.; Wagener, J. Aspergillus fumigatus devoid of cell wall β-1,3-glucan is viable, massively sheds galactomannan and is killed by septum formation inhibitors. Mol. Microbiol. 2014, 95, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Steele, C.; Rapaka, R.R.; Metz, A.; Pop, S.M.; Williams, D.L.; Gordon, S.; Kolls, J.K.; Brown, G.D. The β-Glucan Receptor Dectin-1 Recognizes Specific Morphologies of Aspergillus fumigatus. PLoS Pathog. 2005, 1, e42. [Google Scholar] [CrossRef] [PubMed]
- Carrion, S.D.J.; Leal, S.M.; Ghannoum, M.A.; Aimanianda, V.; Latge, J.P.; Pearlman, E. The RodA Hydrophobin on Aspergillus fumigatus Spores Masks Dectin-1- and Dectin-2-Dependent Responses and Enhances Fungal Survival in vivo. J. Immunol. 2013, 191, 2581–2588. [Google Scholar] [CrossRef] [PubMed]
- Werner, J.L.; Metz, A.E.; Horn, D.; Schoeb, T.R.; Hewitt, M.M.; Schwiebert, L.M.; Faro-Trindade, I.; Brown, G.D.; Steele, C. Requisite Role for the Dectin-1-Glucan Receptor in Pulmonary Defense against Aspergillus fumigatus. J. Immunol. 2009, 182, 4938–4946. [Google Scholar] [CrossRef] [PubMed]
- Mezger, M.; Kneitz, S.; Wozniok, I.; Kurzai, O.; Einsele, H.; Loeffler, J. Proinflammatory Response of Immature Human Dendritic Cells is Mediated by Dectin-1 after Exposure to Aspergillus fumigatus Germ Tubes. J. Infect. Dis. 2008, 197, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Gravelat, F.N.; Beauvais, A.; Liu, H.; Lee, M.J.; Snarr, B.D.; Chen, D.; Xu, W.; Kravtsov, I.; Hoareau, C.M.Q.; Vanier, G.; et al. Aspergillus galactosaminogalactan mediates adherence to host constituents and conceals hyphal β-glucan from the immune system. PLoS Pathog. 2013, 9, e1003575. [Google Scholar] [CrossRef] [PubMed]
- Werner, J.L.; Gessner, M.A.; Lilly, L.M.; Nelson, M.P.; Metz, A.E.; Horn, D.; Dunaway, C.W.; Deshane, J.; Chaplin, D.D.; Weaver, C.T.; et al. Neutrophils Produce Interleukin 17A (IL-17A) in a Dectin-1- and IL-23-Dependent Manner during Invasive Fungal Infection. Infect. Immun. 2011, 79, 3966–3977. [Google Scholar] [CrossRef] [PubMed]
- Gessner, M.A.; Werner, J.L.; Lilly, L.M.; Nelson, M.P.; Metz, A.E.; Dunaway, C.W.; Chan, Y.R.; Ouyang, W.; Brown, G.D.; Weaver, C.T.; et al. Dectin-1-Dependent Interleukin-22 Contributes to Early Innate Lung Defense against Aspergillus fumigatus. Infect. Immun. 2011, 80, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Rivera, A.; Hohl, T.M.; Collins, N.; Leiner, I.; Gallegos, A.; Saijo, S.; Coward, J.W.; Iwakura, Y.; Pamer, E.G. Dectin-1 diversifies Aspergillus fumigatus-specific T cell responses by inhibiting T helper type 1 CD4 T cell differentiation. J. Exp. Med. 2011, 208, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Lilly, L.M.; Gessner, M.A.; Dunaway, C.W.; Metz, A.E.; Schwiebert, L.; Weaver, C.T.; Brown, G.D.; Steele, C. The β-Glucan Receptor Dectin-1 Promotes Lung Immunopathology during Fungal Allergy via IL-22. J. Immunol. 2012, 189, 3653–3660. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, T.; Schlosser, A.; Holmskov, U.; Sorensen, G.L. Ficolins and FIBCD1: Soluble and membrane bound pattern recognition molecules with acetyl group selectivity. Mol. Immunol. 2011, 48, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, T.; Simenel, C.; Dubreucq, G.; Adam, O.; Delepierre, M.; Lemoine, J.; Vorgias, C.E.; Diaquin, M.; Latge, J.P. Molecular organization of the alkali-insoluble fraction of Aspergillus fumigatus cell wall. J. Biol. Chem. 2000, 275, 27594–27607. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.; Lund, K.P.; Christensen, K.B.; Holm, A.T.; Dubey, L.K.; Moeller, J.B.; Jepsen, C.S.; Schlosser, A.; Galgóczy, L.; Thiel, S.; et al. M-ficolin is present in Aspergillus fumigatus infected lung and modulates epithelial cell immune responses elicited by fungal cell wall polysaccharides. Virulence 2017, 1–10. [Google Scholar] [CrossRef]
- Ma, Y.J.; Doni, A.; Hummelshøj, T.; Honoré, C.; Bastone, A.; Mantovani, A.; Thielens, N.M.; Garred, P. Synergy between ficolin-2 and pentraxin 3 boosts innate immune recognition and complement deposition. J. Biol. Chem. 2009, 284, 28263–28275. [Google Scholar] [CrossRef] [PubMed]
- Latge, J.P.; Kobayashi, H.; Debeaupuis, J.P. Chemical and immunological characterization of the extracellular galactomannan of Aspergillus fumigatus. Infect. Immun. 1994, 62, 5424–5433. [Google Scholar] [PubMed]
- Leitao, E.A.; Bittencourt, V.C.B.; Haido, R.M.T.; Valente, A.P.; Peter-Katalinic, J.; Letzel, M.; de Souza, L.M.; Barreto-Bergter, E. β-galactofuranose-containing O-linked oligosaccharides present in the cell wall peptidogalactomannan of Aspergillus fumigatus contain immunodominant epitopes. Glycobiology 2003, 13, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.; Fontaine, T.; Heddergott, C.; Robinet, P.; Aimanianda, V.; Beau, R.; Beauvais, A.; Mouyna, I.; Prevost, M.-C.; Fekkar, A.; et al. Biosynthesis of cell wall mannan in the conidium and the mycelium of Aspergillus fumigatus. Cell. Microbiol. 2016, 18, 1881–1891. [Google Scholar] [CrossRef] [PubMed]
- Paugam, A.; Sarfati, J.; Romieu, R.; Viguier, M.; Dupouy-Camet, J.; Latge, J.P. Detection of Aspergillus galactomannan: Comparison of an enzyme-linked immunoassay and a europium-linked time-resolved fluoroimmunoassay. J. Clin Microbiol. 1998, 36, 3079–3080. [Google Scholar]
- Patterson, T.F.; Thompson, G.R., III; Denning, D.W.; Fishman, J.A.; Hadley, S.; Herbrecht, R.; Kontoyiannis, D.P.; Marr, K.A.; Morrison, V.A.; Nguyen, M.H.; et al. Practice Guidelines for the Diagnosis and Management of Aspergillosis: 2016 Update by the Infectious Diseases Society of America. Clin. Infect. Dis. 2016, 63, e1–e60. [Google Scholar] [PubMed]
- Erwig, L.P.; Gow, N.A.R. Interactions of fungal pathogens with phagocytes. Nat. Rev. Microbiol. 2016, 14, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Gómez, D.; Dominguez-Soto, A.; Ancochea, J.; Jimenez-Heffernan, J.A.; Leal, J.A.; Corbi, A.L. Dendritic Cell-Specific Intercellular Adhesion Molecule 3-Grabbing Nonintegrin Mediates Binding and Internalization of Aspergillus fumigatus Conidia by Dendritic Cells and Macrophages. J. Immunol. 2004, 173, 5635–5643. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Gómez, D.; Antonio Leal, J.; Corbí, A.L. DC-SIGN mediates the binding of Aspergillus fumigatus and keratinophylic fungi by human dendritic cells. Immunobiology 2005, 210, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Xu, X.-Y.; Tian, X.-L.; Shao, H.-T.; Wu, X.-D.; Wang, Q.; Su, X.; Shi, Y. Activation of NF-κB and respiratory burst following Aspergillus fumigatus stimulation of macrophages. Immunobiology 2014, 219, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Loures, F.V.; Röhm, M.; Lee, C.K.; Santos, E.; Wang, J.P.; Specht, C.A.; Calich, V.L.G.; Urban, C.F.; Levitz, S.M. Recognition of Aspergillus fumigatus Hyphae by Human Plasmacytoid Dendritic Cells Is Mediated by Dectin-2 and Results in Formation of Extracellular Traps. PLoS Pathog. 2015, 11, e1004643. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.R.; Roy, S.; Leal, S.M.; Sun, Y.; Howell, S.J.; Cobb, B.A.; Li, X.; Pearlman, E. Activation of neutrophils by autocrine IL-17A–IL-17RC interactions during fungal infection is regulated by IL-6, IL-23, RORγt and dectin-2. Nat. Immunol. 2014, 15, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Xu, X.-Y.; Shao, H.-T.; Su, X.; Wu, X.-D.; Wang, Q.; Shi, Y. Dectin-2 is predominately macrophage restricted and exhibits conspicuous expression during Aspergillus fumigatus invasion in human lung. Cell. Immunol. 2013, 284, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Dumestre-Perard, C.; Lamy, B.; Aldebert, D.; Lemaire-Vieille, C.; Grillot, R.; Brion, J.P.; Gagnon, J.; Cesbron, J.Y. Aspergillus Conidia Activate the Complement by the Mannan-Binding Lectin C2 Bypass Mechanism. J. Immunol. 2008, 181, 7100–7105. [Google Scholar] [CrossRef] [PubMed]
- Neth, O.; Jack, D.L.; Dodds, A.W.; Holzel, H. Mannose-binding lectin binds to a range of clinically relevant microorganisms and promotes complement deposition. Infect. Immun. 2000, 68, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Che, C.-Y.; Zhang, J.-F.; Lee, J.-E.; Lin, J.; Hu, L.-T.; Jiang, N.; Wang, Q.; Xu, Q.; Zhao, G.-Q. Early expression of mannose-binding lectin 2 during Aspergillus fumigatus infection in human corneal epithelial cells. Int. J. Ophthalmol. 2015, 8, 35–38. [Google Scholar] [PubMed]
- Kaur, S.; Gupta, V.K.; Thiel, S.; Sarma, P.U.; Madan, T. Protective role of mannan-binding lectin in a murine model of invasive pulmonary aspergillosis. Clin. Exp. Immunol. 2007, 148, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Clemons, K.V.; Martinez, M.; Tong, A.-J.; Stevens, D.A. Immunology Letters. Immunol. Lett. 2010, 128, 105–107. [Google Scholar] [CrossRef] [PubMed]
- Hogaboam, C.M. Mannose-binding lectin deficiency alters the development of fungal asthma: Effects on airway response, inflammation, and cytokine profile. J. Leukoc. Biol. 2003, 75, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Crosdale, D.J.; Poulton, K.V.; Ollier, W.E.; Thomson, W.; Denning, D.W. Mannose-binding lectin gene polymorphisms as a susceptibility factor for chronic necrotizing pulmonary aspergillosis. J. Infect. Dis. 2001, 184, 653–656. [Google Scholar] [CrossRef] [PubMed]
- Stynen, D.; Sarfati, J.; Goris, A.; Prévost, M.C.; Lesourd, M.; Kamphuis, H.; Darras, V.; Latge, J.P. Rat monoclonal antibodies against Aspergillus galactomannan. Infect. Immun. 1992, 60, 2237–2245. [Google Scholar] [PubMed]
- Luong, M.-L.; Filion, C.; Labbé, A.-C.; Roy, J.; Pépin, J.; Cadrin-Tourigny, J.; Carignan, S.; Sheppard, D.C.; Laverdiere, M. Clinical utility and prognostic value of bronchoalveolar lavagegalactomannan in patients with hematologic malignancies. Diagn. Microbiol. Infect. Dis. 2010, 68, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Lehrnbecher, T.; Robinson, P.D.; Fisher, B.T.; Castagnola, E.; Groll, A.H.; Steinbach, W.J.; Zaoutis, T.E.; Negeri, Z.F.; Beyene, J.; Phillips, B.; et al. Galactomannan, β-D-Glucan, and Polymerase Chain Reaction–Based Assays for the Diagnosis of Invasive Fungal Disease in Pediatric Cancer and Hematopoietic Stem Cell Transplantation: A Systematic Review and Meta-Analysis. Clin. Infect. Dis. 2016, 63, 1340–1348. [Google Scholar] [CrossRef] [PubMed]
- Beauvais, A.; Fontaine, T.; Aimanianda, V.; Latgé, J.-P. Aspergillus cell wall and biofilm. Mycopathologia 2014, 178, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, T.; Beauvais, A.; Loussert, C.; Thevenard, B.; Fulgsang, C.C.; Ohno, N.; Clavaud, C.; Prevost, M.-C.; Latgé, J.-P. Cell wall α1-3glucans induce the aggregation of germinating conidia of Aspergillus fumigatus. Fungal Genet. Biol. 2010, 47, 707–712. [Google Scholar] [CrossRef]
- Latgé, J.-P.; Beauvais, A. Functional duality of the cell wall. Curr. Opin. Microbiol. 2014, 20, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Loussert, C.; Schmitt, C.; Prevost, M.-C.; Balloy, V.; Fadel, E.; Philippe, B.; Kauffmann-Lacroix, C.; Latgé, J.-P.; Beauvais, A. In vivobiofilm composition of Aspergillus fumigatus. Cell. Microbiol. 2010, 12, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Chai, L.Y.A.; Vonk, A.G.; Kullberg, B.J.; Verweij, P.E.; Verschueren, I.; Van der Meer, J.W.M.; Joosten, L.A.B.; Latgé, J.-P.; Netea, M.G. Aspergillus fumigatus cell wall components differentially modulate host TLR2 and TLR4 responses. Microbes Infect. 2011, 13, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Beauvais, A.; Bozza, S.; Kniemeyer, O.; Formosa, C.; Formosa, C.; Balloy, V.; Henry, C.; Roberson, R.W.; Dague, E.; Chignard, M.; et al. Deletion of the α-(1,3)-glucan synthase genes induces a restructuring of the conidial cell wall responsible for the avirulence of Aspergillus fumigatus. PLoS Pathog. 2013, 9, e1003716. [Google Scholar] [CrossRef]
- Komarova, B.S.; Orekhova, M.V.; Tsvetkov, Y.E.; Beau, R.; Aimanianda, V.; Latgé, J.-P.; Nifantiev, N.E. Synthesis of a pentasaccharide and neoglycoconjugates related to fungal α-(1 → 3)-glucan and their use in the generation of antibodies to trace Aspergillus fumigatus cell wall. Chemistry 2015, 21, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Bidula, S.; Kenawy, H.; Ali, Y.M.; Sexton, D.; Schwaeble, W.J.; Schelenz, S. Role of Ficolin-A and Lectin Complement Pathway in the Innate Defense against Pathogenic Aspergillus Species. Infect. Immun. 2013, 81, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Bidula, S.; Sexton, D.W.; Schelenz, S. Serum opsonin ficolin-A enhances host-fungal interactions and modulates cytokine expression from human monocyte-derived macrophages and neutrophils following Aspergillus fumigatus challenge. Med. Microbiol. Immunol. 2016, 205, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Bidula, S.; Sexton, D.W.; Yates, M.; Abdolrasouli, A.; Shah, A.; Wallis, R.; Reed, A.; Armstrong-James, D.; Schelenz, S. H-ficolin binds Aspergillus fumigatus leading to activation of the lectin complement pathway and modulation of lung epithelial immune responses. Immunology 2015, 146, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Bidula, S.; Sexton, D.W.; Abdolrasouli, A.; Shah, A.; Reed, A.; Armstrong-James, D.; Schelenz, S. The Serum Opsonin L-ficolin Is Detected in Lungs of Human Transplant Recipients Following Fungal Infections and Modulates Inflammation and Killing of Aspergillus fumigatus. J. Infect. Dis. 2015, 212, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.L.; Aimanianda, V.; Wang, X.; Gresnigt, M.S.; Ammerdorffer, A.; Jacobs, C.W.; Gazendam, R.P.; Joosten, L.A.B.; Netea, M.G.; Latge, J.P.; et al. Aspergillus Cell Wall Chitin Induces Anti- and Proinflammatory Cytokines in Human PBMCs via the Fc-γ Receptor/Syk/PI3K Pathway. mBio 2016, 7, e01823-15. [Google Scholar] [CrossRef]
- Dubey, L.K.; Moeller, J.B.; Schlosser, A.; Sorensen, G.L.; Holmskov, U. Chitin enhances serum IgE in Aspergillus fumigatus induced allergy in mice. Immunobiology 2015, 220, 714–721. [Google Scholar] [CrossRef] [PubMed]
- O’Dea, E.M.; Amarsaikhan, N.; Li, H.; Downey, J.; Steele, E.; Van Dyken, S.J.; Locksley, R.M.; Templeton, S.P. Eosinophils are Recruited in Response to Chitin Exposure and Enhance Th2-Mediated Immune Pathology in Aspergillus fumigatus infection. Infect. Immun. 2014, 82, 3199–3205. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Gravelat, F.N.; Cerone, R.P.; Baptista, S.D.; Campoli, P.V.; Choe, S.-I.; Kravtsov, I.; Vinogradov, E.; Creuzenet, C.; Liu, H.; et al. Overlapping and distinct roles of Aspergillus fumigatus UDP-glucose 4-epimerases in galactose metabolism and the synthesis of galactose-containing cell wall polysaccharides. J. Biol. Chem. 2014, 289, 1243–1256. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, T.; Delangle, A.; Simenel, C.; Coddeville, B.; van Vliet, S.J.; van Kooyk, Y.; Bozza, S.; Moretti, S.; Schwarz, F.; Trichot, C.; et al. Galactosaminogalactan, a new immunosuppressive polysaccharide of Aspergillus fumigatus. PLoS Pathog. 2011, 7, e1002372. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, D.C. Molecular mechanism of Aspergillus fumigatus adherence to host constituents. Curr. Opin. Microbiol. 2011, 14, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Liu, H.; Barker, B.M.; Snarr, B.D.; Gravelat, F.N.; Al Abdallah, Q.; Gavino, C.; Baistrocchi, S.R.; Ostapska, H.; Xiao, T.; et al. The Fungal Exopolysaccharide Galactosaminogalactan Mediates Virulence by Enhancing Resistance to Neutrophil Extracellular Traps. PLoS Pathog. 2015, 11, e1005187. [Google Scholar] [CrossRef]
- Robinet, P.; Baychelier, F.; Fontaine, T.; Picard, C.; Debré, P.; Vieillard, V.; Latgé, J.-P.; Elbim, C. A Polysaccharide Virulence Factor of a Human Fungal Pathogen Induces Neutrophil Apoptosis via NK Cells. J. Immunol. 2014, 192, 5332–5342. [Google Scholar] [CrossRef] [PubMed]
- Gresnigt, M.S.; Bozza, S.; Becker, K.L.; Joosten, L.A.B.; Abdollahi-Roodsaz, S.; van der Berg, W.B.; Dinarello, C.A.; Netea, M.G.; Fontaine, T.; De Luca, A.; et al. A Polysaccharide Virulence Factor from Aspergillus fumigatus Elicits Anti-inflammatory Effects through Induction of Interleukin-1 Receptor Antagonist. PLoS Pathog. 2014, 10, e1003936. [Google Scholar] [CrossRef]
- Rambach, G.; Blum, G.; Latgé, J.-P.; Fontaine, T.; Heinekamp, T.; Hagleitner, M.; Jeckström, H.; Weigel, G.; Würtinger, P.; Pfaller, K.; et al. Identification of Aspergillus fumigatus surface components that mediate interaction of conidia and hyphae with human platelets. J. Infect. Dis. 2015, 212, 1140–1149. [Google Scholar] [CrossRef]
- Snarr, B.D.; Baker, P.; Bamford, N.C.; Sato, Y.; Liu, H.; Lehoux, M.L.; Gravelat, F.N.; Ostapska, H.; Baistrocchi, S.R.; Cerone, R.P.; et al. Microbial glycoside hydrolases as antibiofilm agents with cross-kingdom activity. Proc. Natl. Acad. Sci. USA 2017, 114, 7124–7129. [Google Scholar] [CrossRef] [PubMed]
- Henriet, S.S.V.; van de Sande, W.W.J.; Lee, M.J.; Simonetti, E.; Momany, M.; Verweij, P.E.; Rijs, A.J.M.M.; Ferwerda, G.; Sheppard, D.C.; de Jonge, M.I.; et al. Decreased Cell Wall Galactosaminogalactan in Aspergillus nidulans Mediates Dysregulated Inflammation in the Chronic Granulomatous Disease Host. J. Interferon Cytokine Res. 2016, 36, 488–498. [Google Scholar] [CrossRef]
- Perfect, J.R.; Cox, G.M.; Lee, J.Y.; Kauffman, C.A.; de Repentigny, L.; Chapman, S.W.; Morrison, V.A.; Pappas, P.; Hiemenz, J.W.; Stevens, D.A.; et al. The impact of culture isolation of Aspergillus species: A hospital-based survey of aspergillosis. Clin. Infect. Dis. 2001, 33, 1824–1833. [Google Scholar] [CrossRef]
- Slesiona, S.; Gressler, M.; Mihlan, M.; Zaehle, C.; Schaller, M.; Barz, D.; Hube, B.; Jacobsen, I.D.; Brock, M. Persistence versus escape: Aspergillus terreus and Aspergillus fumigatus employ different strategies during interactions with macrophages. PLoS ONE 2012, 7, e31223. [Google Scholar] [CrossRef] [PubMed]
- Deak, E.; Nelson, M.; Hernández-Rodríguez, Y.; Gade, L.; Baddley, J.; Momany, M.; Steele, C.; Balajee, S.A. Aspergillus terreus accessory conidia are multinucleated, hyperpolarizing structures that display differential dectin staining and can induce heightened inflammatory responses in a pulmonary model of aspergillosis. Virulence 2011, 2, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Doering, T.L. How does Cryptococcus get its coat? Trends Microbiol. 2000, 8, 547–553. [Google Scholar] [CrossRef]
- Jesus, M.D.; Nicola, A.M.; Chow, S.-K.; Lee, I.R.; Nong, S.; Specht, C.A.; Levitz, S.M.; Casadevall, A. Glucuronoxylomannan, galactoxylomannan, and mannoprotein occupy spatially separate and discrete regions in the capsule of Cryptococcus neoformans. Virulence 2010, 1, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Cross, C.E.; Bancroft, G.J. Ingestion of acapsular Cryptococcus neoformans occurs via mannose and β-glucan receptors, resulting in cytokine production and increased phagocytosis of the encapsulated form. Infect. Immun. 1995, 63, 2604–2611. [Google Scholar] [PubMed]
- Feldmesser, M.; Kress, Y.; Novikoff, P.; Casadevall, A. Cryptococcus neoformans is a facultative intracellular pathogen in murine pulmonary infection. Infect. Immun. 2000, 68, 4225–4237. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.; Casadevall, A. Phagosome extrusion and host-cell survival after Cryptococcus neoformans phagocytosis by macrophages. Curr. Biol. 2006, 16, 2161–2165. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.; Casadevall, A. Cell-to-cell spread and massive vacuole formation after Cryptococcus neoformans infection of murine macrophages. BMC Immunol. 2007, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Zaragoza, O.; Rodrigues, M.L.; De Jesus, M.; Frases, S.; Dadachova, E.; Casadevall, A. The capsule of the fungal pathogen Cryptococcus neoformans. Adv. Appl. Microbiol. 2009, 68, 133–216. [Google Scholar] [PubMed]
- Yauch, L.E.; Mansour, M.K.; Levitz, S.M. Receptor-mediated clearance of Cryptococcus neoformans capsular polysaccharide in vivo. Infect. Immun. 2005, 73, 8429–8432. [Google Scholar] [CrossRef] [PubMed]
- Yauch, L.E.; Mansour, M.K.; Shoham, S.; Rottman, J.B.; Levitz, S.M. Involvement of CD14, toll-like receptors 2 and 4, and MyD88 in the host response to the fungal pathogen Cryptococcus neoformans in vivo. Infect. Immun. 2004, 72, 5373–5382. [Google Scholar] [CrossRef] [PubMed]
- Monari, C.; Bistoni, F.; Casadevall, A.; Pericolini, E.; Pietrella, D.; Kozel, T.R.; Vecchiarelli, A. Glucuronoxylomannan, a microbial compound, regulates expression of costimulatory molecules and production of cytokines in macrophages. J. Infect. Dis. 2005, 191, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Syme, R.M.; Spurrell, J.; Amankwah, E.K.; Green, F.H.Y.; Mody, C.H. Primary dendritic cells phagocytose Cryptococcus neoformans via mannose receptors and Fcγ receptor II for presentation to T lymphocytes. Infect. Immun. 2002, 70, 5972–5981. [Google Scholar] [CrossRef] [PubMed]
- Piccioni, M.; Monari, C.; Bevilacqua, S. A critical role for FcgammaRIIB in up-regulation of Fas ligand induced by a microbial polysaccharide. Clin. Exp. Immunol. 2011, 165, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, F.M.; Fonseca, F.L.; Holandino, C.; Alviano, C.S.; Nimrichter, L.; Rodrigues, M.L. Glucuronoxylomannan-mediated interaction of Cryptococcus neoformans with human alveolar cells results in fungal internalization and host cell damage. Microbes Infect. 2006, 8, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, P.C.; Fonseca, F.L.; Dutra, F.F.; Bozza, M.T.; Frases, S.; Casadevall, A.; Rodrigues, M.L. Cryptococcus neoformans glucuronoxylomannan fractions of different molecular masses are functionally distinct. Future Microbiol. 2014, 9, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Villena, S.N.; Pinheiro, R.O.; Pinheiro, C.S.; Nunes, M.P.; Takiya, C.M.; DosReis, G.A.; Previato, J.O.; Mendonça-Previato, L.; Freire-de-Lima, C.G. Capsular polysaccharides galactoxylomannan and glucuronoxylomannan from Cryptococcus neoformans induce macrophage apoptosis mediated by Fas ligand. Cell. Microbiol. 2008, 10, 1274–1285. [Google Scholar] [CrossRef] [PubMed]
- Chiapello, L.S.; Baronetti, J.L.; Garro, A.P.; Spesso, M.F.; Masih, D.T. Cryptococcus neoformans glucuronoxylomannan induces macrophage apoptosis mediated by nitric oxide in a caspase-independent pathway. Int. Immunol. 2008, 20, 1527–1541. [Google Scholar] [CrossRef] [PubMed]
- Monari, C.; Bistoni, F.; Vecchiarelli, A. Glucuronoxylomannan exhibits potent immunosuppressive properties. FEMS Yeast Res. 2006, 6, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Grijpstra, J.; Tefsen, B.; van Die, I.; de Cock, H. The Cryptococcus neoformans cap10 and cap59 mutant strains, affected in glucuronoxylomannan synthesis, differentially activate human dendritic cells. FEMS Immunol. Med. Microbiol. 2009, 57, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Monari, C.; Pericolini, E.; Bistoni, G.; Casadevall, A.; Kozel, T.R.; Vecchiarelli, A. Cryptococcus neoformans Capsular Glucuronoxylomannan Induces Expression of Fas Ligand in Macrophages. J. Immunol. 2005, 174, 3461–3468. [Google Scholar] [CrossRef] [PubMed]
- Yauch, L.E.; Lam, J.S.; Levitz, S.M. Direct Inhibition of T-Cell Responses by the Cryptococcus Capsular Polysaccharide Glucuronoxylomannan. PLoS Pathog. 2006, 2, e120. [Google Scholar] [CrossRef] [PubMed]
- Moyrand, F.; Klaproth, B.; Himmelreich, U.; Dromer, F.; Janbon, G. Isolation and characterization of capsule structure mutant strains of Cryptococcus neoformans. Mol. Microbiol. 2002, 45, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Janbon, G.; Himmelreich, U.; Moyrand, F.; Improvisi, L.; Dromer, F. Cas1p is a membrane protein necessary for the O-acetylation of the Cryptococcus neoformans capsular polysaccharide. Mol. Microbiol. 2001, 42, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.L.; Fonseca, F.L.; Rodrigues, J.; Guimaraes, A.J.; Cinelli, L.P.; Miranda, K.; Nimrichter, L.; Casadevall, A.; Travassos, L.R.; Rodrigues, M.L. Chitin-Like Molecules Associate with Cryptococcus neoformans Glucuronoxylomannan To Form a Glycan Complex with Previously Unknown Properties. Eukaryot. Cell 2012, 11, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.M.; Murphy, J.W. Cryptococcal polysaccharides bind to CD18 on human neutrophils. Infect. Immun. 1997, 65, 557–563. [Google Scholar] [PubMed]
- Pericolini, E.; Cenci, E.; Monari, C.; De Jesus, M.; Bistoni, F.; Casadevall, A.; Vecchiarelli, A. Cryptococcus neoformans capsular polysaccharide component galactoxylomannan induces apoptosis of human T-cells through activation of caspase-8. Cell. Microbiol. 2006, 8, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Pericolini, E.; Gabrielli, E.; Cenci, E.; De Jesus, M.; Bistoni, F.; Casadevall, A.; Vecchiarelli, A. Involvement of Glycoreceptors in Galactoxylomannan-Induced T Cell Death. J. Immunol. 2009, 182, 6003–6010. [Google Scholar] [CrossRef]
- Pericolini, E.; Gabrielli, E.; Bistoni, G.; Cenci, E.; Perito, S.; Chow, S.-K.; Riuzzi, F.; Donato, R.; Casadevall, A.; Vecchiarelli, A. Role of CD45 Signaling Pathway in Galactoxylomannan-Induced T Cell Damage. PLoS ONE 2010, 5, e12720. [Google Scholar] [CrossRef] [PubMed]
- De Jesus, M.; Nicola, A.M.; Frases, S.; Lee, I.R.; Mieses, S.; Casadevall, A. Galactoxylomannan-Mediated Immunological Paralysis Results from Specific B Cell Depletion in the Context of Widespread Immune System Damage. J. Immunol. 2009, 183, 3885–3894. [Google Scholar] [CrossRef] [PubMed]
- Moyrand, F.; Fontaine, T.; Janbon, G. Systematic capsule gene disruption reveals the central role of galactose metabolism on Cryptococcus neoformans virulence. Mol. Microbiol. 2007, 64, 771–781. [Google Scholar] [CrossRef]
- Teixeira, P.A.C.; Penha, L.L.; Mendonça-Previato, L.; Previato, J.O. Mannoprotein MP84 mediates the adhesion of Cryptococcus neoformans to epithelial lung cells. Front. Cell Infect. Microbiol. 2014, 4, 106. [Google Scholar] [CrossRef] [PubMed]
- Specht, C.A.; Nong, S.; Dan, J.M.; Lee, C.K.; Levitz, S.M. Contribution of glycosylation to T cell responses stimulated by recombinant Cryptococcus neoformans mannoprotein. J. Infect. Dis. 2007, 196, 796–800. [Google Scholar] [CrossRef]
- Levitz, S.M.; Specht, C.A. The molecular basis for the immunogenicity of Cryptococcus neoformans mannoproteins. FEMS Yeast Res. 2006, 6, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.W. Influence of cryptococcal antigens on cell-mediated immunity. Rev. Infect. Dis. 1988, 10, S432–S435. [Google Scholar] [CrossRef] [PubMed]
- Orendi, J.M.; Verheul, A.F.; De Vos, N.M.; Visser, M.R.; Snippe, H.; Cherniak, R.; Vaishnav, V.V.; Rijkers, G.T.; Verhoef, J. Mannoproteins of Cryptococcus neoformans induce proliferative response in human peripheral blood mononuclear cells (PBMC) and enhance HIV-1 replication. Clin. Exp. Immunol. 1997, 107, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Delfino, D.; Cianci, L.; Lupis, E.; Celeste, A.; Petrelli, M.L.; Curró, F.; Cusumano, V.; Teti, G. Interleukin-6 production by human monocytes stimulated with Cryptococcus neoformans components. Infect. Immun. 1997, 65, 2454–2456. [Google Scholar] [PubMed]
- Pietrella, D.; Corbucci, C.; Perito, S.; Bistoni, G.; Vecchiarelli, A. Mannoproteins from Cryptococcus neoformans Promote Dendritic Cell Maturation and Activation. Infect. Immun. 2005, 73, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Pitzurra, L.; Cherniak, R.; Giammarioli, M.; Perito, S.; Bistoni, F.; Vecchiarelli, A. Early induction of interleukin-12 by human monocytes exposed to Cryptococcus neoformans mannoproteins. Infect. Immun. 2000, 68, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Dan, J.M.; Kelly, R.M.; Lee, C.K.; Levitz, S.M. Role of the mannose receptor in a murine model of Cryptococcus neoformans infection. Infect. Immun. 2008, 76, 2362–2367. [Google Scholar] [CrossRef] [PubMed]
- Pietrella, D.; Mazzolla, R.; Lupo, P.; Pitzurra, L.; Gomez, M.J.; Cherniak, R.; Vecchiarelli, A. Mannoprotein from Cryptococcus neoformans Promotes T-Helper Type 1 Anticandidal Responses in Mice. Infect. Immun. 2002, 70, 6621–6627. [Google Scholar] [CrossRef]
- Datta, K.; Bartlett, K.H.; Baer, R.; Byrnes, E.; Galanis, E.; Heitman, J.; Hoang, L.; Leslie, M.J.; MacDougall, L.; Magill, S.S.; et al. Cryptococcus gattii Working Group of the Pacific Northwest Spread of Cryptococcus gattii into Pacific Northwest region of the United States. Emerg. Infect. Dis. 2009, 15, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Angkasekwinai, P.; Sringkarin, N.; Supasorn, O.; Fungkrajai, M.; Wang, Y.-H.; Chayakulkeeree, M.; Ngamskulrungroj, P.; Angkasekwinai, N.; Pattanapanyasat, K. Cryptococcus gattii infection dampens Th1 and Th17 responses by attenuating dendritic cell function and pulmonary chemokine expression in the immunocompetent hosts. Infect. Immun. 2014, 82, 3880–3890. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.-Y.; Sham, A.; Kronstad, J.W. Cryptococcus gattii isolates from the British Columbia cryptococcosis outbreak induce less protective inflammation in a murine model of infection than Cryptococcus neoformans. Infect. Immun. 2009, 77, 4284–4294. [Google Scholar] [CrossRef]
- Urai, M.; Kaneko, Y.; Ueno, K.; Okubo, Y.; Aizawa, T.; Fukazawa, H.; Sugita, T.; Ohno, H.; Shibuya, K.; Kinjo, Y.; et al. Evasion of Innate Immune Responses by the Highly Virulent Cryptococcus gattii by Altering Capsule Glucuronoxylomannan Structure. Front. Cell Infect. Microbiol. 2015, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Washburn, R.G.; Bryant-Varela, B.J.; Julian, N.C.; Bennett, J.E. Differences in Cryptococcus neoformans capsular polysaccharide structure influence assembly of alternative complement pathway C3 convertase on fungal surfaces. Mol. Immunol. 1991, 28, 465–470. [Google Scholar] [CrossRef]
- Holbrook, E.D.; Rappleye, C.A. Histoplasma capsulatum pathogenesis: Making a lifestyle switch. Curr. Opin. Microbiol. 2008, 11, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Schlech, W.F.; Wheat, L.J.; HO, J.L.; French, M.L.; Weeks, R.J.; Kohler, R.B.; Deane, C.E.; Eitzen, H.E.; Band, J.D. Recurrent urban histoplasmosis, Indianapolis, Indiana, 1980–1981. Am. J. Epidemiol. 1983, 118, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Rappleye, C.A.; Engle, J.T.; Goldman, W.E. RNA interference in Histoplasma capsulatum demonstrates a role for α-(1,3)-glucan in virulence. Mol. Microbiol. 2004, 53, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Rappleye, C.A.; Eissenberg, L.G.; Goldman, W.E. Histoplasma capsulatum α-(1,3)-glucan blocks innate immune recognition by the β-glucan receptor. Proc. Natl. Acad. Sci. USA 2007, 104, 1366–1370. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.A.; Alore, E.A.; Rappleye, C.A. The yeast-phase virulence requirement for α-glucan synthase differs among Histoplasma capsulatum chemotypes. Eukaryot. Cell 2011, 10, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Garfoot, A.L.; Shen, Q.; Wüthrich, M.; Klein, B.S.; Rappleye, C.A. The Eng1 β-Glucanase Enhances Histoplasma Virulence by Reducing β-Glucan Exposure. mBio 2016, 7, e01388-15. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, V.E.; Williams, C.L.; Goldman, W.E. Comparison of Phylogenetically Distinct Histoplasma Strains Reveals Evolutionarily Divergent Virulence Strategies. mBio 2014, 5, e01376-14. [Google Scholar] [CrossRef] [PubMed]
- Garfoot, A.L.; Dearing, K.L.; VanSchoiack, A.D.; Wysocki, V.H.; Rappleye, C.A. Eng1 and Exg8 Are the Major β-Glucanases Secreted by the Fungal Pathogen Histoplasma capsulatum. J. Biol. Chem. 2017, 292, 4801–4810. [Google Scholar] [CrossRef] [PubMed]
- Perfect, J.R. The antifungal pipeline: A reality check. Nat. Rev. Drug Discov. 2017. [Google Scholar] [CrossRef] [PubMed]
- Mann, P.A.; McLellan, C.A.; Koseoglu, S.; Si, Q.; Kuzmin, E.; Flattery, A.; Harris, G.; Sher, X.; Murgolo, N.; Wang, H.; et al. Chemical Genomics-Based Antifungal Drug Discovery: Targeting Glycosylphosphatidylinositol (GPI) Precursor Biosynthesis. ACS Infect. Dis. 2015, 1, 59–72. [Google Scholar] [CrossRef]
- Johnson, M.A.; Cartmell, J.; Weisser, N.E.; Woods, R.J.; Bundle, D.R. Molecular Recognition of Candida albicans (1→2)-β-Mannan Oligosaccharides by a Protective Monoclonal Antibody Reveals the Immunodominance of Internal Saccharide Residues. J. Biol. Chem. 2012, 287, 18078–18090. [Google Scholar] [CrossRef]
- Schmidt, C.S.; White, C.J.; Ibrahim, A.S.; Filler, S.G.; Fu, Y.; Yeaman, M.R.; Edwards, J.E., Jr.; Hennessey, J.P., Jr. NDV-3, a recombinant alum-adjuvanted vaccine for Candida and Staphylococcus aureus, is safe and immunogenic in healthy adults. Vaccine 2012, 30, 7594–7600. [Google Scholar] [CrossRef] [PubMed]
- Bamford, N.C.; Snarr, B.D.; Gravelat, F.N.; Little, D.J.; Lee, M.J.; Chabot, J.C.; Geller, A.M.; Baptista, S.D.; Baker, P.; Robinson, H.; et al. Sph3 is a glycoside hydrolase required for the biosynthesis of galactosaminogalactan in Aspergillus fumigatus. J. Biol. Chem. 2015, 290, 27438–27450. [Google Scholar] [CrossRef] [PubMed]
- Pericolini, E.; Alunno, A.; Gabrielli, E.; Bartoloni, E.; Cenci, E.; Chow, S.-K.; Bistoni, G.; Casadevall, A.; Gerli, R.; Vecchiarelli, A. The Microbial Capsular Polysaccharide Galactoxylomannan Inhibits IL-17A Production in Circulating T Cells from Rheumatoid Arthritis Patients. PLoS ONE 2013, 8, e53336. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Snarr, B.D.; Qureshi, S.T.; Sheppard, D.C. Immune Recognition of Fungal Polysaccharides. J. Fungi 2017, 3, 47. https://doi.org/10.3390/jof3030047
Snarr BD, Qureshi ST, Sheppard DC. Immune Recognition of Fungal Polysaccharides. Journal of Fungi. 2017; 3(3):47. https://doi.org/10.3390/jof3030047
Chicago/Turabian StyleSnarr, Brendan D., Salman T. Qureshi, and Donald C. Sheppard. 2017. "Immune Recognition of Fungal Polysaccharides" Journal of Fungi 3, no. 3: 47. https://doi.org/10.3390/jof3030047
APA StyleSnarr, B. D., Qureshi, S. T., & Sheppard, D. C. (2017). Immune Recognition of Fungal Polysaccharides. Journal of Fungi, 3(3), 47. https://doi.org/10.3390/jof3030047