
Co-Expression Network Analysis Suggests PacC Transcriptional Factor Involved in Botryosphaeria dothidea Pathogenicity in Chinese Hickory

Abstract
1. Introduction
2. Results
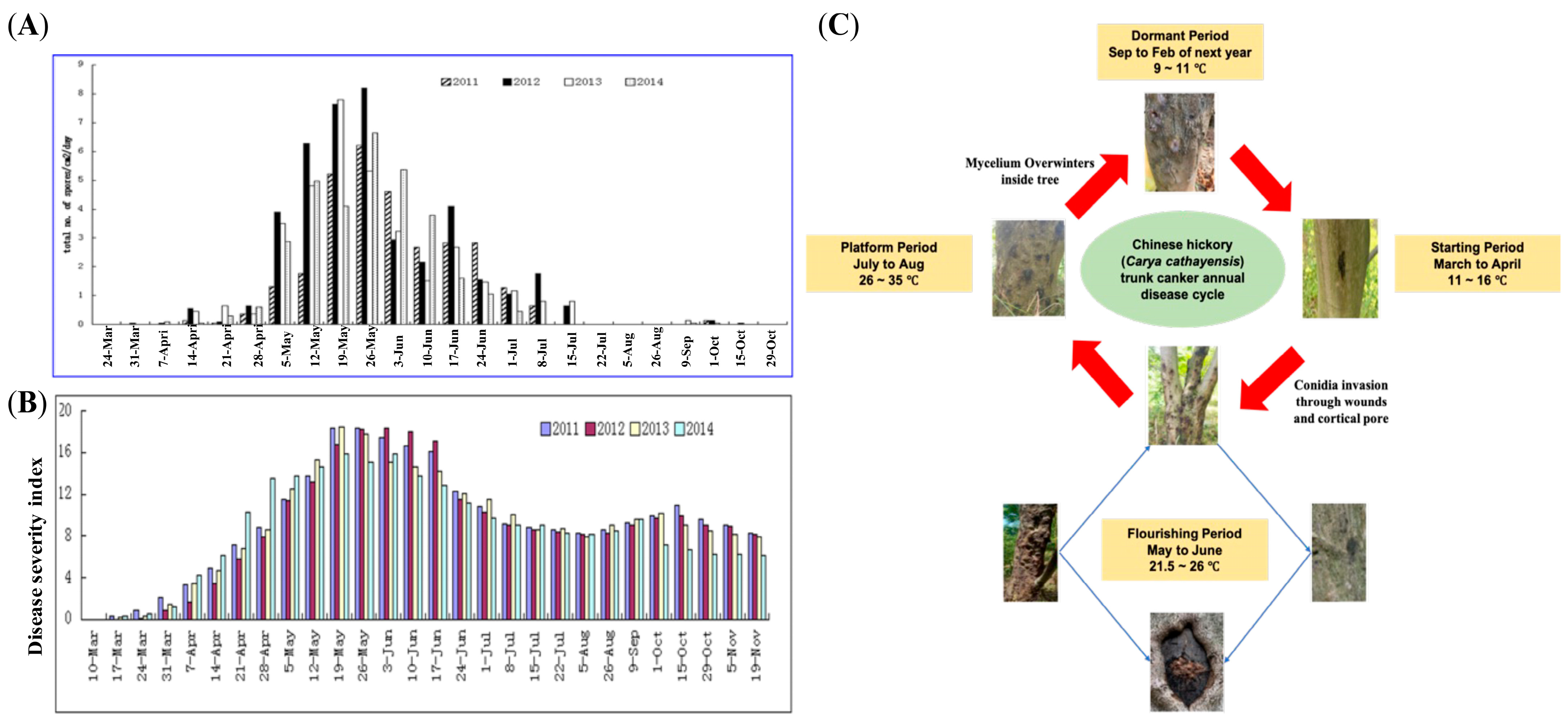
2.1. In Vitro Simulation of B. dothidea’s Latent Infection Phase Using Chinese Hickory
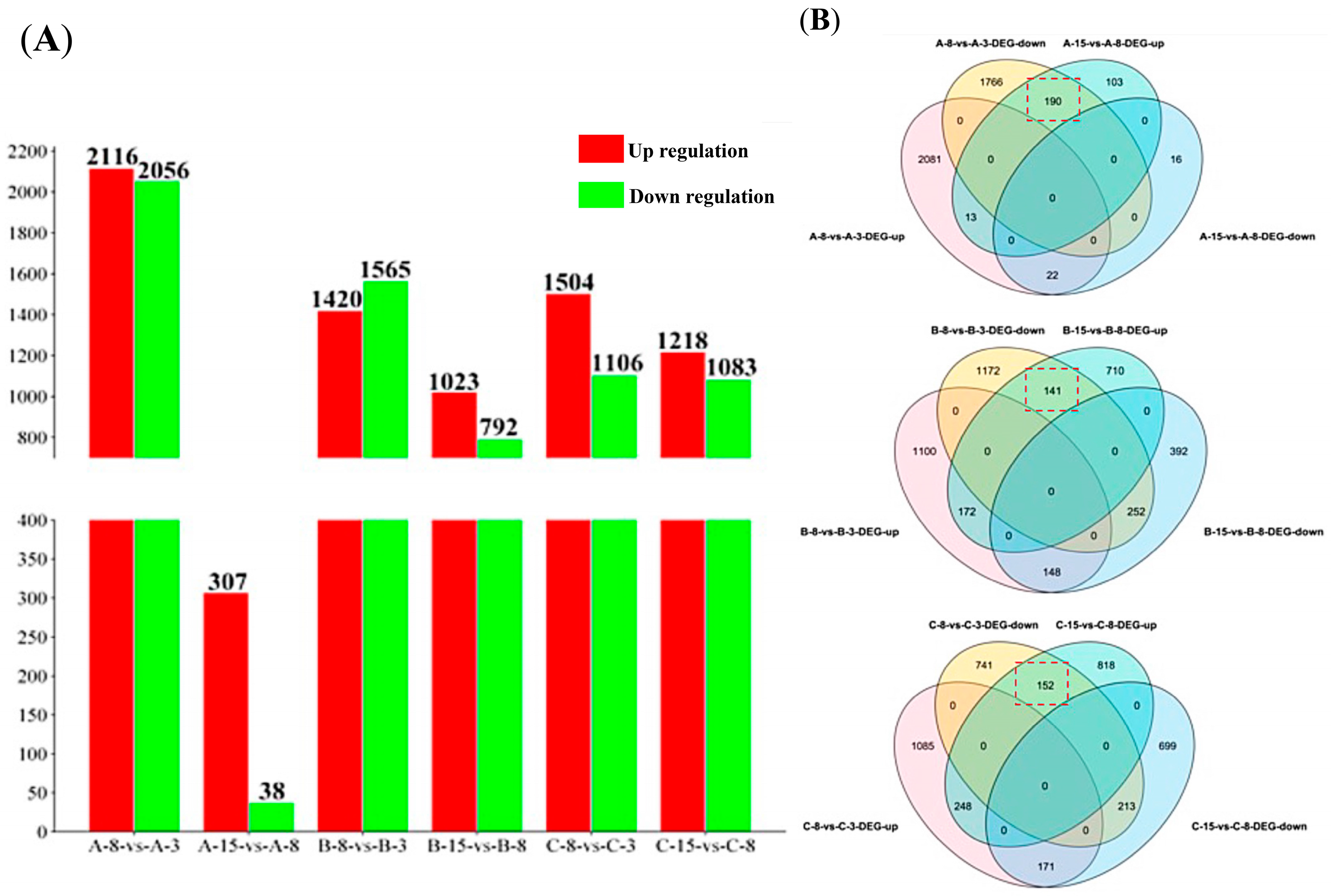
2.2. RNA-Seq Data Processing and Identification of DEGs
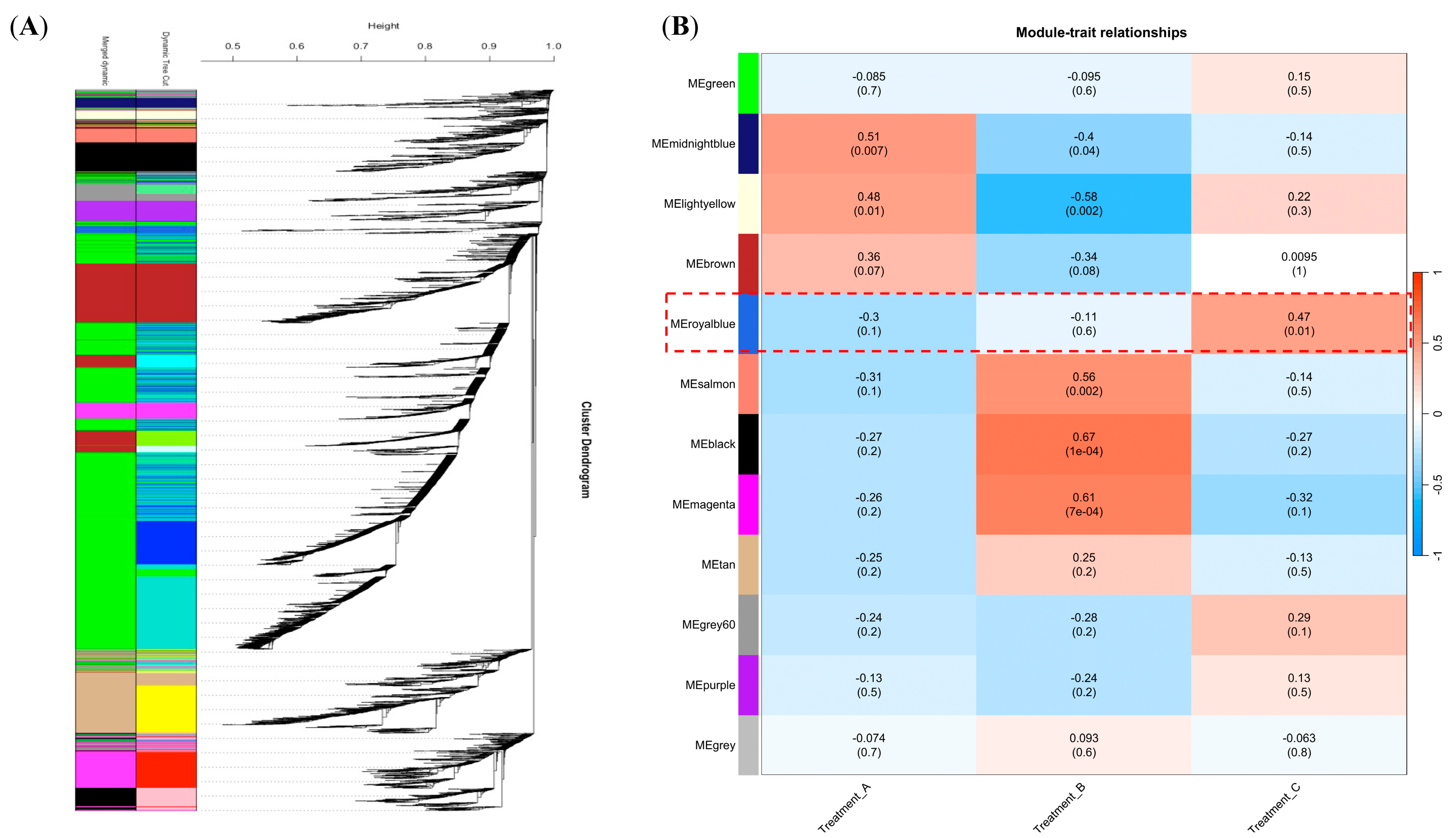
2.3. A Co-Expression Network Construction Revealing the Genes Associated with B. dothidea’s Latent Infection Phase in Chinese Hickory
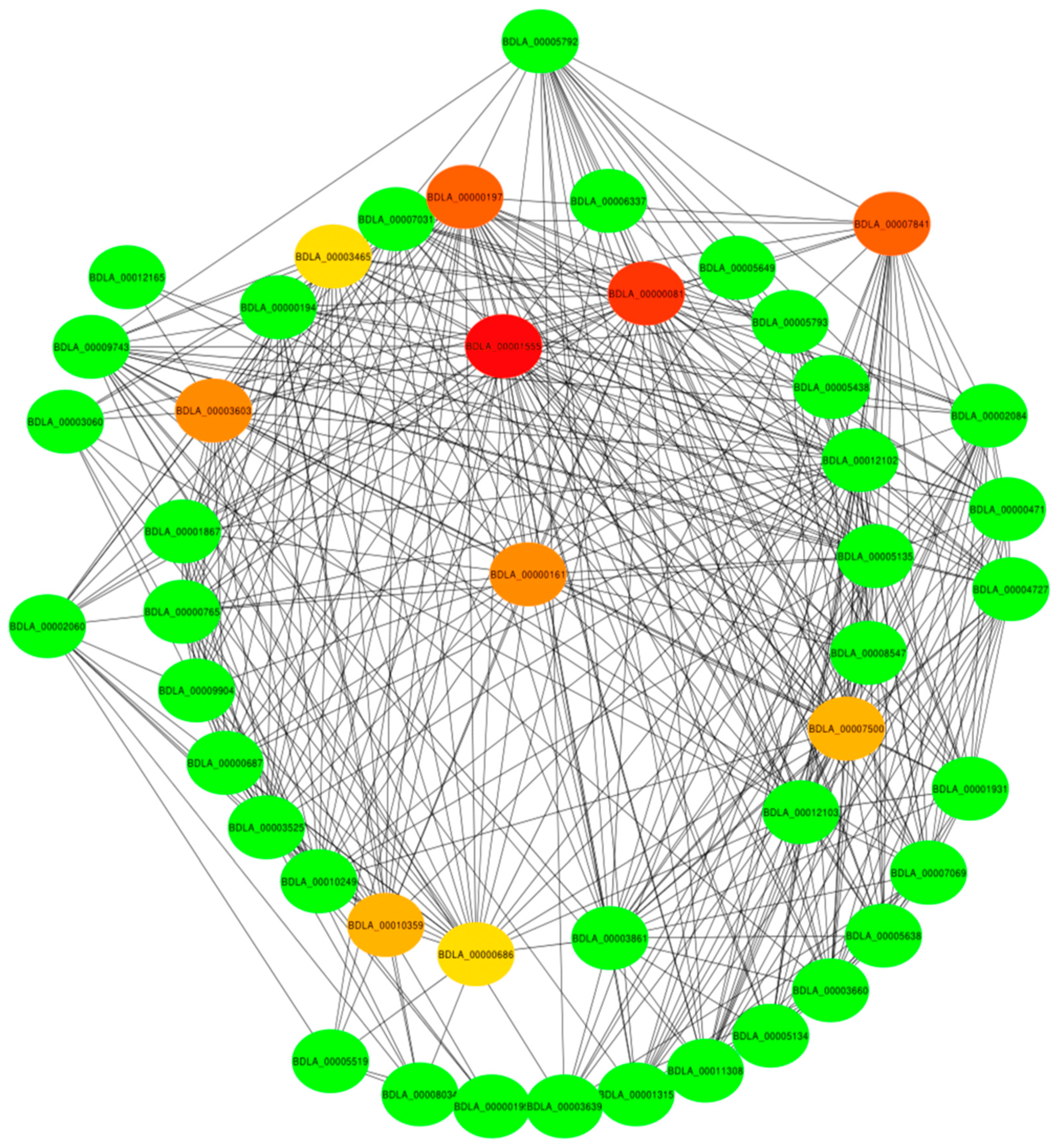
2.4. The Identification of the Hub Genes in the Royal-Blue Module and the Enrichment Analysis
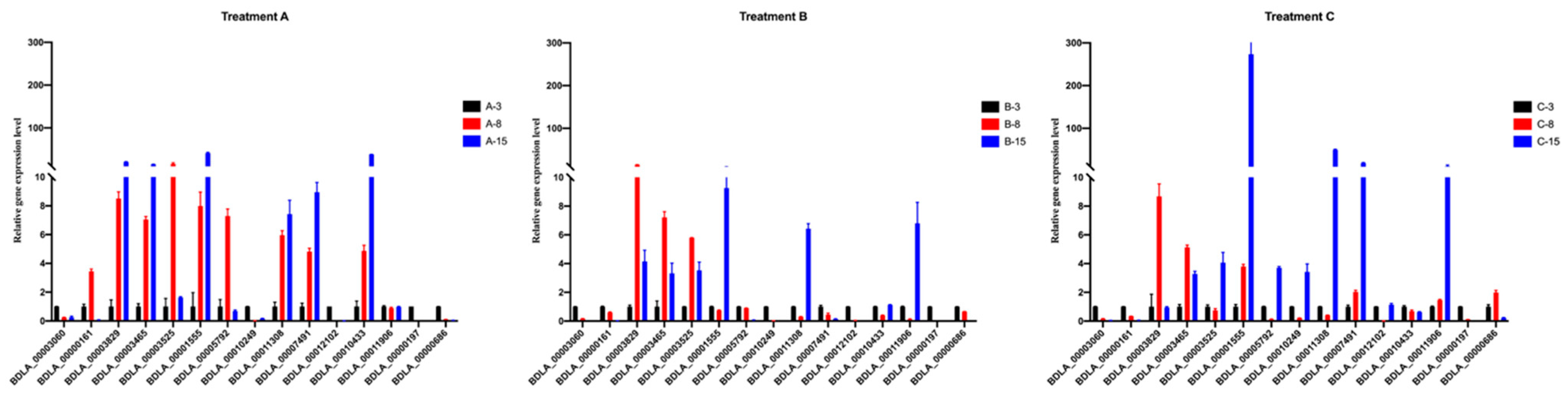
2.5. Validation of Hub Genes Using RT-PCR
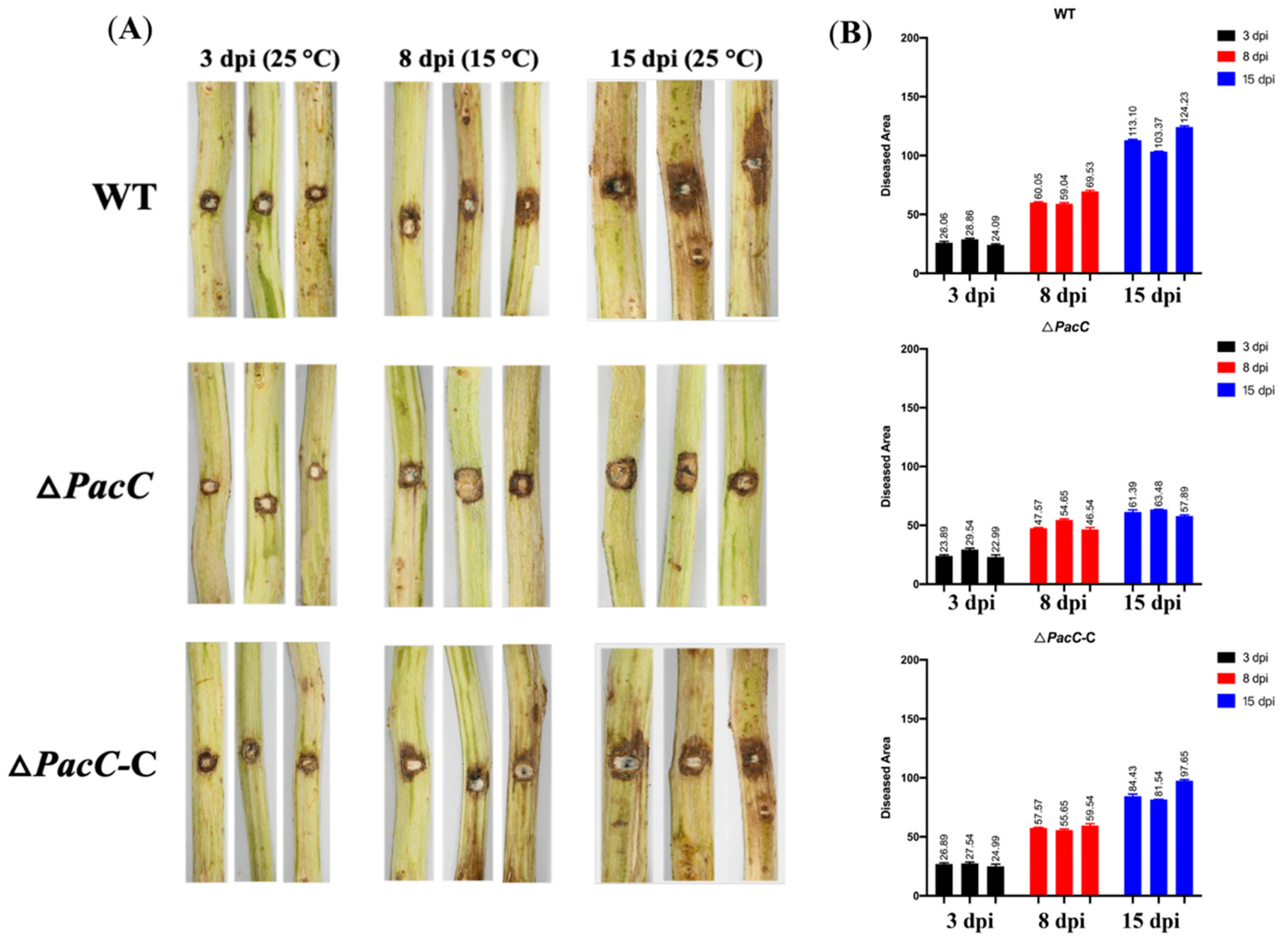
2.6. PacC Transcriptional Factor Is Important for B. dothidea’s Latent Infection Phase
3. Discussion
3.1. Hub Genes Predicted by Co-Expression Analysis Were Associated with B. dothidea’s Infection and Metal Ion Homeostasis or Transport
3.2. PacC Transcription Factor
3.3. Carbohydrate Metabolism Enzymes
4. Conclusions
5. Methods and Materials
5.1. The Plant Material and Pathogens Used for the Inoculation Assays
5.2. RNA Extraction, Transcriptome Sequencing, and Differential Expression Analysis
5.3. The Construction of the Weighted Gene Co-Expression Network
5.4. Selection and Functional Enrichment Analysis of Target Gene Modules
5.5. The Identification and Annotation of Hub Genes in the Target Module
5.6. The Validation of the Hub Genes Using Quantitative Real-Time PCR
5.7. Construction of Deletions and Complemented Mutants
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, C.Q.; Xu, B.C. First report of canker on pecan (Carya cathayensis) caused by Botryosphaeria dothidea in China. Plant Dis. 2011, 95, 1319. [Google Scholar] [CrossRef]
- Marsberg, A.; Kemler, M.; Jami, F.; Nagel, J.H.; Postma-Smidt, A.; Naidoo, S.; Wingfield, M.J.; Crous, P.W.; Spatafora, J.W.; Hesse, C.N.; et al. Botryosphaeria dothidea: A latent pathogen of global importance to woody plant health. Mol. Plant Pathol. 2017, 18, 477–488. [Google Scholar] [CrossRef]
- Michailides, T.J. Pathogenicity, distribution, sources of inoculum, and infection courts of Botryosphaeria dothidea on pistachio. Phytopathology 1991, 81, 566–573. [Google Scholar] [CrossRef]
- Swart, W.J.; Wingfield, M.J. Biology and Control of Sphaeropsis sapinea on Pinus species in South Mrica. Detail 1991, 30, 40. [Google Scholar]
- Smith, H.; Kemp, G.H.J.; Wingfield, M.J. Canker and die-back of Eucalyptus in South Africa caused by Botryosphaeria dothidea. Plant Pathol. 1994, 43, 1031–1034. [Google Scholar] [CrossRef]
- Slippers, B.; Wingfield, M.J. Botryosphaeriaceae as endophytes and latent pathogens of woody plants: Diversity, ecology and impact. Fungal Biol. Rev. 2007, 21, 90–106. [Google Scholar] [CrossRef]
- Zhuang, C.J.; Wang, Q.W.; Wu, Q.Q.; Qiu, Z.L.; Xu, B.C.; Zhang, C.Q. Diversity of Botryosphaeriaceae Species Associated with Chinese Hickory Tree (Carya cathayensis) Trunk Cankers. Plant Dis. 2021, 105, 3869–3879. [Google Scholar] [CrossRef]
- Dai, D.J.; Wang, H.D.; Wang, Y.P.; Zhang, C.Q. Management of Chinese hickory (Carya cathayensis) trunk canker through effective fungicide application programs and baseline sensitivity of Botryosphaeria dothidea to trifloxystrobin. Australas. Plant Pathol. 2017, 46, 75–82. [Google Scholar] [CrossRef]
- Yan, J.Y.; Zhao, W.S.; Chen, Z.; Xing, Q.K.; Zhang, W.; Chethana, K.W.T.; Xue, M.F.; Xu, J.P.; Phillips, A.J.L.; Wang, Y.; et al. Comparative genome and transcriptome analyses reveal adaptations to opportunistic infections in woody plant degrading pathogens of Botryosphaeriaceae. DNA Res. 2018, 25, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Qiao, L.; Gao, X.; Jia, Z.; Liu, X.; Wang, H.; Kong, Y.; Qin, P.; Yang, B. Identification of adult resistant genes to stripe rust in wheat from southwestern China based on GWAS and WGCNA analysis. Plant Cell Rep. 2024, 43, 67. [Google Scholar] [CrossRef]
- Hu, Y.; He, Z.; Kang, Y.; Ye, W.; Cui, L. Identification of a C2H2 transcription factor (PsCZF3) associated with RxLR effectors and carbohydrate-active enzymes in Phytophthora sojae based on WGCNA. J. Fungi 2022, 8, 998. [Google Scholar] [CrossRef]
- Li, B.; Chen, Y.; Tian, S. Function of pH-dependent transcription factor PacC in regulating development, pathogenicity, and mycotoxin biosynthesis of phytopathogenic fungi. FEBS J. 2022, 289, 1723–1730. [Google Scholar] [CrossRef]
- Díez, E.; Álvaro, J.; Espeso, E.A.; Rainbow, L.; Suárez, T.; Tilburn, J.; Arst, H.N., Jr.; Peñalva, M.Á. Activation of the Aspergillus PacC zinc finger transcription factor requires two proteolytic steps. EMBO J. 2002, 21, 1350–1359. [Google Scholar] [CrossRef]
- Hervás-Aguilar, A.; RodrÁguez, J.M.; Tilburn, J.; Arst, H.N.; Peñalva, M.A. Evidence for the direct involvement of the proteasome in the proteolytic processing of the Aspergillus nidulans zinc finger transcription factor PacC. J. Biol. Chem. 2007, 282, 34735–34747. [Google Scholar] [CrossRef] [PubMed]
- Peñas, M.M.; Hervás-Aguilar, A.; Múnera-Huertas, T.; Reoyo, E.; Peñalva, M.Á.; Arst, H.N., Jr.; Tilburn, J. Further characterization of the signaling proteolysis step in the Aspergillus nidulans pH signal transduction pathway. Eukaryot. Cell 2007, 6, 960–970. [Google Scholar] [CrossRef]
- Peñalva, M.A.; Tilburn, J.; Bignell, E.; Arst, H.N. Ambient pH gene regulation in fungi: Making connections. Trends Microbiol. 2008, 16, 291–300. [Google Scholar] [CrossRef]
- Espeso, E.A.; Tilburn, J.; Sánchez-Pulido, L.; Brown, C.V.; Valencia, A.; Arst, H.N., Jr.; Peñalva, M.A. Specific DNA recognition by the Aspergillus nidulans three zinc finger transcription factor PacC. J. Mol. Biol. 1997, 274, 466–480. [Google Scholar] [CrossRef]
- Chen, Y.; Li, B.; Xu, X.; Zhang, Z.; Tian, S. The pH-responsive PacC transcription factor plays pivotal roles in virulence and patulin biosynthesis in Penicillium expansum. Environ. Microbiol. 2018, 20, 4063–4078. [Google Scholar] [CrossRef] [PubMed]
- Barda, O.; Maor, U.; Sadhasivam, S.; Bi, Y.; Zakin, V.; Prusky, D.; Sionov, E. The pH-responsive transcription factor PacC governs pathogenicity and ochratoxin A biosynthesis in Aspergillus carbonarius. Front. Microbiol. 2020, 11, 210. [Google Scholar] [CrossRef] [PubMed]
- Caracuel, Z.; Roncero, M.I.G.; Espeso, E.A.; González-Verdejo, C.I.; García-Maceira, F.I.; Di Pietro, A. The pH signalling transcription factor PacC controls virulence in the plant pathogen Fusarium oxysporum. Mol. Microbiol. 2003, 48, 765–779. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-L.; He, D.; Yin, C.; Yang, J.; Sun, J.; Wang, D.; Xue, M.; Li, Z.; Peng, Z.; Chen, D.; et al. PacC-dependent adaptation and modulation of host cellular pH controls hemibiotrophic invasive growth and disease development by the rice blast fungus. bioRxiv 2020, 2020-06. [Google Scholar] [CrossRef]
- Bertuzzi, M.; Schrettl, M.; Alcazar-Fuoli, L.; Cairns, T.C.; Munoz, A.; Walker, L.A.; Herbst, S.; Safari, M.; Cheverton, A.M.; Chen, D. The pH-Responsive PacC Transcription Factor of Aspergillus fumigatus Governs Epithelial Entry and Tissue Invasion during Pulmonary Aspergillosis. PLoS Pathog. 2014, 10, e1004413. [Google Scholar] [CrossRef]
- Trushina, N.; Levin, M.; Mukherjee, P.K.; Horwitz, B.A. PacC and pH–Dependent Transcriptome of the Mycotrophic Fungus Trichoderma Virens. BMC Genom. 2013, 14, 138. [Google Scholar] [CrossRef]
- Zhuo, R.; Li, G.; Peng, H.; Zong, Y.; Wang, X.; Lu, S.; Chen, Y.; Zhang, Z.; Tian, S.; Li, B. Extensive Regulation of pH-Responsive Transcription Factor PacC on Secondary Metabolism Contributes to Development and Virulence of Botrytis cinerea. Postharvest Biol. Technol. 2023, 197, 112219. [Google Scholar] [CrossRef]
- Úrbez-Torres, J.R.; Bruez, E.; Hurtado, J.; Gubler, W.D. Effect of temperature on conidial germination of Botryosphaeriaceae species infecting grapevines. Plant Dis. 2010, 94, 1476–1484. [Google Scholar] [CrossRef]
- Liang, D.; Jiang, Y.; Zhang, Y.; Mao, C.; Ma, T.; Zhang, C. The Comparative Genomics of Botryosphaeriaceae Suggests Gene Families of Botryosphaeria dothidea Related to Pathogenicity on Chinese Hickory Tree. J. Fungi 2024, 10, 299. [Google Scholar] [CrossRef]
- Gao, X.-M.; Zhou, X.-H.; Jia, M.-W.; Wang, X.-Z.; Liu, D. Identification of Key Genes in Sepsis by WGCNA. Prev. Med. 2023, 172, 107540. [Google Scholar] [CrossRef]
- Li, Z.; Chyr, J.; Jia, Z.; Wang, L.; Hu, X.; Wu, X.; Song, C. Identification of Hub Genes Associated with Hypertension and Their Interaction with miRNA Based on Weighted Gene Coexpression Network Analysis (WGCNA) Analysis. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e923514-1. [Google Scholar] [CrossRef]
- Casci, T. Network fundamentals, via hub genes. Nat. Rev. Genet. 2006, 7, 664–665. [Google Scholar] [CrossRef]
- Yao, J.; Mei, L.; Jiang, H.; Hu, G.; Wang, Y. Evaluation of Carya cathayensis resistance to Botryosphaeria trunk canker using grafting on pecan. Sci. Hortic. 2019, 248, 184–188. [Google Scholar] [CrossRef]
- Ma, T.; Zhang, Y.; Yan, C.; Zhang, C. Phenotypic and genomic difference among four botryosphaeria pathogens in chinese hickory trunk canker. J. Fungi 2023, 9, 204. [Google Scholar] [CrossRef]
- Ma, Z.; Morgan, D.P.; Felts, D.; Michailides, T.J. Sensitivity of Botryosphaeria dothidea from California pistachio to tebuconazole. Crop Prot. 2016, 21, 829–835. [Google Scholar] [CrossRef]
- Brown-Rytlewski, D.E.; McManus, P.S. Virulence of Botryosphaeria dothidea and Botryosphaeria obtusa on apple and management of stem cankers with fungicides. Plant Dis. 2000, 84, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Ding, Z.; Zhou, Z.; Wang, Y.; Guo, L. Phylogenetic and pathogenic analyses show that the causal agent of apple ring rot in China is Botryosphaeria dothidea. Plant Dis. 2012, 96, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Mayorquin, J.S.; Wang, D.H.; Twizeyimana, M.; Eskalen, A. Identification, distribution, and pathogenicity of Diatrypaceae and Botryosphaeriaceae associated with citrus branch canker in the southern California desert. Plant Dis. 2016, 100, 2402–2413. [Google Scholar] [CrossRef] [PubMed]
- Na, L.; Sen, L.; Zhou, S.; Wang, C.; Ren, W.; Li, B. Involvement of the autophagy-related gene BdATG8 in development and pathogenicity in Botryosphaeria dothidea. J. Integr. Agric. 2022, 21, 2319–2328. [Google Scholar]
- Ren, W.; Zhang, Y.; Zhu, M.; Liu, Z.; Lian, S.; Wang, C.; Li, B.; Liu, N.; Atanasova, L. The Phosphatase Cascade Nem1/Spo7-Pah1 Regulates Fungal Development, Lipid Homeostasis, and Virulence in Botryosphaeria dothidea. Microbiol. Spectr. 2023, 11, e03881-22. [Google Scholar] [CrossRef]
- Gerwien, F.; Skrahina, V.; Kasper, L.; Hube, B.; Brunke, S. Metals in fungal virulence. FEMS Microbiol. Rev. 2018, 42, fux050. [Google Scholar] [CrossRef]
- López-Berges, M.S.; Capilla, J.; Turrà, D.; Schafferer, L.; Matthijs, S.; Jöchl, C.; Cornelis, P.; Guarro, J.; Haas, H.; Di Pietro, A. HapX-mediated iron homeostasis is essential for rhizosphere competence and virulence of the soilborne pathogen Fusarium oxysporum. Plant Cell 2012, 24, 3805–3822. [Google Scholar] [CrossRef] [PubMed]
- Pecherina, A.; Grinberg, M.; Ageyeva, M.; Zdobnova, T.; Ladeynova, M.; Yudintsev, A.; Vodeneev, V.; Brilkina, A. Whole-plant measure of temperature-induced changes in the cytosolic pH of potato plants using genetically encoded fluorescent sensor Pt-GFP. Agriculture 2021, 11, 1131. [Google Scholar] [CrossRef]
- Grams, T.E.; Lautner, S.; Felle, H.H.; Matyssek, R.; Fromm, J. Heat-induced electrical signals affect cytoplasmic and apoplastic pH as well as photosynthesis during propagation through the maize leaf. Plant Cell Environ. 2009, 32, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Qi, L.; Li, Y.; Chu, J.; Li, C. PIF4–mediated activation of YUCCA8 expression integrates temperature into the auxin pathway in regulating Arabidopsis hypocotyl growth. PLoS Genet. 2012, 8, e1002594. [Google Scholar] [CrossRef]
- Tsai, H.-H.; Schmidt, W. The enigma of environmental pH sensing in plants. Nat. Plants 2021, 7, 106–115. [Google Scholar] [CrossRef]
- Hückelhoven, R. Cell wall–associated mechanisms of disease resistance and susceptibility. Annu. Rev. Phytopathol. 2007, 45, 101–127. [Google Scholar] [CrossRef]
- Cantu, D.; Vicente, A.R.; Labavitch, J.M.; Bennett, A.B.; Powell, A.L. Strangers in the matrix: Plant cell walls and pathogen susceptibility. Trends Plant Sci. 2008, 13, 610–617. [Google Scholar] [CrossRef]
- Hansen, J.E.; Kharecha, P.; Sato, M.; Tselioudis, G.; Kelly, J.; Bauer, S.E.; Ruedy, R.; Jeong, E.; Jin, Q.; Rignot, E. Global Warming Has Accelerated: Are the United Nations and the Public Well-Informed? Environ. Sci. Policy Sustain. Dev. 2025, 67, 6–44. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Yu, J.-H.; Hamari, Z.; Han, K.-H.; Seo, J.-A.; Reyes-Domínguez, Y.; Scazzocchio, C. Double-joint PCR: A PCR-based molecular tool for gene manipulations in filamentous fungi. Fungal Genet. Biol. 2004, 41, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Kohl, M.; Wiese, S.; Warscheid, B. Cytoscape: Software for visualization and analysis of biological networks. Data Min. Proteom. Stand. Appl. 2011, 696, 291–303. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads | Mapped Reads |
|---|---|---|---|
| A-3-rep1 | 33,063,322 | 28,434,457 (86%) | 1,031,575 (3.12%) |
| A-3-rep2 | 33,100,103 | 29,459,092 (89%) | 1,112,163 (3.36%) |
| A-3-rep3 | 33,093,090 | 28,129,127 (85%) | 1,068,906 (3.23%) |
| A-8-rep1 | 37,309,993 | 32,459,694 (87%) | 1,385,662 (3.71%) |
| A-8-rep2 | 36,218,469 | 32,958,807 (91%) | 1,355,090 (3.74%) |
| A-8-rep3 | 35,317,069 | 29,666,338 (84%) | 1,293,980 (3.66%) |
| A-15-rep1 | 38,881,872 | 34,216,047 (88%) | 577,038 (1.48%) |
| A-15-rep2 | 37,450,144 | 33,705,130 (9%) | 563,034 (1.50%) |
| A-15-rep3 | 36,578,848 | 30,360,444 (83%) | 517,571 (1.41%) |
| B-3-rep1 | 33,663,537 | 28,614,006 (85%) | 1,190,962 (3.54%) |
| B-3-rep2 | 33,746,234 | 29,359,224 (87%) | 1,257,647 (3.73%) |
| B-3-rep3 | 33,754,092 | 30,041,142 (89%) | 1,287,632 (3.81%) |
| B-8-rep1 | 39,204,555 | 32,931,826 (84%) | 410,609 (1.05%) |
| B-8-rep2 | 41,068,181 | 35,318,636 (86%) | 428,930 (1.04%) |
| B-8-rep3 | 36,002,844 | 33,122,616 (92%) | 372,589 (1.03%) |
| B-15-rep1 | 35,088,723 | 30,878,076 (88%) | 946,741 (2.70%) |
| B-15-rep2 | 36,922,243 | 30,645,462 (83%) | 981,773 (2.66%) |
| B-15-rep3 | 40,480,611 | 34,408,519 (85%) | 1,077,289 (2.66%) |
| C-3-rep1 | 33,210,243 | 28,892,911 (87%) | 1,212,173 (3.65%) |
| C-3-rep2 | 33,421,176 | 29,744,847 (89%) | 1,293,399 (3.87%) |
| C-3-rep3 | 33,514,076 | 28,822,105 (86%) | 1,203,155 (3.59%) |
| C-8-rep1 | 39,289,045 | 33,002,798 (84%) | 2,488,240 (6.33%) |
| C-8-rep2 | 34,610,850 | 31,495,874 (91%) | 2,053,054 (5.93%) |
| C-8-rep3 | 36,050,384 | 31,724,338 (88%) | 2,211,313 (6.13%) |
| C-15-rep1 | 38,819,866 | 34,937,879 (90%) | 1,976,784 (5.09%) |
| C-15-rep2 | 37,800,346 | 31,374,287 (83%) | 1,991,694 (5.27%) |
| C-15-rep3 | 37,947,859 | 32,255,680 (85%) | 1,967,094 (5.18%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, D.; Jiang, Y.; Ai, W.; Zhang, Y.; Mao, C.; Ma, T.; Zhang, C. Co-Expression Network Analysis Suggests PacC Transcriptional Factor Involved in Botryosphaeria dothidea Pathogenicity in Chinese Hickory. J. Fungi 2025, 11, 580. https://doi.org/10.3390/jof11080580
Liang D, Jiang Y, Ai W, Zhang Y, Mao C, Ma T, Zhang C. Co-Expression Network Analysis Suggests PacC Transcriptional Factor Involved in Botryosphaeria dothidea Pathogenicity in Chinese Hickory. Journal of Fungi. 2025; 11(8):580. https://doi.org/10.3390/jof11080580
Chicago/Turabian StyleLiang, Dong, Yiru Jiang, Wei Ai, Yu Zhang, Chengxing Mao, Tianlin Ma, and Chuanqing Zhang. 2025. "Co-Expression Network Analysis Suggests PacC Transcriptional Factor Involved in Botryosphaeria dothidea Pathogenicity in Chinese Hickory" Journal of Fungi 11, no. 8: 580. https://doi.org/10.3390/jof11080580
APA StyleLiang, D., Jiang, Y., Ai, W., Zhang, Y., Mao, C., Ma, T., & Zhang, C. (2025). Co-Expression Network Analysis Suggests PacC Transcriptional Factor Involved in Botryosphaeria dothidea Pathogenicity in Chinese Hickory. Journal of Fungi, 11(8), 580. https://doi.org/10.3390/jof11080580