
Telomere-to-Telomere Assembly of the Cordyceps militaris CH1 Genome and Integrated Transcriptomic and Metabolomic Analyses Provide New Insights into Cordycepin Biosynthesis Under Light Stress


Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling Information
2.2. Genome Sequencing and Assembly
2.3. Genome Size Estimation
2.4. Gene Prediction and Annotation
2.5. Gene Family Clustering and Phylogenetic Analysis
2.6. Comparative Genomic Analysis
2.7. Identification of CAZymes, CYP450s, and Secondary Metabolite Clusters
2.8. Transcriptome Analysis
2.9. Metabolome Analysis
2.10. qRT-PCR Analysis Method
3. Results
3.1. Genome Sequencing and Assembly Analysis
3.2. Genome Annotation
3.3. Evolutionary Analysis of C. militaris CH1
3.4. Comparative Genomic Analysis of C. militaris CH1
3.5. The Secondary Metabolism in C. militaris CH1
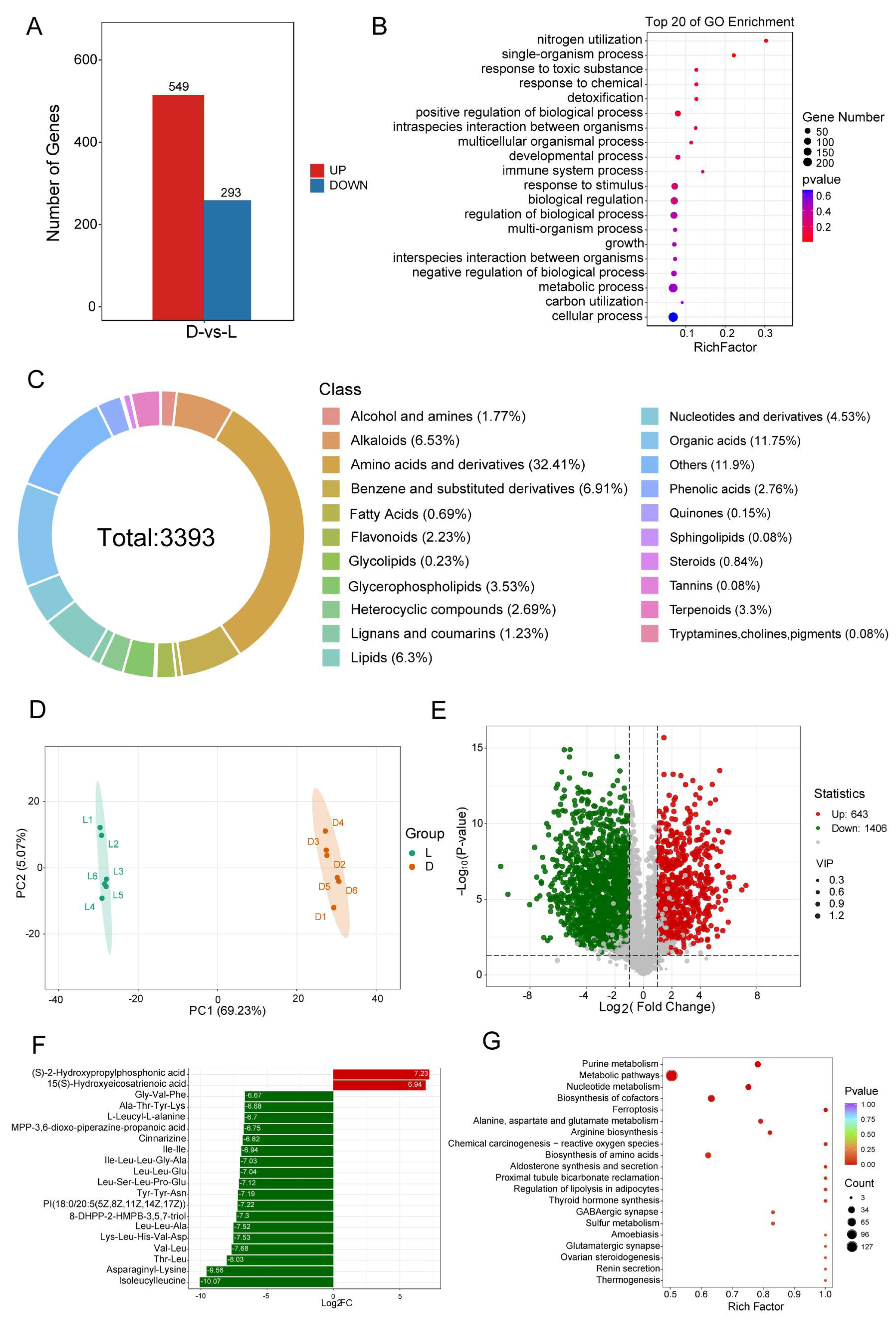
3.6. The Transcriptome and Metabolome Revealed the Effects of Light and Dark Conditions on the Mycelia of C. militaris CH1
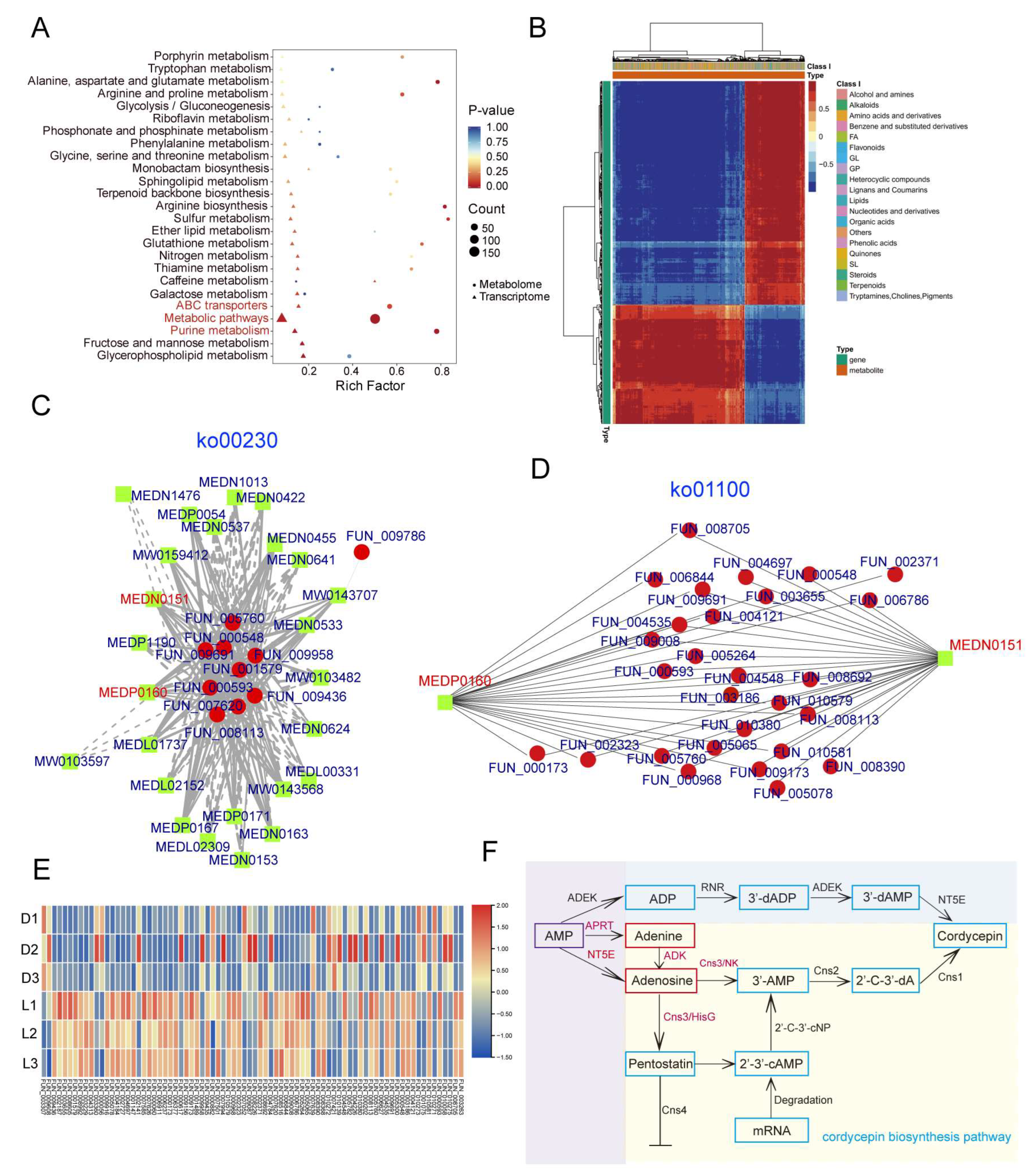
3.7. The Combined Analysis of Transcriptomics and Metabolomics Elucidated the Cordycepin Biosynthesis Pathway in C. militaris CH1
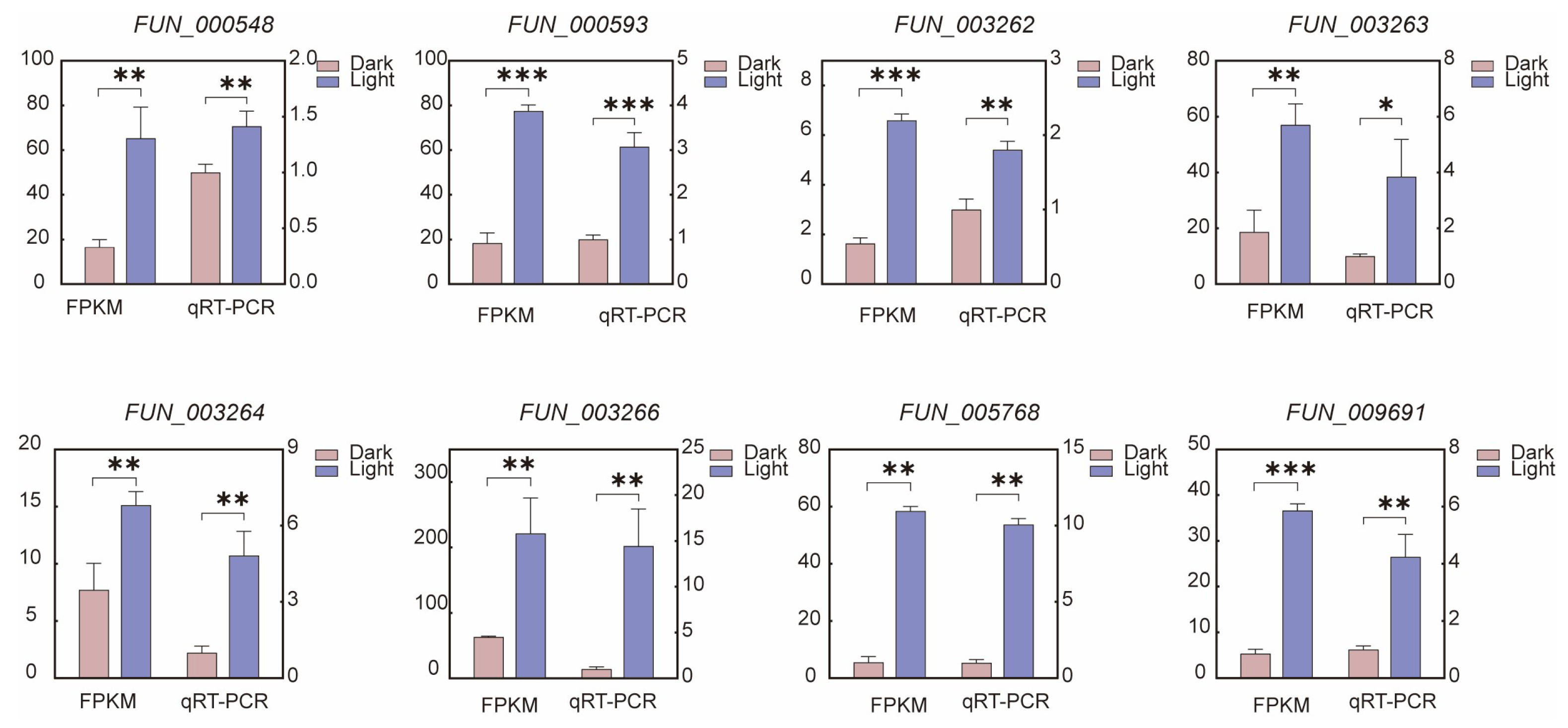
3.8. qRT-PCR Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Olatunji, O.J.; Tang, J.; Tola, A.; Auberon, F.; Oluwaniyi, O.; Ouyang, Z. The genus Cordyceps: An extensive review of its traditional uses, phytochemistry and pharmacology. Fitoterapia 2018, 129, 293–316. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, B.; Zhang, W.; Zhang, Y.; Liu, X. The medicinal fungus Cordyceps militaris: Research and development. Mycol. Prog. 2012, 11, 599–614. [Google Scholar] [CrossRef]
- Cui, J.D. Biotechnological production and applications of Cordyceps militaris, a valued traditional Chinese medicine. Crit. Rev. Biotechnol. 2015, 35, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Masuda, M.; Sakurai, A.; Sakakibara, M. Medicinal uses of the mushroom Cordyceps militaris: Current state and prospects. Fitoterapia 2010, 81, 961–968. [Google Scholar] [CrossRef]
- Zhang, J.; Wen, C.; Duan, Y.; Zhang, H.; Ma, H. Advance in Cordyceps militaris (Linn) Link polysaccharides: Isolation, structure, and bioactivities: A review. Int. J. Biol. Macromol. 2019, 132, 906–914. [Google Scholar] [CrossRef] [PubMed]
- Basith, M.; Madelin, M. Studies on the production of perithecial stromata by Cordyceps militaris in artificial culture. Can. J. Bot. 1968, 46, 473–480. [Google Scholar] [CrossRef]
- Dong, C.; Guo, S.; Wang, W.; Liu, X. Cordyceps industry in China. Mycology 2015, 6, 121–129. [Google Scholar] [CrossRef]
- Jędrejko, K.J.; Lazur, J.; Muszyńska, B. Cordyceps militaris: An overview of its chemical constituents in relation to biological activity. Foods 2021, 10, 2634. [Google Scholar] [CrossRef]
- Cunningham, K.; Manson, W.; Spring, F.; Hutchinson, S. Cordycepin, a metabolic product isolated from cultures of Cordyceps militaris (Linn.) Link. Nature 1950, 166, 949. [Google Scholar] [CrossRef]
- Xia, Y.; Luo, F.; Shang, Y.; Chen, P.; Lu, Y.; Wang, C. Fungal cordycepin biosynthesis is coupled with the production of the safeguard molecule pentostatin. Cell Chem. Biol. 2017, 24, 1479–1489.e4. [Google Scholar] [CrossRef]
- Yang, L.; Li, G.; Chai, Z.; Gong, Q.; Guo, J. Synthesis of cordycepin: Current scenario and future perspectives. Fungal Genet. Biol. 2020, 143, 103431. [Google Scholar] [CrossRef]
- Duan, X.; Yang, H.; Wang, C.; Liu, H.; Lu, X.; Tian, Y. Microbial synthesis of cordycepin, current systems and future perspectives. Trends Food Sci. Technol. 2023, 132, 162–170. [Google Scholar] [CrossRef]
- Jiaojiao, Z.; Fen, W.; Kuanbo, L.; Qing, L.; Ying, Y.; Caihong, D. Heat and light stresses affect metabolite production in the fruit body of the medicinal mushroom Cordyceps militaris. Appl. Microbiol. Biotechnol. 2018, 102, 4523–4533. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Xia, Y.; Xiao, G.; Xiong, C.; Hu, X.; Zhang, S.; Zheng, H.; Huang, Y.; Zhou, Y.; Wang, S. Genome sequence of the insect pathogenic fungus Cordyceps militaris, a valued traditional Chinese medicine. Genome Biol. 2012, 12, R116. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, Y.; Liu, L.; Feng, J.; Zhang, T.; Qin, S.; Zhao, X.; Wang, C.; Li, D.; Han, W. Study of the whole genome, methylome and transcriptome of Cordyceps militaris. Sci. Rep. 2019, 9, 898. [Google Scholar] [CrossRef] [PubMed]
- Kramer, G.J.; Nodwell, J.R. Chromosome level assembly and secondary metabolite potential of the parasitic fungus Cordyceps militaris. BMC Genom. 2017, 18, 912. [Google Scholar] [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Naish, M.; Alonge, M.; Wlodzimierz, P.; Tock, A.J.; Abramson, B.W.; Schmücker, A.; Mandáková, T.; Jamge, B.; Lambing, C.; Kuo, P. The genetic and epigenetic landscape of the Arabidopsis centromeres. Science 2021, 374, eabi7489. [Google Scholar] [CrossRef]
- Wei, C.; Gao, L.; Xiao, R.; Wang, Y.; Chen, B.; Zou, W.; Li, J.; Mace, E.; Jordan, D.; Tao, Y. Complete telomere-to-telomere assemblies of two sorghum genomes to guide biological discovery. iMeta 2024, 3, e193. [Google Scholar] [CrossRef]
- Song, J.-M.; Xie, W.-Z.; Wang, S.; Guo, Y.-X.; Koo, D.-H.; Kudrna, D.; Gong, C.; Huang, Y.; Feng, J.-W.; Zhang, W. Two gap-free reference genomes and a global view of the centromere architecture in rice. Mol. Plant 2021, 14, 1757–1767. [Google Scholar] [CrossRef]
- Wang, M.; Meng, G.; Yang, Y.; Wang, X.; Xie, R.; Dong, C. Telomere-to-Telomere Genome Assembly of Tibetan Medicinal Mushroom Ganoderma leucocontextum and the First Copia Centromeric Retrotransposon in Macro-Fungi Genome. J. Fungi 2023, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, L.; Yang, Q.; Dong, J.; Wang, Y.; Duan, Y.; Yin, W.; Zheng, L.; Sun, W.; Fan, J. Gap-free nuclear and mitochondrial genomes of Ustilaginoidea virens JS60-2, a fungal pathogen causing rice false smut. Mol. Plant-Microbe Interact. 2022, 35, 1120–1123. [Google Scholar] [CrossRef]
- Huang, X.; Duan, N.; Xu, H.; Xie, T.; Xue, Y.-R.; Liu, C.-H. CTAB-PEG DNA extraction from fungi with high contents of polysaccharides. Mol. Biol. 2018, 52, 621–628. [Google Scholar] [CrossRef]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Fan, J.; Sun, Z.; Liu, S. NextPolish: A fast and efficient genome polishing tool for long-read assembly. Bioinformatics 2020, 36, 2253–2255. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Yi, Z.; Zhang, X.; Du, Y.; Li, Y.; Zhou, Z.; Chen, S.; Zhao, H.; Yang, S.; Wang, Y. Chromosome-level scaffolding of haplotype-resolved assemblies using Hi-C data without reference genomes. Nat. Plants 2024, 10, 1184–1200. [Google Scholar] [CrossRef]
- Durand, N.C.; Robinson, J.T.; Shamim, M.S.; Machol, I.; Mesirov, J.P.; Lander, E.S.; Aiden, E.L. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 2016, 3, 99–101. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, 5, 4.10.1–4.10.14. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Keilwagen, J.; Hartung, F.; Grau, J. GeMoMa: Homology-based gene prediction utilizing intron position conservation and RNA-seq data. In Gene Prediction: Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2019; pp. 161–177. [Google Scholar]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Madden, T. The BLAST sequence analysis tool. NCBI Handb. 2013, 2, 425–436. [Google Scholar]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Mendes, F.K.; Vanderpool, D.; Fulton, B.; Hahn, M.W. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 2020, 36, 5516–5518. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Marçais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-h.; Jin, H.; Marler, B.; Guo, H. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39 (Suppl. S2), W29–W37. [Google Scholar] [CrossRef]
- Cortés-Maldonado, L.; Marcial-Quino, J.; Gómez-Manzo, S.; Fierro, F.; Tomasini, A. A method for the extraction of high quality fungal RNA suitable for RNA-seq. J. Microbiol. Methods 2020, 170, 105855. [Google Scholar] [CrossRef]
- Chen, S. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. iMeta 2023, 2, e107. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Shumate, A.; Wong, B.; Pertea, G.; Pertea, M. Improved transcriptome assembly using a hybrid of long and short reads with StringTie. PLoS Comput. Biol. 2022, 18, e1009730. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Chan, C.-K.K. Analysis of RNA-Seq data using TopHat and Cufflinks. In Plant Bioinformatics: Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2016; pp. 339–361. [Google Scholar]
- Li, G.; Jian, T.; Liu, X.; Lv, Q.; Zhang, G.; Ling, J. Application of metabolomics in fungal research. Molecules 2022, 27, 7365. [Google Scholar] [CrossRef]
- Smedsgaard, J.; Nielsen, J. Metabolite profiling of fungi and yeast: From phenotype to metabolome by MS and informatics. J. Exp. Bot. 2005, 56, 273–286. [Google Scholar] [CrossRef]
- Singh, V.K.; Mangalam, A.K.; Dwivedi, S.; Naik, S. Primer premier: Program for design of degenerate primers from a protein sequence. Biotechniques 1998, 24, 318–319. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Chung, D.; Kwon, Y.M.; Yang, Y. Telomere-to-telomere genome assembly of asparaginase-producing Trichoderma simmonsii. BMC Genom. 2021, 22, 830. [Google Scholar] [CrossRef]
- Chen, X.; Wei, Y.; Meng, G.; Wang, M.; Peng, X.; Dai, J.; Dong, C.; Huo, G. Telomere-to-Telomere Haplotype-Resolved Genomes of Agrocybe chaxingu Reveals Unique Genetic Features and Developmental Insights. J. Fungi 2024, 10, 602. [Google Scholar] [CrossRef]
- Chai, L.; Li, J.; Guo, L.; Zhang, S.; Chen, F.; Zhu, W.; Li, Y. Genomic and Transcriptome Analysis Reveals the Biosynthesis Network of Cordycepin in Cordyceps militaris. Genes 2024, 15, 626. [Google Scholar] [CrossRef]
- Dong, J.Z.; Lei, C.; Zheng, X.J.; Ai, X.R.; Wang, Y.; Wang, Q. Light wavelengths regulate growth and active components of c ordyceps militaris fruit bodies. J. Food Biochem. 2013, 37, 578–584. [Google Scholar] [CrossRef]
- Dong, J.; Liu, M.; Lei, C.; Zheng, X.; Wang, Y. Effects of selenium and light wavelengths on liquid culture of Cordyceps militaris Link. Appl. Biochem. Biotechnol. 2012, 166, 2030–2036. [Google Scholar] [CrossRef]
- Toldi, D.; Gyugos, M.; Darkó, É.; Szalai, G.; Gulyás, Z.; Gierczik, K.; Székely, A.; Boldizsár, Á.; Galiba, G.; Müller, M. Light intensity and spectrum affect metabolism of glutathione and amino acids at transcriptional level. PLoS ONE 2019, 14, e0227271. [Google Scholar] [CrossRef]
- Oliveira, I.; Brenner, E.; Chiu, J.; Hsieh, M.-H.; Kouranov, A.; Lam, H.-M.; Shin, M.; Coruzzi, G. Metabolite and light regulation of metabolism in plants: Lessons from the study of a single biochemical pathway. Braz. J. Med. Biol. Res. 2001, 34, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Fischer, R. Light sensing and responses in fungi. Nat. Rev. Microbiol. 2019, 17, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Radhi, M.; Ashraf, S.; Lawrence, S.; Tranholm, A.A.; Wellham, P.A.D.; Hafeez, A.; Khamis, A.S.; Thomas, R.; McWilliams, D.; De Moor, C.H. A systematic review of the biological effects of cordycepin. Molecules 2021, 26, 5886. [Google Scholar] [CrossRef]
- Zhou, X.; Luo, L.; Dressel, W.; Shadier, G.; Krumbiegel, D.; Schmidtke, P.; Zepp, F.; Meyer, C.U. Cordycepin is an immunoregulatory active ingredient of Cordyceps sinensis. Am. J. Chin. Med. 2008, 36, 967–980. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Value |
|---|---|
| Total genes | 10,749 |
| Avg. gene length | 1976.97 |
| Avg. gene cds length | 1540.18 |
| Total cds | 29,460 |
| Total cds length | 16,555,424 |
| Avg. cds length | 561.96 |
| Avg. cds per gene | 2.74 |
| Total exons | 30,942 |
| Total exon length | 22,116,766 |
| Avg. exon length | 714.78 |
| Avg. exons per gene | 2.88 |
| Total genes with introns | 7826 |
| Total introns | 19,367 |
| Total intron length | 1,538,583 |
| Avg. intron length | 79.44 |
| Avg. introns per gene | 1.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Huang, J.; Dong, G.; Hu, X. Telomere-to-Telomere Assembly of the Cordyceps militaris CH1 Genome and Integrated Transcriptomic and Metabolomic Analyses Provide New Insights into Cordycepin Biosynthesis Under Light Stress. J. Fungi 2025, 11, 461. https://doi.org/10.3390/jof11060461
Yang Y, Huang J, Dong G, Hu X. Telomere-to-Telomere Assembly of the Cordyceps militaris CH1 Genome and Integrated Transcriptomic and Metabolomic Analyses Provide New Insights into Cordycepin Biosynthesis Under Light Stress. Journal of Fungi. 2025; 11(6):461. https://doi.org/10.3390/jof11060461
Chicago/Turabian StyleYang, Yang, Jingjing Huang, Gangqiang Dong, and Xuebo Hu. 2025. "Telomere-to-Telomere Assembly of the Cordyceps militaris CH1 Genome and Integrated Transcriptomic and Metabolomic Analyses Provide New Insights into Cordycepin Biosynthesis Under Light Stress" Journal of Fungi 11, no. 6: 461. https://doi.org/10.3390/jof11060461
APA StyleYang, Y., Huang, J., Dong, G., & Hu, X. (2025). Telomere-to-Telomere Assembly of the Cordyceps militaris CH1 Genome and Integrated Transcriptomic and Metabolomic Analyses Provide New Insights into Cordycepin Biosynthesis Under Light Stress. Journal of Fungi, 11(6), 461. https://doi.org/10.3390/jof11060461