1. Introduction
The commercial production of high-THC-containing plants of cannabis (
Cannabis sativa L.) in Canada following legalization in 2018 for recreational and medicinal uses has been steadily increasing. Production occurs mostly in environmentally controlled indoor rooms and in greenhouses, although a small proportion of the production, approximately 10%, occurs outdoors. One of the major challenges to the large-scale production of cannabis plants is the potential exposure to infection by pathogens, which may originate from various sources. These sources include infected propagation materials, contaminated seeds, or inoculum from adjacent fields in which the pathogens may be infecting unrelated crops, which subsequently provide a source of inoculum for cannabis plants [
1]. There is previously published evidence that a large number of fungal pathogens with wide host ranges are able to spread and infect cannabis plants under the appropriate environmental conditions [
1,
2].
There are currently no regulatory restrictions to prevent the spread of pathogens within or between cannabis crops or across geographical regions where cannabis production is permitted. Hence, the unregulated movement of plant materials from one region to another has been shown to result in the spread of a number of pathogens contained in that material [
1]. Regulations are imposed on the occurrence of specific fungal pathogens that have the potential to produce mycotoxins within the cannabis inflorescences that are harvested and marketed for sale [
3]. These regulations specify the tolerance levels for certain species of
Aspergillus and
Penicillium species and their associated mycotoxins. In some jurisdictions, these tolerance levels may be zero. There is currently little published information on the extent to which these mycotoxins may be present and the corresponding levels in different jurisdictions [
3,
4].
Recently, reports of infection by a range of mycotoxin-producing species of
Fusarium on both greenhouse-grown cannabis plants as well as on field-grown hemp plants have raised concerns over the potential for mycotoxin production within the inflorescences, and hence their potential to cause harm to humans and animals exposed to these contaminating species [
5,
6,
7,
8]. In hemp plants grown outdoors, several mycotoxins produced by
Fusarium graminearum have been reported to be present in inflorescences [
7]. These plants were generally exposed to prolonged periods of wet weather outdoors that provided conducive conditions for growth of these pathogens. The sources of inoculum were identified to be adjacent wheat or cereal fields harboring these pathogens [
6,
8]. In greenhouse-grown cannabis plants in Canada, the occurrence of potential mycotoxin-producing species of
Fusarium that include
F. graminearum and
F. sporotrichiodes has been reported [
1,
2,
9], but there are no previous reports of the occurrence of associated mycotoxins in the infected inflorescences. In addition, the potential sources of inoculum of
Fusarium species that are able to infect these tissues and the environmental conditions favoring disease development are unknown.
In 2024, cannabis plants showing pinkish-white mycelial growth on the inflorescences were observed in the greenhouse environment of a producer in British Columbia (
Figure 1a,b). Samples of dried cannabis inflorescences after the drying process was completed and before packaging had occurred also showed evidence of infection. These samples showed pinkish-white mycelial growth indicative of the presence of a
Fusarium sp. on the exterior of the sample as well as internally (
Figure 1c,d). The objectives of this study were to (i) confirm the species of
Fusarium that were present on these inflorescences and to determine if the tissues contained any mycotoxins; (ii) to conduct artificial inoculation studies to demonstrate pathogenicity of the isolates recovered and to determine if there were any mycotoxins produced; and (iii) to investigate possible sources of inoculum that could explain the occurrence of infection on greenhouse-grown inflorescences.
2. Materials and Methods
Plant and fungal sources. During July 2024, visible symptoms of infection of the inflorescences of cannabis plants of genotype AP were observed in a licensed commercial greenhouse facility situated in the Fraser Valley region of British Columbia. The affected inflorescences developed a whitish-pink mycelium (
Figure 1a,b) that was visibly different from that commonly caused by
Botrytis cinerea, the gray mold pathogen that causes bud rot [
5,
6]. Small tissue pieces measuring 0.5 mm
2 were dissected from the affected inflorescences and surface-sterilized for 90 s in a 10% solution of household bleach (containing 5.25% NaOCl) followed by a 30 s dip in 70% EtOH and then rinsed thrice in sterile distilled water. The pieces were blotted dry and plated onto potato dextrose agar containing 130 mg/L of streptomycin sulfate (PDA+S) and incubated under ambient laboratory conditions (temperature range of 21–23 °C and 10–12 h/day fluorescent lighting) for 5–7 days. Emerging colonies were sub-cultured onto fresh media and kept for further identification using morphological and molecular criteria.
In August of 2024, samples of cannabis inflorescences of genotype AD from the same greenhouse facility were observed to contain a whitish-pink mycelium on the surface of the tissues as well as internally, causing visible decay (
Figure 1c,d). These samples had been harvested and dried following commercial practices and were being sorted after trimming and before packaging. The tissues were subjected to the isolation procedure described above. Additional sampling was conducted during August of 2024 of the florets (inflorescences) of tall fescue (
Festuca arundinacea Schreb.) plants that were growing outside the greenhouse in which diseased cannabis inflorescences were observed. These plants were situated at a distance of approximately 30 m away from the greenhouse (
Figure 2a,b). Samples of inflorescences and leaves were obtained from these plants at random and surface-sterilized as above and plated onto PDA+S and select fungal colonies were transferred to fresh medium after 5–7 days. The weather conditions during sampling were overcast skies with a light drizzle and a temperature of 18 °C.
Molecular identification. A number of fungal colonies that were consistently recovered on Petri dishes from the tissue samples of cannabis and tall fescue inflorescences were sub-cultured and examined for colony color and growth pattern. Most colonies produced a reddish-brown pigment and resembled
Fusarium spp. Others were darkly pigmented and resembled
Alternaria species. Six representative isolates of
Fusarium spp. as well as three unidentified cultures from diseased inflorescences or leaves were transferred from hyphal-tipped colonies and sent to the University of Guelph Laboratory Services, Agriculture and Food Laboratory, Guelph, ON (
www.guelphlabservices.com, accessed on 4 August 2024) for identification to the species level by PCR using the primers ITS1–ITS4 (ITS1-F CTTGGTCATTTAGAGGAAGTAA and ITS4 TCCTCCGCTTATTGATATGC). The elongation factor 1α (EF-1α) primer set EF-1 (5′ ATG GGT AAG GAG GAC AAG AC 3′) and EF-2 (5′ GGA GGT ACC AGT GAT CAT GTT 3′) [
2] was also used for further speciation of
Fusarium spp. as follows. Cultures of representative isolates from different tissue sources were grown in potato dextrose broth at room temperature for 7 days and DNA was extracted from harvested mycelium using the QIAGEN DNeasy Plant Mini Kit (QIAGEN, Montreal, QC, Canada). Aliquots of 1 μL containing 5–20 ng DNA were used for PCR in a 25 μL reaction volume consisting of 2.5 μL 10x buffer (containing 15 mM MgCl
2), 0.5 μL 10 mM dNTP, 0.25 μL Taq DNA Polymerase (Qiagen, Montreal, QC, Canada), 0.25 μL 10 mM forward and reverse primers, as well as 20.25 μL DNAse- and RNAse-free water (Invitrogen, Waltham, MA, USA). All PCR amplifications were performed in a MyCycler thermocycler (BioRad, Hercules, CA, USA) with the following program: 3 min at 94 °C; 30 s at 94 °C, 30 s at 60 °C, 3 min at 72 °C (35 cycles); and 7 min at 72 °C. PCR products were separated on 1% agarose gels and bands of the expected size (700 bp) were purified with the QIAquick Gel Extraction Kit (QIAGEN, Montreal, QC, Canada) and sent to Eurofins Genomics (Eurofins MWG Operon LLC 2016, Louisville, KY, USA) for sequencing. The resulting sequences were compared to the corresponding ITS1-5.8S-ITS4 or EF-1α sequences from the National Center for Biotechnology Information (NCBI) GenBank database (
www.ncbi.nlm.nih.gov, accessed on 4 September 2024). Multiple sequence alignment of the respective isolates was performed using the CLUSTAL W program (
http://www.genome.jp/tools/clustalw, accessed on 10 September 2024). Species identities were also confirmed with species-specific DNA markers following the methods as described by Bamforth et al. [
10] for both DNA samples isolated from pure fungal cultures and from naturally and artificially infected plant tissues.
Pathogenicity tests. Following the identification of the fungal isolates to the species level, five
Fusarium isolates representing two species were used for pathogenicity tests. The five isolates included three
F. avenaceum isolates—two from cannabis inflorescences (fresh and dried, FB1 and FB2) and the other isolate from tall fescue inflorescences (FB3)—one isolate of
F. graminearum from tall fescue plants (FG), and a
F. sporotrichiodes isolate (FS) from a diseased cannabis inflorescence obtained in 2021 [
2,
9]. The five isolates were grown on PDA+S for 2 weeks and mycelial plugs (8 mm diameter) were placed mycelial side down onto freshly harvested detached cannabis inflorescences of two cannabis genotypes—AK and AD. The characteristic features of these two genotypes are described in
Supplementary Figure S1. Three mycelial plugs were placed equidistant from one another on each inflorescence sample. The inflorescences were harvested from 7-week-old flowering plants in the same greenhouse facility. They were chosen at random but ensuring there were no visible signs of any fungal infection. They were placed inside plastic bags and transported to the laboratory and stored at 4 °C overnight. Two inflorescences were each placed inside plastic containers (25 cm × 12 cm) lined with wet paper towels. Following inoculation, the tissues were misted thrice with sterile distilled water from a hand-held spray bottle and the lids were closed. There were two containers for each of the five isolates (
n = 4 per isolate) and they were incubated under ambient laboratory conditions. Control inflorescences were similarly misted but not inoculated. In addition to the five
Fusarium isolates, an isolate each of
Penicillium citrinum and
Aspergillus ochraceous was also inoculated onto a duplicate set of inflorescences and placed under the same conditions. These cultures were obtained from cannabis inflorescence tissues in a previous study [
11]. All containers were examined at 4 and 7 days after inoculation, and each inoculated sample was photographed to show the extent of mycelial development of the respective fungi. The experiment was repeated a second time in September 2024 using a different set of inflorescences of the same two genotypes harvested from 7-week-old flowering plants and inoculated as described previously. Thus, each fungal isolate was tested for pathogenicity in 8 replications on cannabis inflorescences of 2 genotypes from 2 independent trials.
Mycotoxin analysis. After 7 days of incubation, all inflorescence samples were gently transferred to large trays and placed inside a forced air incubator set at 35 °C for 5 days or until all moisture had been removed and the samples felt dry to the touch. They were then transferred to Ziploc bags and sealed and stored inside a dark cabinet for 3 months. Prior to mycotoxin analysis, one inflorescence sample from each of the repeated experiments was combined to represent the specific fungal species inoculated. Control samples were combined in the same manner. The samples were crumbled by hand and tissue pieces were crushed using a mortar with a pestle until a comminuted mixture of small fragments was obtained. They were transferred into a 50 mL Falcon tube and stored for 2 weeks in darkness. The contents were thoroughly shaken by hand and a 5 g subsample was transferred to a small screw-cap vial and prepared for analysis for mycotoxins as described by Brown et al. [
12]. A sub-sample of 0.1 g of the ground dried cannabis tissue was transferred to a 30 mL glass test tube to which 1 mL of acetonitrile was added. The tubes were capped and rolled vertically by hand for 30 s to completely coat and submerge the tissues with solvent, followed by sonication for 5 min. The tubes were rolled vertically by hand once again for 30 s, and the solvent was injected directly into an LC vial and subjected to the HPLC-MS/MS protocol described below.
A method based on high-performance liquid chromatography (HPLC) with tandem mass spectrometry (HPLC-MS/MS) was used for analyte (mycotoxin) determination [
13]. The HPLC-MS/MS method covered 28 analytes, and the limits of quantitation are shown in
Table 1. Stable isotope labeled
13C internal standards were added to the diluted sample extract prior to HPLC-MS/MS analysis to mitigate the impact of matrix effects. Quality control samples were processed and analyzed alongside the 12 cannabis samples to monitor analytical method performance.
Analysis was performed on an AB Sciex 5500 TQ tandem mass spectrometer (AB Sciex, Concord, ON, Canada) coupled to a Waters Acquity I Class ultra-high-pressure liquid chromatography system consisting of an Acquity binary solvent manager, an Acquity FTN sample manager, and an Acquity column manager fitted with a Phenomenex Kinetex (Phenomenex, Torrance, CA, USA) 2.6 μm XB-C18 (50 × 2.1 mm) column held at 40 °C. A linear gradient HPLC program using 95.5% 5 mM ammonium acetate in water/0.5% acetic acid (
v/
v) and 95.5% 5 mM ammonium acetate in methanol with 0.5% acetic acid (
v/
v) as mobile phases A and B, respectively, separated the mycotoxin analytes shown in
Table 1. The initial mobile phase was 98% A, changing to 30% A at 15 min and 2% A at 15.7 min. The mobile phase was changed back to the initial 98% A at 18.2 min and held there until the end of the run at 20 min. Mass spectrometric analysis was performed using electrospray ionization with polarity switching. The following mass spectrometer parameters were used: ion spray voltage of ±4500 V (depending on analyte), source temperature of 550 °C, curtain gas (N
2) at 25 psi, ion source gases (N
2) at 60 and 70 psi, and collision gas (N
2) at 9 psi. The transitions monitored and used to identify and quantify each of the mycotoxin analytes are described by Tittlemier et al. [
13].
Mycotoxin analytes were quantified using a calibration curve constructed from seven external standards. Peak areas from quantitation transitions were normalized to the peak area of 13C-labeled internal standard during data analysis in order to counter matrix effects on quantitation prior to the interpolation of analyte concentration from the calibration curve. Isotope-labeled structural analogs were not commercially available for AOH, AME, ALT, TENT, BEAU, ENN A, ENN A1, ENN B, and ENN B1; therefore, peak areas from their quantitation transitions were normalized to 13C34-FB1. Analyte recoveries were calculated from pre-extraction fortified blanks analyzed with each sample batch. Mycotoxin concentrations in samples were corrected for recovery using the batch-specific recoveries determined for the pre-extraction fortified blanks. Analytes were considered to be identified and were quantified in samples when their retention time was within 0.1 min of the mean retention time in external standards used to construct the calibration curve, the ratio of qualifier to quantitation transition was within ±30% of the mean ratio in external standards used to construct the calibration curve, and the peak area signal-to-noise ratio was at least 10:1. Limits of quantitation were determined as the concentration in sample that produced a chromatographic peak with a signal-to-noise ratio of 10:1.
3. Results
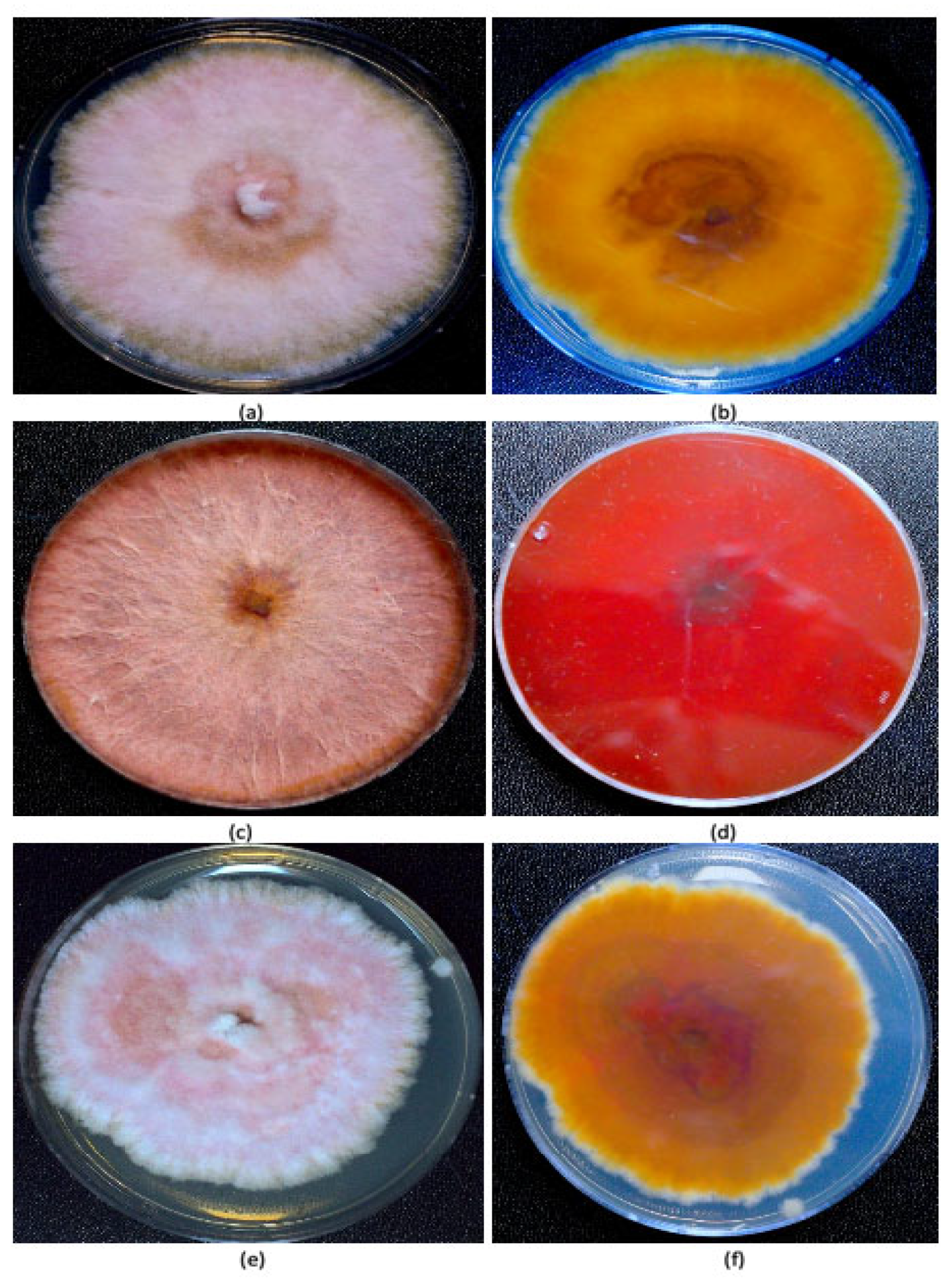
Fungal isolation from plants and identification. Fungal colonies recovered from dried cannabis inflorescences and sub-cultured onto PDA+S were represented by two major morphological types. The first were slow-growing colonies with irregular margins and cottony aerial mycelia, and the underside of the colonies were beige with a tint of red (
Figure 2a,b). The second type was represented by fast-growing red-pigmented colonies with some aerial mycelia (
Figure 2c,d). Many fungal colonies recovered from tall fescue plants, when sub-cultured to fresh medium, resembled these two colony types (
Figure 2e,f), in addition to other colonies that were either pigmented red or black, or were not pigmented. The tall fescue plants sampled and the colonies recovered from them are shown in
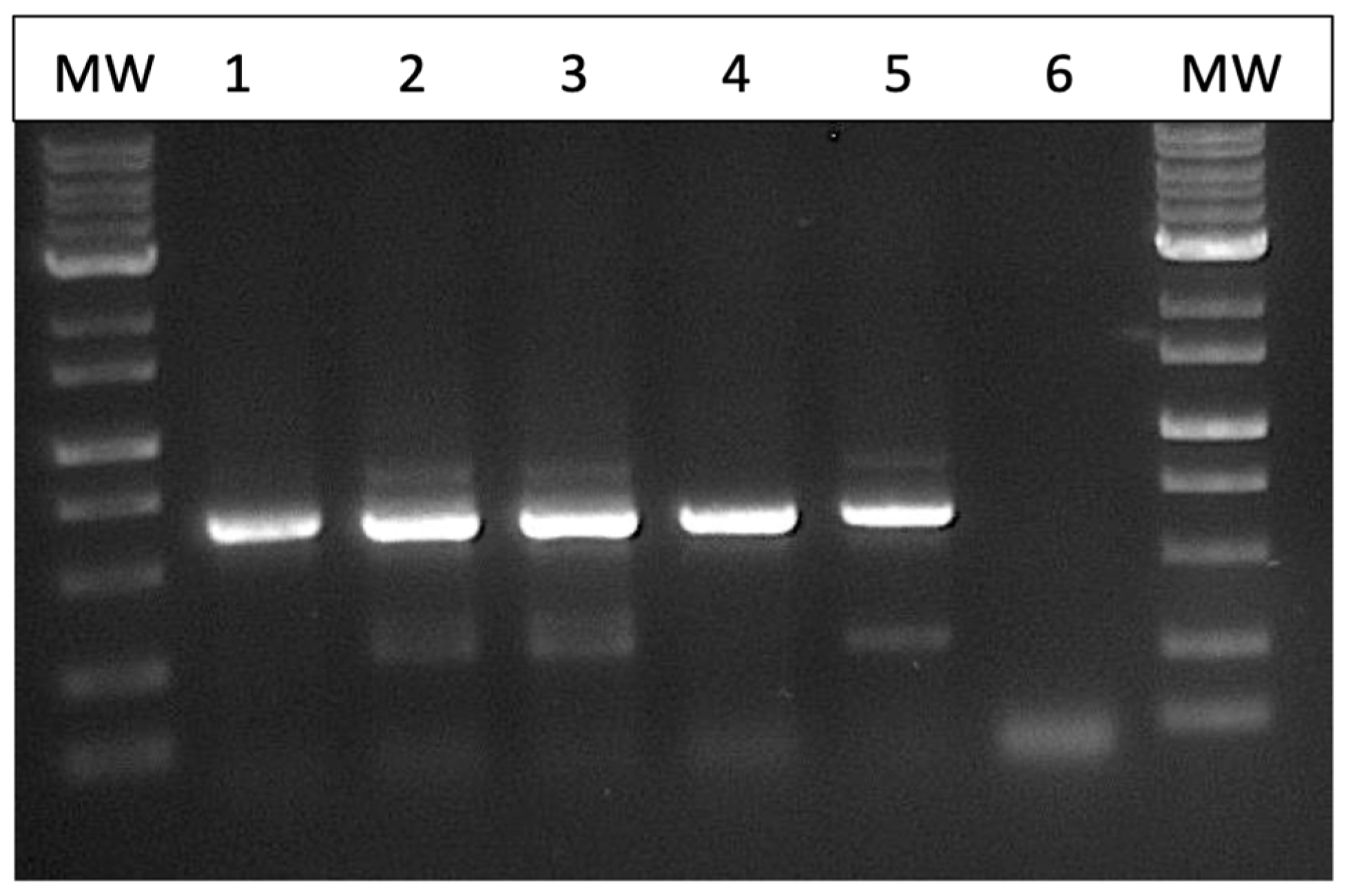
Figure 3. Among the six isolates of
Fusarium spp. that were subjected to PCR of the ITS1-ITS4 region of rDNA and of the EF-1α region, two species were identified—
F. avenaceum and
F. graminearum (
Figure 4). Both species were recovered from cannabis and tall fescue inflorescences, with homologies in the range of 99.4–99.6% for both gene regions according to BLAST analysis (
https://blast.ncbi.nlm.nih.gov/Blast.cgi, accessed on 4 September 2024). Three additional cultures that were pigmented red or black were identified as
Epicoccum nigrum,
Sordaria macrospora, and
Alternaria alternata, with 100%, 99.6%, and 99.4% homology, respectively, to the ITS region, and all of them were recovered from tall fescue leaves.
Pathogenicity tests. Following the inoculation of freshly harvested detached cannabis inflorescences with mycelial plugs of
F. avenaceum and
F. graminearum, extensive mycelial growth was observed after 4 and 7 days on each of the two cannabis genotypes (AK, AD) due to the high humidity conditions within the sealed plastic containers (
Figure 5). The
F. avenaceum isolates from cannabis and tall fescue plants (FB1, FB2, FB3) grew similarly and there were no observable differences in the extent of mycelial colonization (
Figure 5a–h). After 7 days, a ball of mycelium formed at each inoculation site. In contrast, inoculation with
F. graminearum resulted in the complete colonization of the inflorescences after 7 days, resulting in a mummified appearance (
Figure 5i–l). When these samples were dried, they retained the physical appearance of a mummified structure (
Figure 6a). Inflorescences inoculated with
F. sporotrichiodes were also covered by mycelial growth after 7 days, but to a much lesser extent compared to
F. graminearum (
Figure 6b). Inoculations with
A. ochraceus and
P. citrinum resulted in considerable sporulation by these species as shown in
Figure 6c,d. These samples were similarly dried and used for mycotoxin analysis.
Mycotoxin occurrence. From the 8 samples of cannabis tissues that were naturally infected or artificially infected by 3
Fusarium species on 2 cannabis genotypes, a total of 14 mycotoxins were detected as shown in
Table 2. From the naturally infected, dried cannabis inflorescences (buds) that were confirmed to contain
F. avenaceum by isolation in pure culture and PCR identification, the mycotoxins BEAU and enniatins A1, B, and B1 were present at concentrations ranging from 0.063 to 1.78 μg/g. In cannabis inflorescences inoculated with an isolate of
F. graminearum recovered from cannabis plants, the mycotoxins 3ADON and DON accumulated at levels of 0.34 and 1.91 μg/g, respectively, on cannabis genotype AK. In addition, ZEAR was present at a level of 56.2 μg/g (
Table 2).
On cannabis genotype AD, these mycotoxin levels were 0.13, 1.18, and 31.8 μg/g, which were much lower than in genotype AK under the same conditions. In addition, the mycotoxin culmorin was present at a level of 0.38 μg/g in genotype AD.
In cannabis inflorescences inoculated with
F. avenaceum originating from cannabis, the mycotoxins enniatins A, A1, B, and B1 were produced at varying levels. These levels were lower in tissues of genotype AD compared to AK, as observed previously with the mycotoxins 3ADON, DON, and ZEAR. In addition, BEAU was produced on genotype AD. In cannabis inflorescences inoculated with
F. avenaceum originating from tall fescue spikes, the mycotoxins enniatins A, A1, B, and B1 were produced at varying levels, with higher levels detected in genotype AK compared to AD as before. BEAU was present in both genotypes, with higher levels in genotype AD. Lastly, in
F. sporotrichiodes-inoculated tissues, the mycotoxins HT2 and T2 accumulated at levels of 13.9 and 10.9 μg/g, respectively. In addition, BEAU was also present at 0.133 μg/g (
Table 2).
There were some unexpected results regarding the mycotoxins that were detected. For example, the mycotoxin alternariol was present in tissues of cannabis genotype AK inoculated with F. graminearum (FG) and F. avenaceum FB3 from tall fescue. As well, the mycotoxin tentoxin was present in tissues of genotypes AK and AD inoculated with F. avenaceum from tall fescue. In addition, the mycotoxins 3ADON and DON were present in tissues of both cannabis genotypes inoculated with F. avenaceum isolates from cannabis (FB1, FB2) and tall fescue (FB3). Control (uninoculated) cannabis tissues were devoid of mycotoxins, except for culmorin that was present at 0.38 μg/g. Inflorescences inoculated with A. ochraceus contained no mycotoxins with those receiving P. citrinum containing 6.1 μg/g of BEAU.
To explain the possible causes of the unexpected results above, tissues from which the mycotoxins were obtained were subsequently used for DNA extraction and were subjected to RT-qPCR using species-specific primers for
F. graminearum and
F. avenaceum [
10]. In tissues inoculated with these two species, the corresponding DNA sequences were present (
Table 2). However, in tissues that were inoculated with
F. avenaceum only, the presence of
F. graminearum was confirmed, suggesting some background infection had potentially occurred from natural inoculum at the time the inflorescences were harvested for use in this study. These findings can explain the spurious presence of mycotoxins that were likely to be associated with incipient infections by
F. graminearum in samples where the tissues were either not inoculated or were inoculated with a different species.
4. Discussion
Fusarium species are widespread pathogens of crop plants and cause a broad range of disease symptoms. Up to 16 species of
Fusarium have been reported to be associated with cannabis plants and are classified in 6 species complexes:
Fusarium oxysporum, F. solani,
F. incarnatum-equiseti, F. sambucinum, F. tricinctum, and
F. fujikuroi. Among these, a sub-set of the species that cause diseases on cereal crops are known to produce a range of mycotoxins that can cause problems following ingestion by humans and animals [
14,
15]. In particular, the
Fusarium species causing head blight or scab symptoms on wheat are the most well characterized with regard to the range of mycotoxins produced [
14,
15,
16]. On cannabis plants, infection by two cereal-infecting
Fusarium species, namely
F. graminearum and
F. sporotrichiodes, has been previously reported [
1,
2,
9]. However, the potential of these species to produce mycotoxins in cannabis tissues has not been previously investigated.
In the present study, isolates of
F. graminearum and
F. sporotrichioides and a species previously unreported from cannabis,
F. avenaceum, all isolated from symptomatic fresh and dried inflorescences, were tested for mycotoxin production under controlled conditions. It should be noted that the inoculated cannabis tissues were incubated under high relative humidity (>90%) for a prolonged period (7 days) prior to mycotoxin analyses being conducted. Similarly, asymptomatic control tissues were exposed to these incubation conditions. Where present, these conditions would have allowed undetected incipient infections to progress before the assays were performed. Under normal greenhouse-growing conditions, these environmental conditions would not be expected to occur unless air circulation was compromised and temperature and relative humidity levels exceeded the normal range (55% relative humidity and temperatures in the range of 27–30 °C). Such conducive conditions (70% relative humidity and temperatures in the range of 34–36 °C) can occur in greenhouses at certain times of the year, resulting in outbreaks of diseases, such as Botrytis bud rot, caused by
B. cinerea. This disease is favored by humid conditions that occur from July to October, resulting in disease outbreaks [
17]. In the present study, the development of pinkish-white mycelium of
Fusarium was observed on some plants growing under greenhouse conditions in late summer, suggesting the environmental conditions were conducive for infection to occur at these time periods. In addition, the relative humidity and temperature within inflorescences are reported to be higher than the ambient environment [
10], indicating that microclimatic conditions may be conducive for infection by
Fusarium spp. as they are for
B. cinerea [
17]. Such conditions can result in the development of
F. avenaceum and
F. graminearum in inflorescence tissues prior to packaging, a situation also similarly encountered with
B. cinerea infections [
17]. The incidence of visible infections due to
Fusarium, however, appears to be less common on greenhouse-grown cannabis plants when contrasted with outdoor-grown hemp plants. Outdoors,
Fusarium spp. were reported to infect inflorescences and produce mycotoxins, and prolonged periods of rain and high humidity (75–80%) were associated with outbreaks of disease [
6,
7,
8]. The incidence of detection of various
Fusarium spp. within these samples was 95%, indicating that the potential for contamination by mycotoxins in outdoor-grown hemp is significantly higher than for indoor-grown cannabis. In addition, the post-harvest drying and storage conditions for hemp can lead to the extensive growth of
Fusarium spp. [
6,
8]. While the sources of inoculum for hemp may include spores from adjacent fields planted to cereal crops, the inoculum sources for indoor-grown cannabis have not been previously identified.
In the present study, tall fescue plants that were grown as a part of the urban landscape within a 30 m distance from the greenhouse were observed to harbor both
F. avenaceum and
F. graminearum. Isolates of both species recovered from spikelet tissues of this host were capable of infecting cannabis inflorescences under laboratory conditions and produced mycotoxins in these tissues. The mycotoxins accumulated were reflective of the infecting species, with some exceptions, and were produced at significant levels (
Table 2). The importance of wild grass species, including tall fescue plants, growing adjacent to wheat fields in providing a source of inoculum for initiation of disease has been confirmed in several previous studies. At a field site located in Germany, Gerling et al. [
18] reported that grasses belonging to
Lolium spp. growing next to wheat production fields impacted the
Fusarium species composition and mycotoxin accumulation in wheat plants, particularly those located adjacent to the grasses. The most prevalent species of
Fusarium recovered from the grasses, and shown to be pathogenic on wheat plants, were
F. graminearum,
F. culmorum, and
F. sporotrichioides. A low level of recovery of
F. avenaceum was also reported. Wheat kernels produced on plants located next to the infected grasses had higher
Fusarium abundance and mycotoxin accumulation compared to those located at further distances [
18]. The use of irrigation enhanced the abundance of
Fusarium spp. on both the grasses and on the wheat plants when compared to non-irrigated sides of the field, with
F. graminearum being recovered at the highest frequency. Sampling at various distances from the grasses revealed a gradient in percent recovery from the wheat plants, with the highest recovery observed at a 1 m distance and lowest at 33 m. These results demonstrate that the grasses can be a reservoir of inoculum for
Fusarium spp. that subsequently spread into the wheat field [
18].
Similar findings were obtained from field studies conducted in New York State, where
F. graminearum was shown to be prevalent within spikes of various wild grass species, including tall fescue, especially in irrigated sites adjacent to agricultural fields [
19].
Fusarium avenaceum was also recovered from a number of different grass species [
20] and it has been shown to be seed-borne in tall fescue production sites in Oregon [
21], and was shown to infect tall fescue and orchard grass plants grown for pasture [
22]. These observations support the hypothesis that
Fusarium inoculum produced in infected spikes on tall fescue plants growing adjacent to a cannabis greenhouse facility could potentially have spread and infected cannabis inflorescences within the greenhouse. While both
F. graminaearum and
F. sporotrichioides have been previously recovered from diseased cannabis and hemp inflorescences [
5,
6,
9,
10], they are not reported to infect other parts of the cannabis plant, such as roots and stems, and hence do not provide a potential source of inoculum. Since there are strict biosecurity protocols in place for indoor production of cannabis to meet regulatory guidelines [
17], other inoculum sources of these cereal-infecting species, such as worker’s clothing or boots, are unlikely. Air-borne inoculum from external sources is the most likely source, which can enter through open greenhouse vents.
In addition to the mycotoxins associated with the three
Fusarium spp. included in this study, other mycotoxins were detected in dried cannabis inflorescences, such as alternariol and tentoxin, which were present in three samples. The likely source of these mycotoxins is from
Alternaria spp., which are known to infect cannabis plants [
23]; they are also commonly present in grain samples similarly colonized by this pathogen [
13,
24]. While the inflorescence samples collected for artificial inoculation studies did not show external symptoms due to any pathogen, pre-existing latent infections were likely to have been present that continued to progress during the high relative humidity conditions under which the tissues were incubated. Similar
Alternaria toxins were also reported to be present at high concentrations in dried hop flowers [
25] and in hemp seeds [
26]. In hop flowers, DON accumulation from
Fusarium spp. was detected in all the samples tested. Mycotoxins normally associated with
Aspergillus or
Penicillium species colonizing plant tissues, including aflatoxins and ochratoxins, were not detected in hop flowers. They were also absent in a large-scale sampling of dried cannabis flowers conducted in California [
27].
In the present study, artificial inoculations conducted with isolates of
A. ochraceus and
P. citrinum derived from cannabis inflorescences did not give rise to any of the ochratoxins normally produced by these species [
16,
28] in the inoculated tissues, although additional studies may be needed to determine the mycotoxin potential of these species in cannabis. Other aflatoxin-producing species of
Aspergillus were not included in this study as they were unavailable at the time this study was initiated. Currently, there is not a strong body of evidence to support that aflatoxins and ochratoxins are prevalent in cannabis samples originating from production sites that follow regulatory guidelines during production. However, these mycotoxins have attracted attention from regulatory agencies as their known carcinogenic properties can present potential risks to immunocompromised humans if exposed to levels that exceed certain thresholds [
3]. There is a growing body of evidence, including data from the current study, which indicates that
Fusarium and
Alternaria toxins may actually be more ubiquitous in cannabis tissues. In future studies, these mycotoxins should receive more attention to determine their presence and if the establishment of threshold limits is required and if monitoring for their presence in cannabis tissues should be enforced.
Incipient infections by
F. graminearum were confirmed to be present in asymptomatic fresh inflorescences samples used for inoculation with
F. avenaceum by RT-qPCR. Pathogen presence explains the unexpected detection of mycotoxins associated with this species. It should be noted that these tissues were tested for mycotoxin presence after 7 days of incubation under high relative humidity conditions. By this time, mycelial growth had encompassed the cannabis tissues, a situation not commonly observed in commercial growing environments. It is not certain whether natural infections would have proceeded to develop and accumulate mycotoxins under normal cultivation practices. In the absence of a known source of
Fusarium inoculum, such as the tall fescue plants identified in the present study, the risk levels for mycotoxin accumulation would appear to be low. While
Fusarium mycotoxins and various fungal metabolites were reported to be present in seized samples from illicit growing operations in Arizona and California [
4], the growing environments depicted do not reflect the production environments encountered in legalized production sites.
In the future, a large-scale systematic study of cannabis inflorescences for the presence of
Fusarium mycotoxins in licensed production facilities should shed light on the extent to which they accumulate in inflorescence tissues produced under greenhouse conditions. It also remains to be determined what levels of relative humidity conditions could promote the development of
Aspergillus and
Penicillium species to the level of colonization that would be associated with ochratoxin and aflatoxin accumulation. Based on the observations from the present study, the risks of contamination with these toxins remain low within licensed production facilities. The current findings, also supported by the recent published literature, suggest that the potential for
Alternaria and
Fusarium colonization of cannabis and hop tissues is more likely to lead to mycotoxin presence, and this could represent a moderate risk level. Some of these toxins—enniatins, BEAU, and
Alternaria toxins—can be classified as “emerging mycotoxins”, defined as “mycotoxins which are neither routinely determined nor legislatively regulated”. However, the evidence of their incidence is rapidly increasing according to Chiotta et al. [
16]. Future studies should target these groups of mycotoxins to determine whether or not the current regulations surrounding mycotoxins in cannabis should be modified to include them.
The two cannabis genotypes included in this study that were artificially inoculated produced comparable rates of tissue colonization by mycelium but differed in the levels of mycotoxins that were present, in some cases differing by as much as four-fold. These differences could be attributed to the different genetic backgrounds of these genotypes (
Supplementary Figure S1), but the mechanism remains unexplained. However, they are highly interesting as they suggest there may be differences in the extent to which certain genotypes of cannabis may accumulate these mycotoxins, with some apparently showing lower levels. In wheat samples, differences in the accumulation of mycotoxins in harvested grains were reported to be influenced by the cultivar, fungal isolate, and geographic location, suggesting that the complex interactions between the pathogens, different cultivars, and the environment are important in determining disease outcomes and mycotoxin accumulation [
13].
In summary, this study indicates the potential for disease development and mycotoxin accumulation in cannabis inflorescence tissues following infection by
Fusarium species that commonly infect graminaceous hosts, including cereal crops and other grasses. In view of the findings from this study, there may be a need to develop best practices that limit the spread of inoculum and introduction of
Fusarium species from neighboring alternate hosts. These may include the elimination of these hosts if they are adjacent to a greenhouse facility to provide a clear buffer zone, as well as spore-monitoring studies to confirm inoculum presence. In addition, research is needed to better understand the interactions between cannabis cultivars and
Fusarium fungal isolates, since there are known to be differences in cultivar responses to mycotoxin accumulation in cereal grains [
29,
30,
31]. Furthermore, studies on the environmental conditions leading to disease development and mycotoxin accumulation in cannabis are needed, both in greenhouse-grown and outdoor-grown crops, since these factors can influence mycotoxin accumulation in cereal crops [
29,
32]. Lastly, targeted disease management approaches and assessing the potential for breeding for resistance to infection by
Fusarium species and mycotoxin accumulation in cannabis and hemp should be explored to better manage this unique host–pathogen interaction.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}