
Local-Level Genetic Diversity and Structure of Matsutake Mushroom (Tricholoma matsutake) Populations in Nagano Prefecture, Japan, Revealed by 15 Microsatellite Markers

Abstract
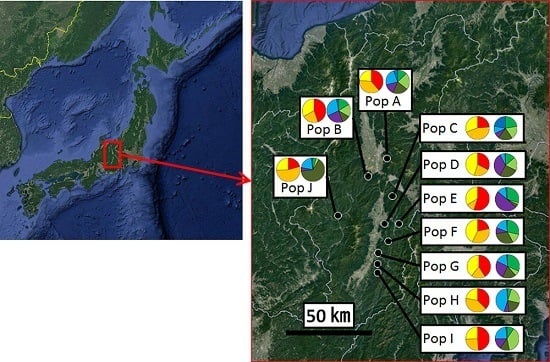
:
1. Introduction
2. Materials and Methods
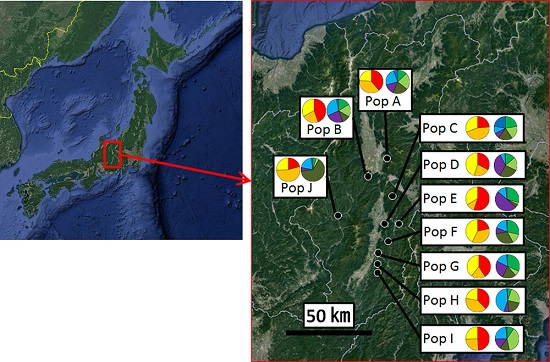
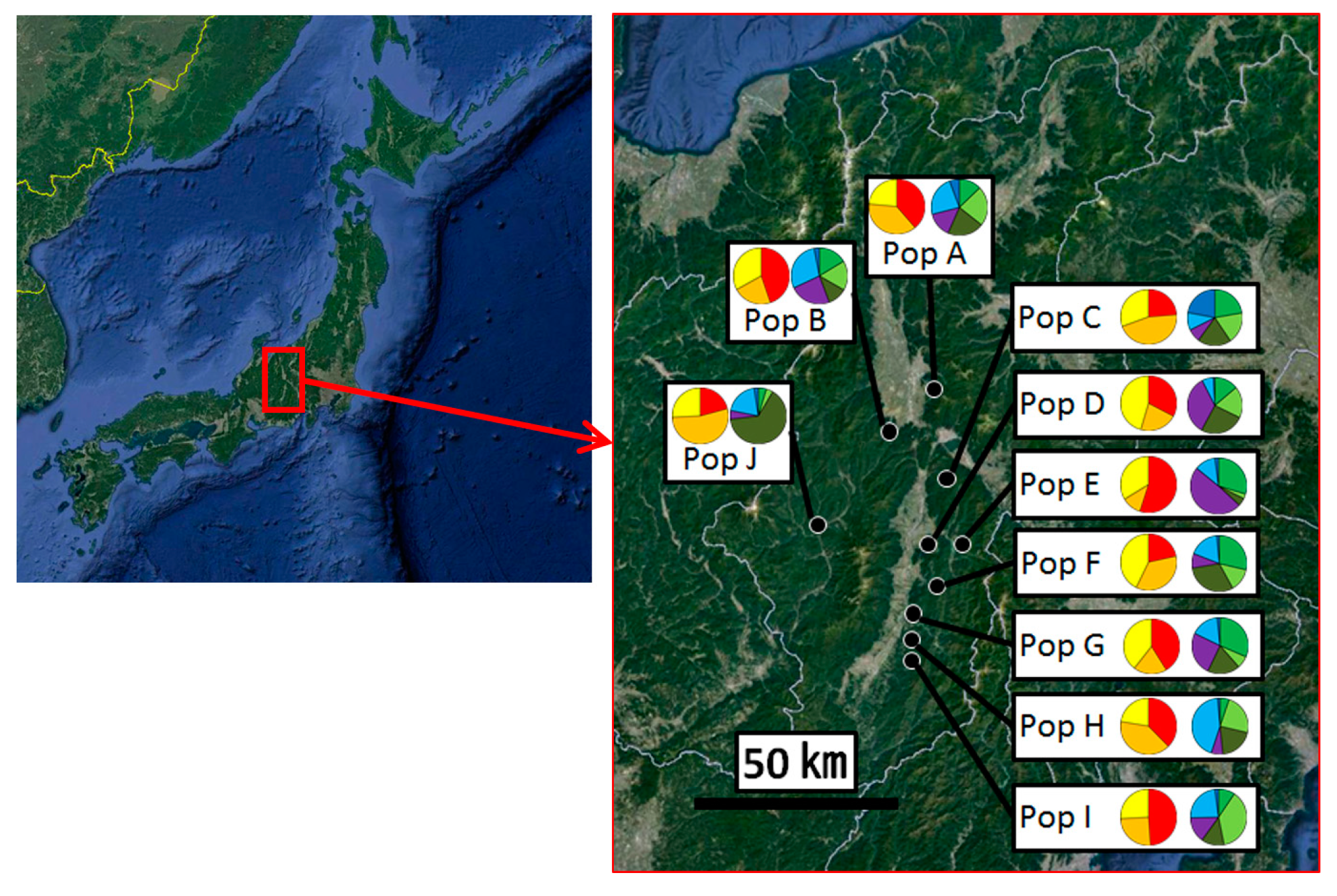
2.1. Sample Collection
2.2. Marker Development
2.3. Genotyping
2.4. Statistical Analysis
3. Results
3.1. Genotypes and Genetic Diversity in Matsutake Mushroom Populations
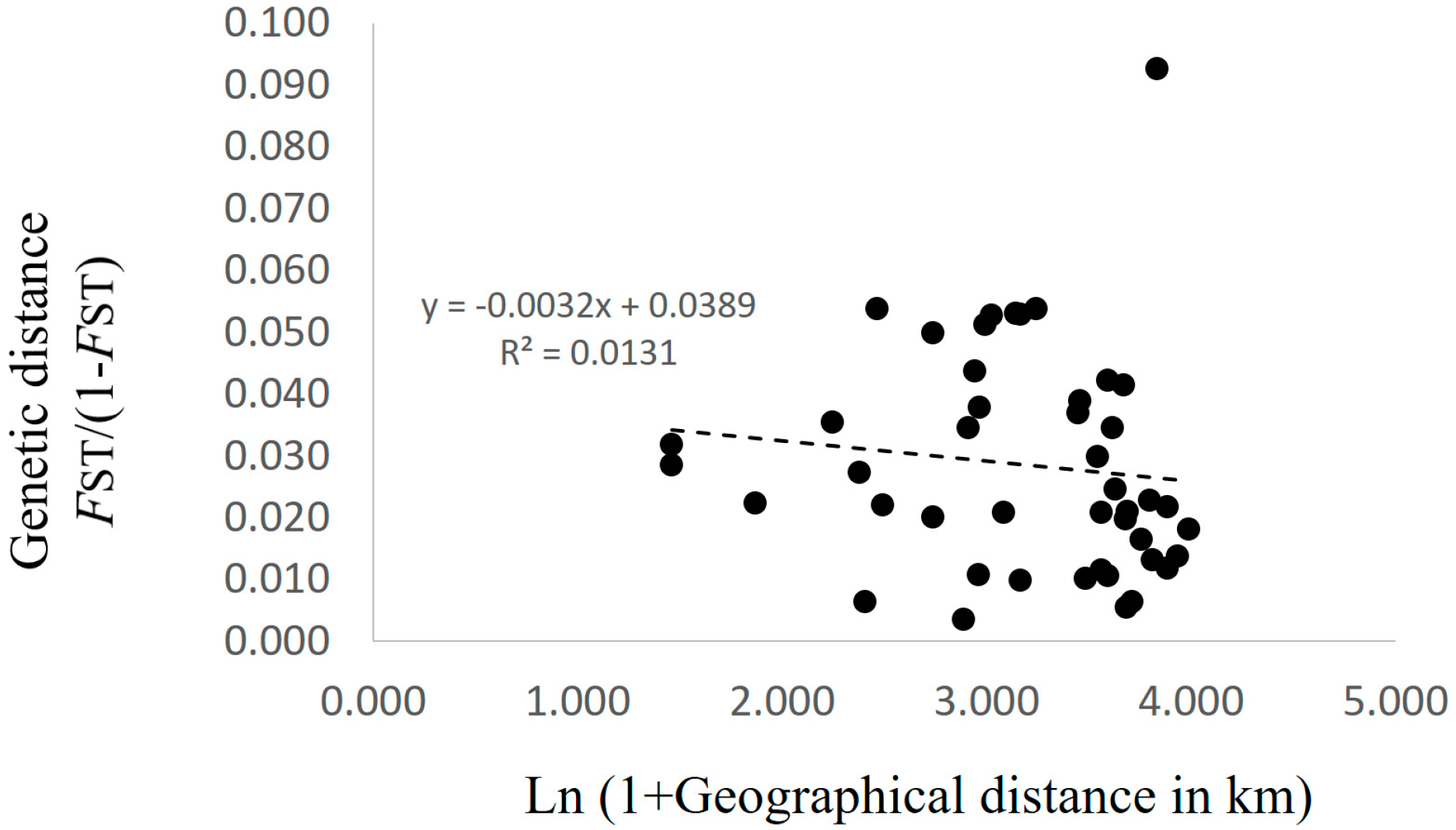
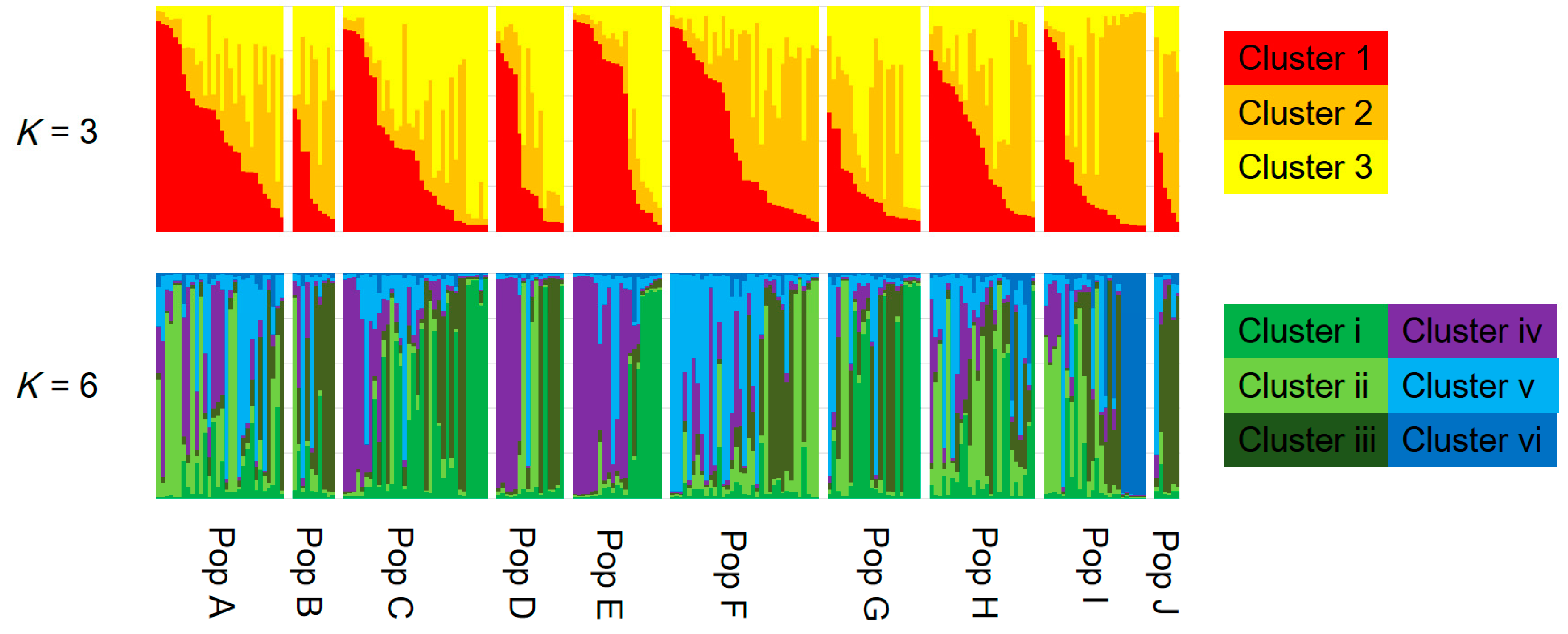
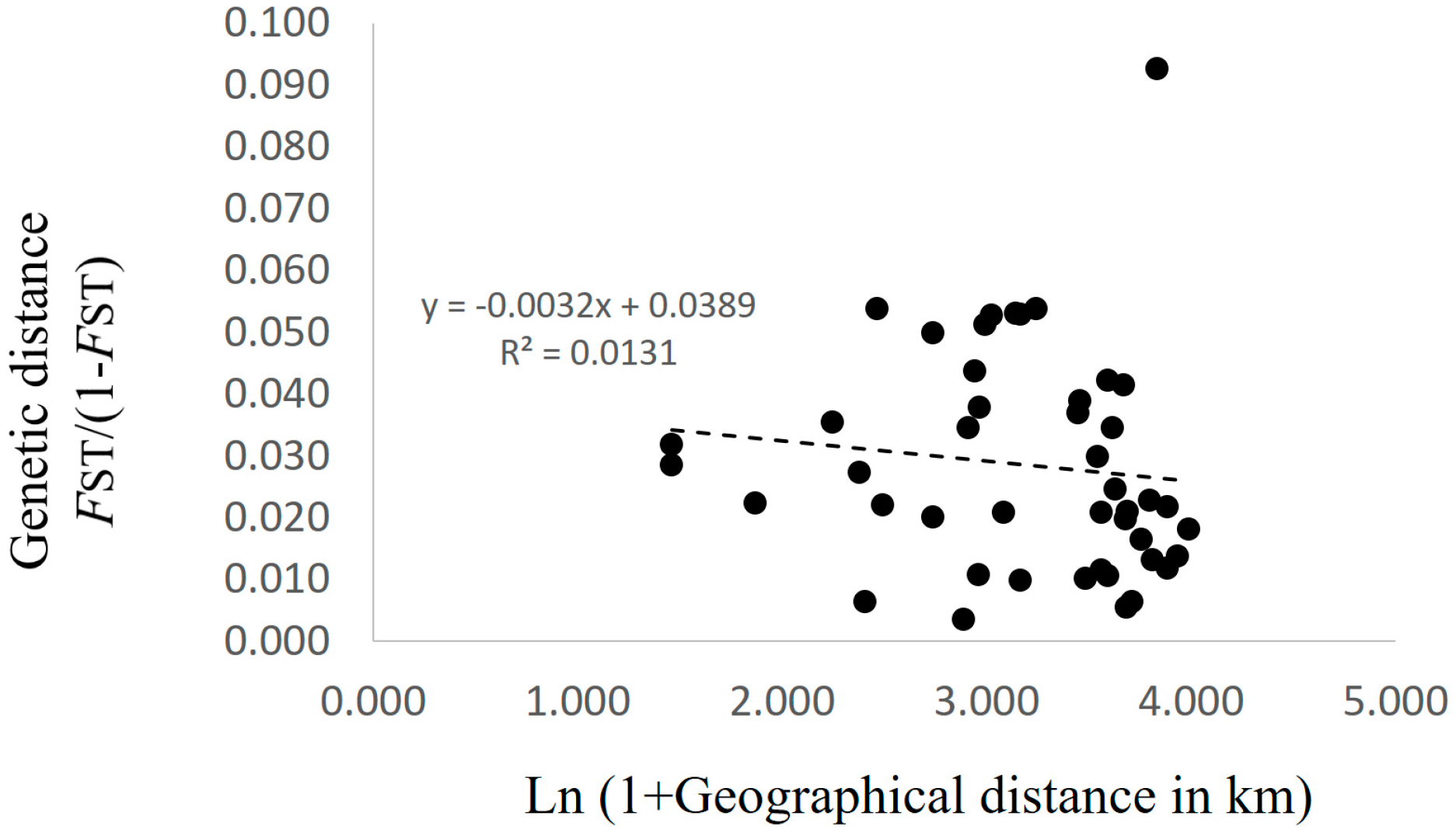
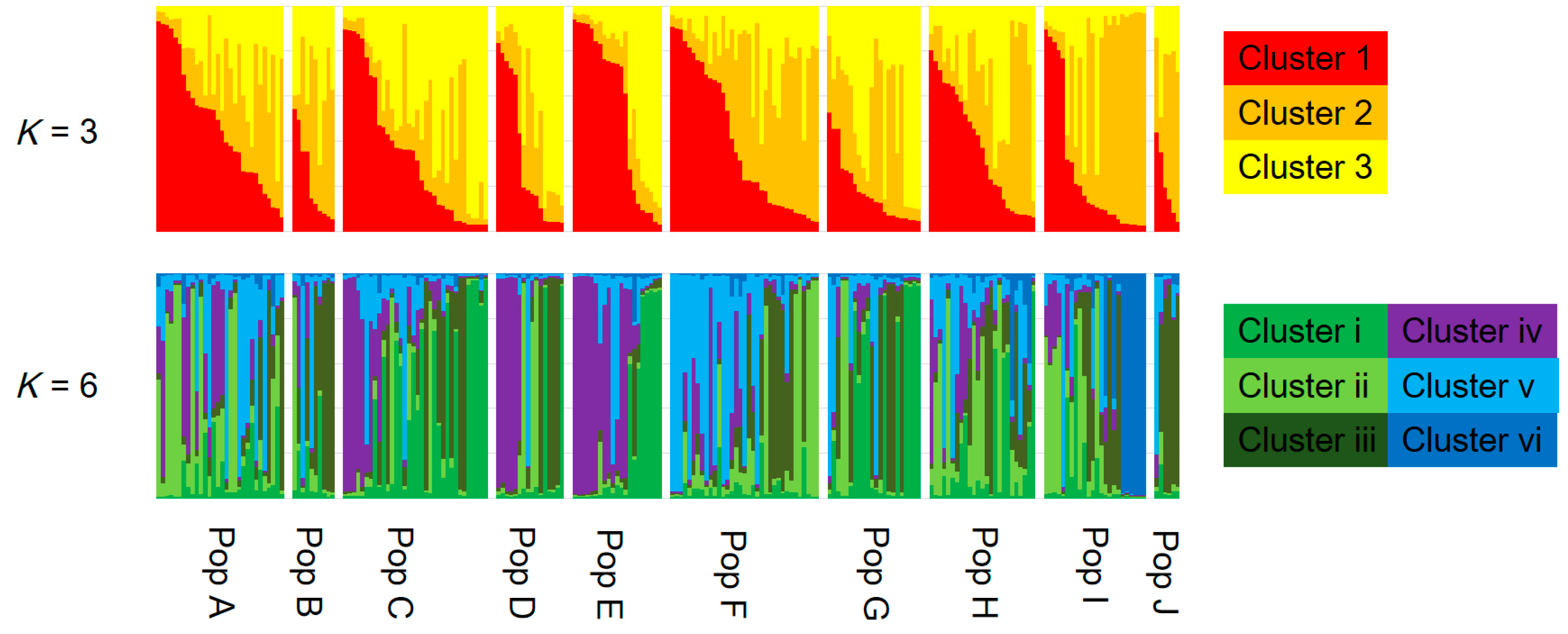
3.2. Genetic Structure and Connectivity
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yamanaka, K.; Aimi, T.; Wan, J.; Cao, H.; Chen, M. Species of host trees associated with Tricholoma matsutake and close allies in Asia. Mushroom Sci. Biotechnol. 2011, 19, 79–87. [Google Scholar]
- Kobayashi, F.; Ito, T.; Fujita, H. Cultivation of Matsutake in field in Kyoto prefecture: Standards of selection for suitable site and controlling of environments. Jpn. Soc. For. Environ. 1980, 22, 1–7. [Google Scholar]
- Matsushita, N.; Kikuchi, K.; Sasaki, Y.; Guerin-Laguette, A.; Lapeyrie, F.; Vaario, L.M.; Suzuki, K. Genetic relationship of Tricholoma matsutake and T. nauseosum from the Northern Hemisphere based on analyses of ribosomal DNA spacer regions. Mycoscience 2005, 46, 90–96. [Google Scholar] [CrossRef]
- Murata, H.; Babasaki, K.; Saegusa, T.; Takemoto, K.; Yamada, A.; Ohta, A. Traceability of Asian Matsutake, specialty mushrooms produced by the ectomycorrhizal basidiomycete Tricholoma matsutake, on the basis of retroelement-based DNA markers. Appl. Environ. Microbiol. 2008, 74, 2023–2031. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Kiuke, A.; Yamanaka, K.; Saotome, K.; Morinaga, T.; Tanaka, C.; Terashima, Y.; Aimi, T. Genetic diversity of Tricholoma matsutake and close allies associated with broad-leaved trees in Asia. Mushroom Sci. Biotechnol. 2012, 19, 167–174. [Google Scholar]
- Lian, C.; Narimatsu, M.; Nara, K.; Hogetsu, T. Tricholoma matsutake in a natural Pinus densiflora forest: Correspondence between above-and below-ground genets, association with multiple host trees and alteration of existing ectomycorrhizal communities. New Phytol. 2006, 171, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Narimatsu, M.; Koiwa, T.; Sakamoto, Y.; Natsume, S.; Kurokochi, H.; Lian, C.; Nakajima, Y.; Nakade, K.; Yoshida, K.; Tawaraya, K. Estimation of novel colony establishment and persistence of the ectomycorrhizal basidiomycete Tricholoma matsutake in a Pinus densiflora forest. Fungal Ecol. 2016, 24, 35–43. [Google Scholar] [CrossRef]
- Kurokochi, H.; Toyama, K.; Hogetsu, T. Regeneration of Robinia pseudoacacia riparian forests after clear-cutting along the Chikumagawa River in Japan. Plant Ecol. 2010, 210, 31–41. [Google Scholar] [CrossRef]
- Kurokochi, H.; Hogetsu, T. Fine-scale initiation of non-native Robinia pseudoacacia riparian forests along the Chikumagawa River in central Japan. J. Ecol. Environ. 2014, 37, 21–29. [Google Scholar] [CrossRef]
- Kurokochi, H.; Saito, Y.; Ide, Y. Genetic structure of the introduced heaven tree (Ailanthus altissima) in Japan: Evidence for two distinct origins with limited admixture. Botany 2014, 93, 133–139. [Google Scholar] [CrossRef]
- Chuman, M.; Kurokochi, H.; Saito, Y.; Ide, Y. Expansion of an invasive species, Ailanthus altissima, at a regional scale in Japan. J. Ecol. Environ. 2015, 38, 47–56. [Google Scholar] [CrossRef]
- Arriesgado, D.M.; Kurokochi, H.; Nakajima, Y.; Matsuki, Y.; Uy, W.H.; Fortes, M.D.; Lian, C. Population genetic diversity and structure of a dominant tropical seagrass, Cymodocea rotundata, in the Western Pacific region. Mar. Ecol. 2016, 37, 786–800. [Google Scholar] [CrossRef]
- Kurokochi, H.; Matsuki, Y.; Nakajima, Y.; Fortes, M.D.; Uy, W.H.; Campos, W.L.; Lian, C. A baseline for the genetic conservation of tropical seagrasses in the western North Pacific under the influence of the Kuroshio Current: The case of Syringodium isoetifolium. Conserv. Genet. 2016, 17, 103–110. [Google Scholar] [CrossRef]
- Kurokochi, H.; Tan, E.; Asakawa, S.; Sukartiningsih Saito, Y.; Ide, Y. Development of 16 microsatellite markers in Eusideroxylon zwageri by next-generation sequencing. Conserv. Genet. Resour. 2014, 6, 593–595. [Google Scholar] [CrossRef]
- Kurokochi, H.; Tan, E.; Asakawa, S.; Lian, C. Development of 18 microsatellite markers in Pieris japonica, a poisonous tree insulated from the browsing pressure of herbivores, using a next-generation sequencer. J. For. Res. 2015, 20, 244–247. [Google Scholar] [CrossRef]
- Kurokochi, H.; Nurtjahjaningsih, I.L.G.; Tan, E.; Asakawa, S.; Saito, Y.; Ide, Y. Development of polymorphic chloroplast DNA markers for the endangered tree Eusideroxylon zwageri through chloroplast isolation and next-generation sequencing. Conserv. Genet. Resour. 2015, 7, 845–850. [Google Scholar] [CrossRef]
- Sugiura, N.; Kurokochi, H.; Tan, E.; Asakawa, S.; Sato, N.; Saito, Y.; Ide, Y. Development of 13 polymorphic chloroplast DNA markers in Quercus gilva, a regionally endemic species in Japan. Conserv. Genet. Resour. 2014, 6, 961–965. [Google Scholar] [CrossRef]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007, 35, W71–W74. [Google Scholar] [CrossRef] [PubMed]
- Kurokochi, H.; Saito, Y.; Chuman, M.; Ide, Y. Low chloroplast diversity despite of phylogenetically divergent haplotypes in Japanese populations of Ailanthus altissima (Simaroubaceae). Botany 2013, 91, 148–154. [Google Scholar] [CrossRef]
- Schuelke, M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 2000, 18, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Marshall, T.C.; Slate, J.; Kruuk, L.E.B.; Pemberton, J.M. Statistical confidence for likelihood-based paternity inference in natural populations. Mol. Ecol. 1998, 7, 639–655. [Google Scholar] [CrossRef] [PubMed]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- El Mousadik, A.; Petit, R.J. High level of genetic differentiation for allelic richness among populations of the argan tree (Argania spinosa (L.) Skeels) endemic to Morocco. Theor. Appl. Genet. 1996, 92, 832–839. [Google Scholar] [CrossRef] [PubMed]
- Goudet, J. FSTAT, A Program to Estimate and Test Gene Diversities and fixation Indices Version 2.9.3. Available online: https://www2.unil.ch/popgen/softwares/fstat.htm (accessed on 1 January 2017).
- Peakall, R.; Smouse, P.E. GenAlEx 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evol. Bioinform. Online 2005, 1, 47–50. [Google Scholar]
- Pritchard, J.K.; Stephens, S.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software Structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Bao, D.; Koike, A.; Yao, F.; Yamanaka, K.; Aimi, T.; Kitamoto, Y. Analyses of the genetic diversity of matsutake isolates collected from different ecological environments in Asia. J. Wood Sci. 2007, 53, 344–350. [Google Scholar] [CrossRef]
- Xu, J.; Sha, T.A.O.; Li, Y.C.; Zhao, Z.W.; Yang, Z.L. Recombination and genetic differentiation among natural populations of the ectomycorrhizal mushroom Tricholoma matsutake from southwestern China. Mol. Ecol. 2008, 17, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
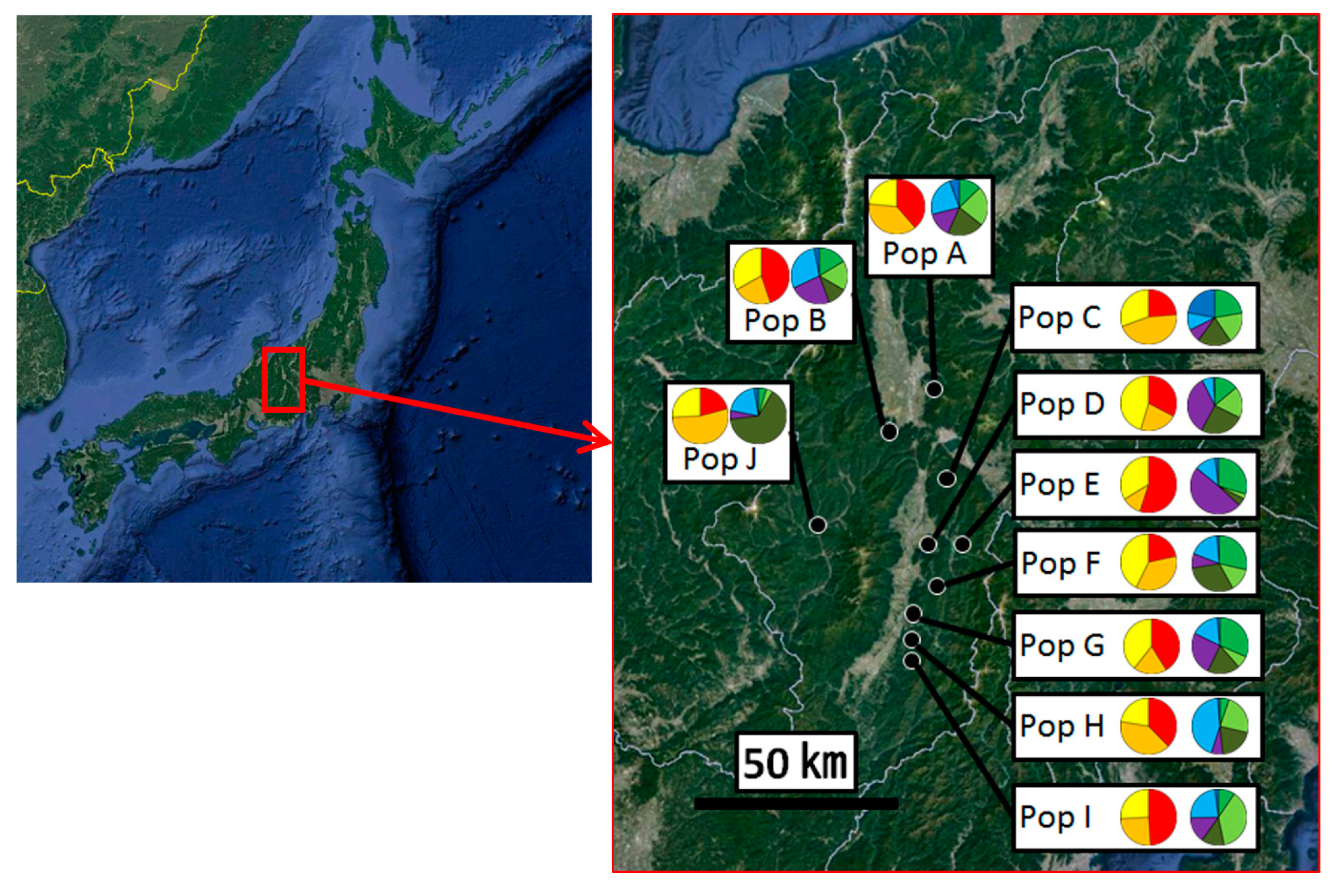

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Longitude | Latitude | N | G | HE | AR | PA | FIS |
|---|---|---|---|---|---|---|---|---|
| Pop A | 138°02′ | 36°11′ | 42 | 30 | 0.68 | 4.30 | 4 | 0.090 |
| Pop B | 137°54′ | 36°05′ | 21 | 10 | 0.66 | 4.09 | 2 | −0.032 |
| Pop C | 138°04′ | 35°58′ | 41 | 34 | 0.70 | 4.11 | 3 | 0.090 |
| Pop D | 138°00′ | 35°49′ | 25 | 16 | 0.60 | 3.67 | 4 | 0.133 |
| Pop E | 138°05′ | 35°45′ | 37 | 21 | 0.66 | 3.74 | 1 | 0.119 |
| Pop F | 138°01′ | 35°42′ | 43 | 35 | 0.68 | 4.21 | 6 | 0.086 * |
| Pop G | 137°57′ | 35°37′ | 38 | 22 | 0.67 | 4.03 | 6 | 0.142 * |
| Pop H | 137°58′ | 35°35′ | 33 | 25 | 0.67 | 4.05 | 2 | 0.165 * |
| Pop I | 137°57′ | 35°33′ | 29 | 24 | 0.69 | 4.18 | 4 | 0.066 |
| Pop J | 137°41′ | 35°51′ | 19 | 6 | 0.65 | 4.14 | 2 | −0.006 |
| (Total) | (Average) | |||||||
| 328 | 223 | 0.67 | 4.05 | 3.4 | 0.085 | |||
| Source of Variation | D.F. | Sum of Squares | Variance Components | Percentage of Variation |
|---|---|---|---|---|
| Among populations | 9 | 44.5 | 0.06 * | 2.91 |
| Among genets within populations | 213 | 484.7 | 0.24 * | 11.62 |
| Within genets | 233 | 399.0 | 1.79 * | 85.47 |
| Total | 445 | 928.2 | 2.09 | 100 |
| Population | Pop A | Pop B | Pop C | Pop D | Pop E | Pop F | Pop G | Pop H | Pop I | Pop J |
|---|---|---|---|---|---|---|---|---|---|---|
| Pop A | −0.003 | 0.012 | 0.055 | 0.045 | 0.003 | 0.001 | 0.015 | 0.020 | 0.037 | |
| Pop B | 0.040 | 0.088 | 0.032 | 0.017 | 0.008 | 0.019 | 0.027 | 0.074 | ||
| Pop C | 0.058 | 0.060 | 0.015 | 0.009 | 0.036 | 0.020 | 0.023 | |||
| Pop D | 0.085 | 0.048 | 0.048 | 0.070 | 0.097 | 0.095 | ||||
| Pop E | 0.043 | 0.053 | 0.029 | 0.061 | 0.127 | |||||
| Pop F | 0.000 | 0.025 | 0.033 | 0.036 | ||||||
| Pop G | 0.019 | 0.007 | 0.029 | |||||||
| Pop H | 0.038 | 0.081 | ||||||||
| Pop I | 0.040 | |||||||||
| Pop J |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurokochi, H.; Zhang, S.; Takeuchi, Y.; Tan, E.; Asakawa, S.; Lian, C. Local-Level Genetic Diversity and Structure of Matsutake Mushroom (Tricholoma matsutake) Populations in Nagano Prefecture, Japan, Revealed by 15 Microsatellite Markers. J. Fungi 2017, 3, 23. https://doi.org/10.3390/jof3020023
Kurokochi H, Zhang S, Takeuchi Y, Tan E, Asakawa S, Lian C. Local-Level Genetic Diversity and Structure of Matsutake Mushroom (Tricholoma matsutake) Populations in Nagano Prefecture, Japan, Revealed by 15 Microsatellite Markers. Journal of Fungi. 2017; 3(2):23. https://doi.org/10.3390/jof3020023
Chicago/Turabian StyleKurokochi, Hiroyuki, Shijie Zhang, Yoshie Takeuchi, Engkong Tan, Shuichi Asakawa, and Chunlan Lian. 2017. "Local-Level Genetic Diversity and Structure of Matsutake Mushroom (Tricholoma matsutake) Populations in Nagano Prefecture, Japan, Revealed by 15 Microsatellite Markers" Journal of Fungi 3, no. 2: 23. https://doi.org/10.3390/jof3020023
APA StyleKurokochi, H., Zhang, S., Takeuchi, Y., Tan, E., Asakawa, S., & Lian, C. (2017). Local-Level Genetic Diversity and Structure of Matsutake Mushroom (Tricholoma matsutake) Populations in Nagano Prefecture, Japan, Revealed by 15 Microsatellite Markers. Journal of Fungi, 3(2), 23. https://doi.org/10.3390/jof3020023