1. Introduce
The Chinese caterpillar fungus,
Ophiocordyceps sinensis (Berk.) Sung et al. (Ascomycota, Ophiocordycipitaceae), is traditional Chinese medicine, known for its enormous medicinal and commercial value [
1,
2]. The adenosine, polysaccharides, proteins, and other bioactive compounds abundantly present in it significantly contribute to human health and disease prevention. Research has demonstrated that
O. sinensis possesses various health benefits, including the ability to lower blood sugar levels, reduce blood lipids, exhibit anti-tumor properties, provide antioxidant effects, combat aging, and promote skin whitening [
3,
4,
5,
6,
7,
8]. Consequently, both the nutritional and medicinal values of
O. sinensis have garnered recognition among consumers, leading to a year-on-year increase in demand. However, human activities such as farming, grazing, and excessive harvesting have severely compromised its natural habitat. Additionally, environmental challenges such as global warming have further diminished the reserves of wild
O. sinensis. These pressing supply and demand issues have prompted researchers to explore alternatives to
O. sinensis to mitigate the resource depletion crisis [
9,
10,
11].
The results showed that the nutrient quality of
O. sinensis mycelia was affected by the change in culture temperature. When the growth temperature range is between 15 °C and 25 °C, the growth cycle of the fungi is relatively short, allowing for a faster completion of the growth and development process. For instance, the optimal pH for mycelium growth and dry matter weight of
O. sinensis pupa is 7.0, while the optimal temperature for mycelium growth and dry matter weight is 22 °C [
12]. The ideal combination of temperature, pH, and culture time to maximize the yield of cordycepin is 25 °C, 5.5, and 21 days, respectively [
13]. When the ambient temperature exceeds 25 °C, the enzyme activity of the fungus significantly decreases and may become deactivated, resulting in changes to the nutrient content of the fungus. Li et al. reported that raising the temperature from 25 °C to 30 °C and 35 °C during the cultivation of the filamentous fungus Aspergillus niger B1-D generally enhanced cellular metabolism and led to the transient accumulation of protein hydroxyl compounds, indicating that the antioxidant function of cells was impaired at high temperatures [
14]. Under heat stress, 45.45% of
O. sinensis DnaJ are differentially expressed, suggesting their involvement in the response to temperature stress and their regulatory role in growth and development [
15]. Fungal proteome analysis under high temperature stress revealed that the heat shock protein (HSP) was up-regulated at 20 °C, potentially activating the unfolded protein response (UPR) [
16]. Changes in transcriptional responses induced by low temperatures (T < 15 °C) affect several metabolic processes related to nutrient assimilation and energy sources, including amino acid and lipid metabolism, with the lipid biosynthesis pathway being regulated through the transcriptional up-regulation of membrane function [
17]. Consequently, the degradation of strains significantly impacts the yield and quality of filamentous fungi. Additionally, degradation causes the accumulation of large amounts of reactive oxygen species (ROS) within the strain. Concurrently, the activity of antioxidant enzymes diminishes, and the levels of exopolysaccharides, cellulase, and amylase become unbalanced, which disrupts the normal physiological processes of the strain and induces oxidative stress reactions [
18]. It has been reported that fungi are frequently employed for large-scale industrial production due to their adaptability to various substrates and multifunctional characteristics [
19]. Metabolomics is the research object of all metabolites in organisms or cells. The purpose for this field is to reveal the metabolic characteristics and metabolic laws of organisms in different physiological states by studying the changes in metabolites. The changes in these metabolites reflect the metabolic process and physiological state inside the host [
20,
21]. Zhou et al. determined the matrix and sclertium of wild
O. sinensis through a wide range of targeted metabolomics and non-targeted metabolomics, and found that niacin and niacinamide metabolism, thiamine metabolism, riboflavin metabolism, glycine, serine and threonine metabolism, and arginine biosynthesis may be key aspects in the differentiation of the matrix and sclertium [
22]. Zhong et al. conducted a transcriptomic analysis of pathogenic fungi before and after
O. sinensis infection, and gene ontology (GO) analysis showed that differentially expressed genes (DEGs) were particularly abundant in aspects related to biological processes and molecular functions [
23]. He et al. analyzed changes in Cordyceps sinensis fungus for different fermentation time durations using metabolomics and transcriptomics, revealing that the increase in the contents of DEGs and DAMs promoted the increase in enzyme and non-enzyme substance levels in Cordyceps sinensis, ultimately enhancing the antioxidant capacity of Cordyceps sinensis [
24]. However, strain degradation during temperature culture or preservation can result in substantial economic losses [
25,
26]. Therefore, determining the optimal culture temperature of
O. sinensis mycelium is not only crucial for the effective preparation of its mycelium but also for large-scale industrial production in the future.
In this study, we established five distinct fungal fermentation temperature treatments at 8 °C, 13 °C, 18 °C, 23 °C, and 28 °C. By analyzing morphological, physiological, and biochemical characteristics, as well as metabolic profiles and gene expression at these varying temperatures, we identified the optimal temperature range for culturing O. sinensis. This research provides theoretical support for the industrial-scale production of O. sinensis and enhances our understanding of how temperature fluctuations influence the nutrient quality of O. sinensis mycelium.
2. Materials and Methods
2.1. Test Strain and Medium
O. sinensis was provided by the O. sinensis Laboratory, Academy of Animal Science and Veterinary Science, Qinghai University (Xining City, China), and the strain was stored in the germplasm bank of O. sinensis Laboratory of Qinghai Province (no. CX-HN-9).
The solid potato dextrose agar (PDA) medium was composed of 20% potato, 2% glucose, 0.3% peptone, 0.2% potassium dihydrogen phosphate, 0.02% magnesium sulfate, and 1.6% agar, with a pH of 7. This medium was autoclaved at 121 °C for 30 min before use. The liquid PDA medium consisted of the same components: 20% potato, 2% glucose, 0.3% peptone, 0.2% potassium dihydrogen phosphate, and 0.02% magnesium sulfate, with a pH of 7. This medium was dispensed into triangular bottles and also autoclaved at 121 °C for 30 min before use.
2.2. Treatment and Collection of Mycelia
The culture of solid mycelia was initiated by using a hole punch with a diameter of 0.25 cm to extract an equal amount of mycelia from the center of a solid culture dish. This mycelial sample was then inoculated at the center of a PDA solid culture medium and incubated at temperatures of 8 °C, 13 °C, 18 °C, 23 °C, and 28 °C, respectively.
Culture of liquid mycelium: An equal amount of strain was extracted from the previous operation, homogenized using an inoculating shovel, and subsequently inoculated into PDA liquid medium. The cultures were maintained under five different temperature conditions, with a shaking speed of 135 rpm. The culture was deemed complete when 80% of the mycelium had proliferated within the liquid medium at the specified temperature. Following centrifugation at 4 °C, the supernatant was discarded (10,000 g), yielding the cultured mycelium, which was then stored in a cryogenic freezer at −80 °C.
2.3. Growth Rate, Apparent Morphology, SEM Analysis, Dry and Fresh Weight, and Conidial Weight Were Measured
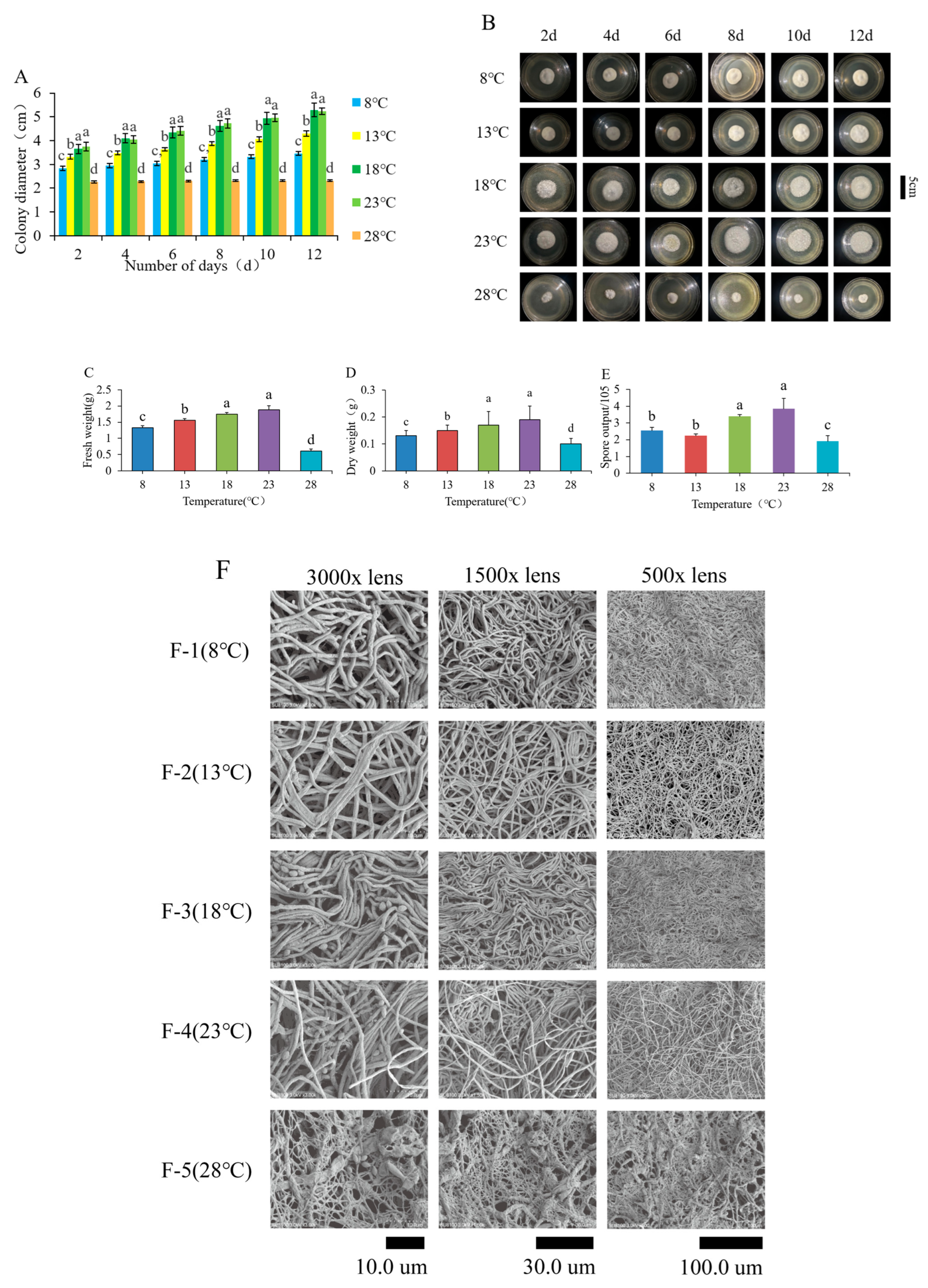
Scanning electron microscopy (SEM) analysis was conducted to observe the mycelial growth morphology at various temperatures. Mycelia were collected at 8 °C, 13 °C, 18 °C, 23 °C, and 28 °C, and fixed in 2.5% glutaraldehyde at 4 °C for 12 h. The samples were rinsed three times with a 0.1 mol/L phosphate buffer, with each rinse lasting 15 min. They were then dehydrated using a series of ethanol solutions (30%, 50%, 70%, 90%, and 95%), followed by two rounds of dehydration in 100% ethanol, each lasting 20 min. The samples were subsequently treated with a mixture of ethanol and isoamyl acetate (in a 1:1 volume ratio) for 30 min, followed by treatment with isoamyl acetate alone for 2 h. Finally, the samples were placed in a freeze dryer for 15 h. The dried fungi were affixed to the sample table for gold-spraying treatment and observed under scanning electron microscopy to assess the thickness, curvature, branching, and surface characteristics of the mycelia at each temperature gradient, after which photographs were taken and recorded.
Three biological replicates were established for each measurement. After ten days of culture, the colony diameter was measured using a ruler and the cross-crossing method every two days. The growth of colonies at different temperatures was observed, and their morphology was described in terms of colony growth rate, mycelium status on the colony surface, mycelium texture, colony color, and colony shape. Photographs were taken and recorded for documentation. At the end of 22 days of culture, the entire mycelium was removed along with the medium, which was subsequently washed off with hot sterile water. The residual water on the colony surface was absorbed using filter paper (Hangzhou City, China), and the fresh weight of the colony was determined using an electronic balance. Finally, the colonies were placed in a constant temperature drying box set at 55 °C, where they were weighed every 30 min until the mass stabilized over three consecutive measurements. The average of these three measurements was recorded as the dry weight of the colonies. To prepare a spore suspension, 5 mL of sterile water was added to the colony plate, and the colony surface was gently brushed to dislodge the conidia. The mycelium, media, and other impurities were filtered out using gauze rinsed with sterile water. A 0.05% Tween-80 solution was used to create a spore suspension with a fixed volume of 10 mL, which was thoroughly mixed using a vortex oscillator (Shanghai City, China). A hemocytometer was employed to count the number of spores produced by a single colony. The results were subsequently calculated using the formula provided below.
Conidial number (each/mL) = total number of small square spores (80)/80 × 400 × 104 × dilution.
2.4. Determination of Antioxidant Enzymes
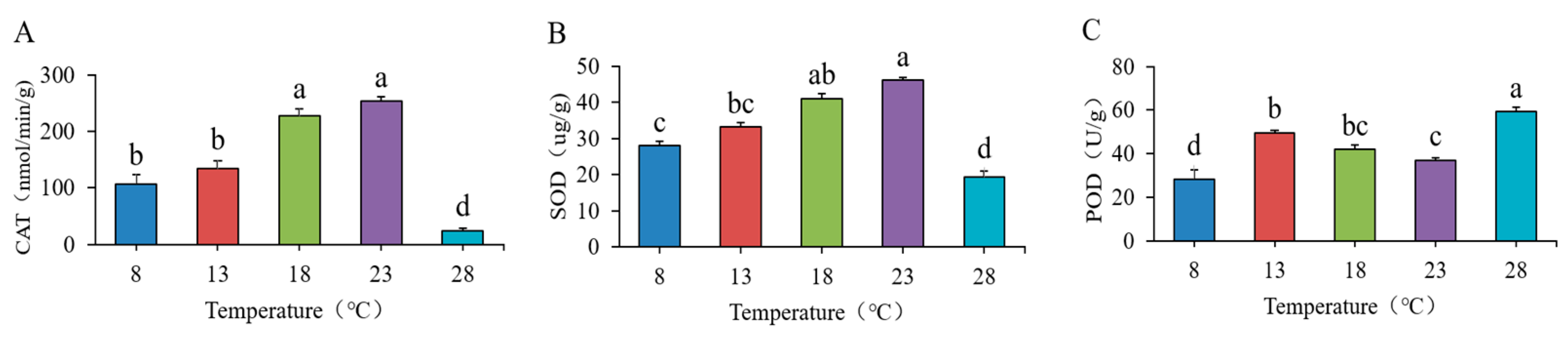
This study measured several antioxidant enzymes, specifically catalase (CAT), superoxide dismutase (SOD), and peroxidase (POD). The measurement methods strictly adhered to the protocols established by Suzhou Keming Biotechnology Co., Ltd. (Suzhou City, China). The assays were performed following the detailed instructions provided in the corresponding kit.
2.5. Extraction of Metabolites
The extraction method for metabolites was refined based on our laboratory’s research findings and relevant published studies [
27,
28]. Initially, 100 mg of the sample was placed in a 2 mL centrifuge tube, to which 6 mm diameter glass beads were added, and the mixture was ground at a low temperature (4 °C). Subsequently, 80 mg of the pulverized sample was combined with 1000 μL of extractant (composed of 40% methanol, 40% acetonitrile, and 20% water) and swirled for 30 s. A tissue grinder (JXFSTPRP-CLN-24, Shanghai, China) was employed for mixing, with parameters set to −10 °C, 55 Hz, for 2 min. The mixture was then subjected to ultrasonic extraction using a cell comminuter (JC-12L, Guangdong, China) at 4 °C, applying a force of 13,000 g for 10 min, after which it was removed. The liquid mixture was centrifuged at 12,000 rpm for 10 min at 4 °C, and the supernatant was collected, concentrated, and dried. Finally, the supernatant was filtered through a 0.22 μm membrane and injected into the test bottle.
2.6. LC-MS/MS
The metabolomics analysis was performed using the UPLC-ESI-Q-orbital well MS system (UHPLC, Shimazu Nexera X2 LC-30AD, Shimazu Corporation, Kyoto, Japan) in conjunction with Q-Exactive Plus (Thermo Scientific, San Jose, CA, USA). For liquid chromatography (LC) separation, samples were analyzed using the ACQUITY UPLC and HSS T3 column (2.1 × 100 mm, 1.8 μm) (Waters, Milford, MA, USA). During the preparation of the experiment, we mixed all samples in equal amounts to obtain the quality control (QC) samples. During data preprocessing, we adopted QC sample-based normalization for substance quantification: support vector regression correction based on QC samples to eliminate systematic errors. Subsequently, substances with a coefficient of variance (CV) of less than 30% in the QC samples were retained for further analysis. The parameters were set as follows: flow rate of 0.3 mL/min, column temperature of 40 °C, and sample size of 2 μL. The mobile phase consisted of A (0.1% formic acid in water) and B (0.1% formic acid in acetonitrile). Linear gradient elution was conducted on the samples in both negative (NEG) and positive (POS) modes, with the following gradient: 0–1 min at 8% B; 1–8 min from 8% to 98% B; 8–10 min at 98% B; 10–10.1 min from 98% to 8% B; and 10.1–12 min at 8% B. The parameters for the mass spectrometer were configured as follows: the positive ion spray voltage was set to 3.50 kV, the negative ion spray voltage was set to −2.50 kV, the sheath gas flow was maintained at 40 arb, and the auxiliary gas flow was maintained at 10 arb. The capillary temperature was set to 325 °C, with first-level full scanning conducted at a resolution of 60,000. The first-level ion scanning range was set from 100 to 1000 m/z, and second-level fragmentation was performed using HCD with a collision energy of 30%. The second-level resolution was set to 15,000, and the first four ions were fragmented following signal acquisition, with unnecessary MS/MS data excluded via dynamic exclusion. MS-DIAL was employed to conduct peak alignment, retention time correction, and peak area extraction on the original mass spectrometry data. Metabolites were identified based on mass (mass tolerance < 10 ppm) and MS/MS data (mass tolerance < 0.02 Da), which were accurately matched with public databases such as the Human Metabolome Database (HMDB), Mass Bank, and our self-constructed metabolite standard library. Among the extracted ion features, only variables with more than 50% non-zero measurements in at least one dataset were retained.
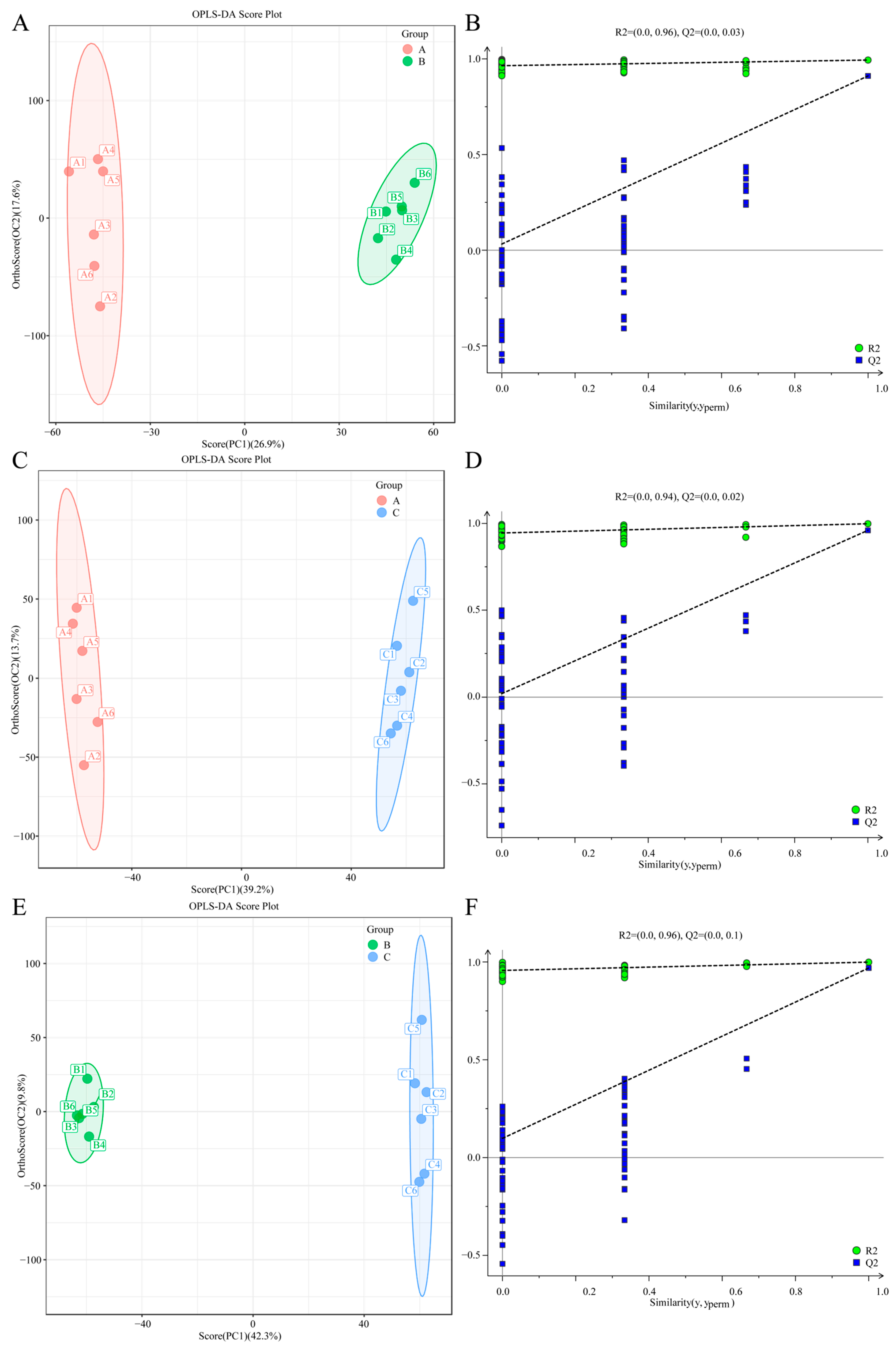
2.7. Multivariate Statistical Analysis
The R software (v4.3.1) package was utilized for conducting principal component analysis (PCA), partial least squares discriminant analysis (PLS-DA), and orthogonal partial least squares discriminant analysis (OPLS-DA) for dimensionality reduction in the sample data. The model’s overfitting was assessed using the permutation test method. R
2X and R
2Y denote the proportion of variance explained by the model for the X and Y matrices, respectively, while Q
2 indicates the model’s predictive capability. Values closer to 1 indicate a better fit of the model. The
p value was computed based on statistical tests, the variable importance in projection (VIP) was derived from the OPLS-DA dimensionality reduction method, and the fold change between groups was calculated using log
2. The influence and interpretative capacity of each metabolite on sample classification and discrimination were evaluated, facilitating the selection of metabolites. Metabolites with a
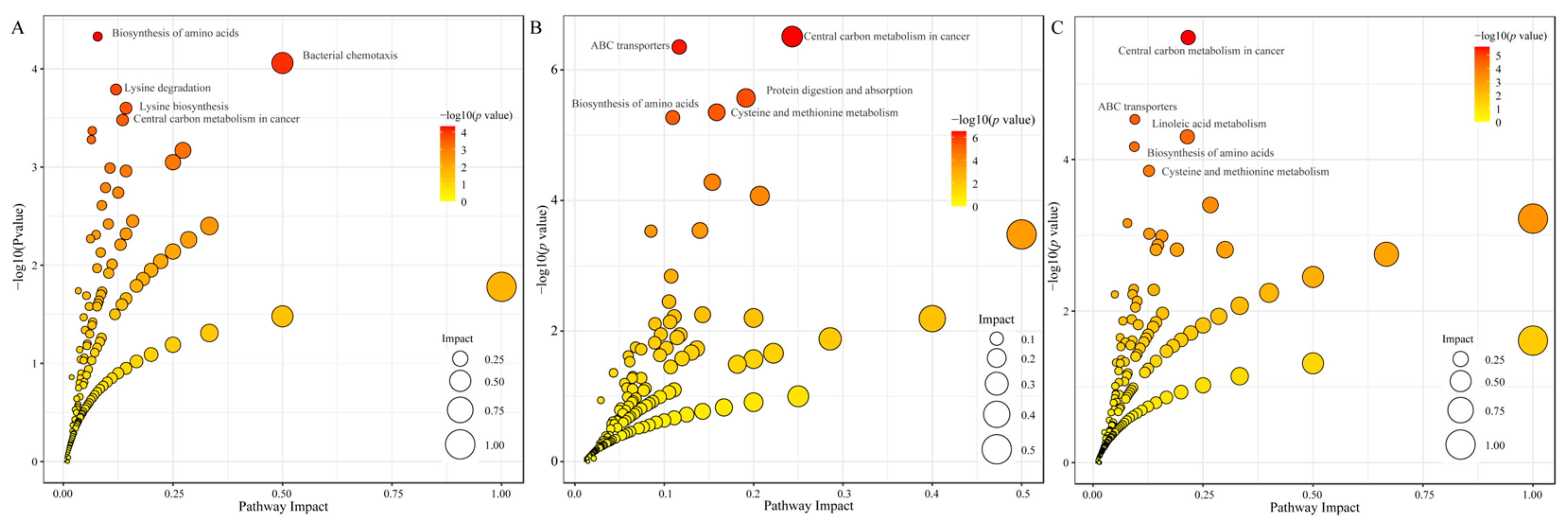
p value 1 were deemed to exhibit statistically significant differences, and the identified differential metabolites were subjected to cluster analysis using the R package. KEGG pathway analysis was conducted on the differential metabolite data using the KEGG database (
http://www.kegg.jp, accessed on 9 October 2024). The Fisher precision test was employed for KEGG enrichment analysis, with false discovery rate (FDR) correction applied for multiple comparisons. The enriched KEGG pathways were considered statistically significant at a
p < 0.05 level.
2.8. RNASeq Process
Total RNA was enriched for mRNA with a polyA structure using Oligo (dT) magnetic beads. The RNA was fragmented into approximately 300 bp pieces through ion interruption, and these fragments were selected for further analysis. Using RNA as a template, the first strand of cDNA was synthesized with 6-base random primers and reverse transcriptase, followed by the synthesis of the second strand of cDNA using the first strand as a template. PCR amplification was employed to enrich the library fragments, resulting in a library size of 450 bp. The library was assessed using the Agilent 2100 Bioanalyzer (100 nm × 2.1 nm, 1.7 μm, Beijing City, China) to determine both the total and effective concentrations. Based on the effective concentration and the required data amount, libraries containing different index sequences were mixed proportionally and diluted to 2 nM. An alkaline denaturation process was then applied to form a single-stranded library. Paired-end sequencing was conducted on the sample library after RNA extraction, purification, and library construction, utilizing Illumina’s Next-Generation sequencing technology. Reads that were spliced, shorter than 50 bp, or had an average quality below Q20 were discarded. The remaining high-quality sequences were assembled from scratch to obtain transcript sequences. The transcripts were clustered, and the longest transcript was designated as Unigene for subsequent Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) annotation. Concurrently, the filtered sequences were compared to Unigene to obtain the Reads Count for each Unigene, followed by differential expression and enrichment analysis. Finally, the differentially expressed genes were analyzed to elucidate changes in gene expression under various conditions and to uncover potential biological significance.
2.9. Real-Time Quantitative Fluorescence (qRT-PCR) Verification
To validate the RNA-seq data, we selected 15 differentially expressed genes (DEGs) for quantitative reverse transcription polymerase chain reaction (qRT-PCR) validation. Primers were designed using the Primer Quest Tool (Oligo v7.56), with a preference for those located near the 3′ end of the genes. The reaction mixture consisted of 5 µL of 2× PerfectStart® Green (Beijing Quanshijin Biotechnology Co., Ltd., Beijing City, China) qPCR Super Mix, 0.2 µL of forward primer (10 µM), 0.2 µL of reverse primer (10 µM), and 1 µL of cDNA. The cycling conditions were set at 94 °C for 30 s, followed by 94 °C for 5 s and 60 °C for 30 s. The relative expression levels of the target genes were analyzed using the 2−ΔΔCt method, with three biological replicates for each treatment.
2.10. Data Processing and Analysis
Microsoft Office Excel 2019 was utilized to organize the data, while IBM SPSS Statistics 26.0 software was employed to perform one-way ANOVA on the data that satisfied the assumptions of normal distribution and homogeneity of variance. The Waller–Duncan test was then applied to analyze the significance of differences among multiple sample groups. The data analysis in this work was supported by Allbioknow Biotechnology Co., Ltd. (Chongqing, China).
4. Discuss
O. sinensis has been widely recognized as a traditional Chinese medicinal herb for the treatment of various diseases. Its notable biological activities, including anti-cancer properties, blood sugar regulation, and immune modulation, have attracted considerable attention and favor from the public [
29,
30]. However, temperature is a critical factor influencing the growth rate of
O. sinensis; excessively high or low temperatures can inhibit its growth. Under varying temperature conditions, the enzymatic activity of strain, as well as the synthesis and decomposition of metabolites, undergo significant changes [
31]. Therefore, temperature adjustments can impact the metabolic pathways of
O. sinensis, making the optimization of product quality and yield essential for enhancing its efficiency. This study examined the effects of temperature stress on the growth of
O. sinensis mycelium. Significant differences were observed in various parameters, including mycelium growth rate, colony surface characteristics, dry and fresh weight, and conidia count, with an optimal temperature range for mycelium growth identified as 18 °C to 23 °C. Additionally, at 28 °C, mycelium growth was monitored over 12 days, revealing an increase in colony diameter of only 0.05 to 0.1 cm, indicating extremely slow growth or even cessation. This suggests that
O. sinensis is unsuitable for growth at elevated temperatures, which can inhibit mycelial development. Previous studies have indicated that the optimal temperature for fungal colony growth varies, with P. suffultus thriving at 15 °C and C. farinosa and C. coleopterorum preferring 20 °C. Notably, spore germination is most vigorous at 25 °C, whereas temperatures as low as 5 °C completely halt spore germination across all fungal species [
32].
Mycelium growth is influenced by various environmental conditions, with temperature being a critical factor. Numerous enzymes are involved in the growth of mycelia; however, these enzymes can only be utilized by the mycelia after undergoing enzymatic decomposition [
33]. In an optimal environment, the strain enters the growth phase and initiates conidial germination. The germination and subsequent growth of slender mycelia occur only under suitable temperatures, humidity, and adequate nutritional conditions. At this stage, the mycelia grow at their fastest rate, continuously forming new branches through division and extension. This underscores the close relationship between mycelial growth and environmental factors, as well as nutrient availability [
34].
Antioxidant enzymes constitute a class of enzymes that play a crucial role in mitigating oxidative damage in living organisms. They are capable of reducing the production of oxygen free radicals and other ROS, thereby regulating the REDOX balance and protecting cells from damage [
35]. Oxidation is a fundamental process that many organisms undergo during energy conversion. Uncontrolled sources of oxygen, including ROS, can be categorized into two main types, namely free radicals and peroxides, both of which can generate free radicals that lead to oxidative damage in organisms. This oxidative damage can further accelerate degenerative processes, such as aging [
8,
36]. Therefore, the essence of antioxidation lies in delaying or preventing the oxidative degradation of cellular substrates [
37]. Reactive oxygen species within organisms typically require various enzymes for decomposition to achieve the effect of delaying aging. In the lower temperature range of 8–13 °C, all three antioxidant enzymes exhibited an increasing trend. This suggests that when the strain detects potential environmental changes due to the gradual rise in temperature, it actively enhances the activity of antioxidant enzymes to manage the anticipated increase in reactive oxygen species (ROS) production. ROS are continuously generated during the metabolic processes of the strain, and excessive accumulation can lead to damage to cellular structures and functions. The increased activity of antioxidant enzymes during this period can effectively eliminate these ROS, thereby maintaining redox balance within the cells. When the temperature ranged from 13 to 23 °C, the activity changes in catalase (CAT), superoxide dismutase (SOD), and peroxidase (POD) exhibited notable differences. CAT and SOD gradually increased their activity within this range, indicating the strain’s strong adaptability to these temperatures. Although POD activity also increased, its magnitude and rate of change diverged from those of the former two enzymes, reflecting the unique response patterns of different antioxidant enzymes as the strain adjusts to temperature changes. The enzymes cooperate with one another while focusing on different aspects, collectively contributing to the formation of an antioxidant defense network. However, when the temperature reaches 28 °C, the activities of catalase (CAT) and superoxide dismutase (SOD) significantly decrease. This decrease indicates that elevated temperatures can disrupt the spatial structure of enzyme proteins, leading to alterations in the active sites of the enzymes, which drastically reduces their catalytic efficiency. Consequently, the strain’s ability to scavenge reactive oxygen species diminishes, resulting in a substantial accumulation of these species within the cells and triggering oxidative stress. This oxidative stress inflicts severe damage on the strain’s cells, compromising the integrity of cell membranes and impairing the functions of organelles, ultimately threatening the normal growth and development of the plant strain. The observed increase in peroxidase (POD) activity suggests that it may possess a more stable protein structure, or that structural changes at elevated temperatures have a lesser impact on its activity, thereby allowing it to maintain a higher level of activity.
The study of metabolomics involves the investigation of endogenous metabolic changes induced by both internal factors within organisms and external environmental stimuli. This field allows for the quantitative and qualitative analysis of key compounds that describe the metabolic cycle, facilitating the exploration of the relationship between metabolites and physiological as well as pathological changes [
38,
39]. Fungal metabolites possess the ability to counteract various biological and abiotic stresses and play a significant role in human health due to their diverse biological properties [
40]. Temperature is one of the most critical factors influencing fungal growth. In this study, we employed metabolomics to analyze and compare the metabolite profiles of
O. sinensis cultivated under different temperature conditions. The analysis using principal component analysis (PCA) and orthogonal partial least squares discriminant analysis (OPLS-DA) models revealed the differences in metabolites produced by
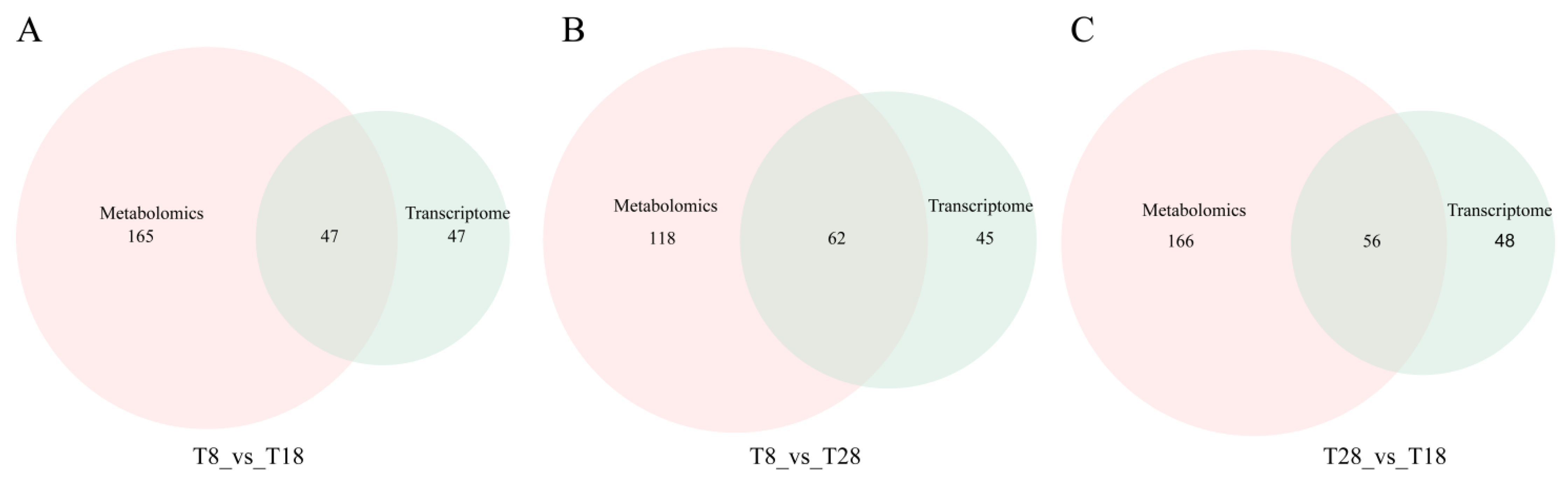
O. sinensis under low, moderate, and high temperature cultivation conditions. By comparing metabolites between T8 vs. T18, T8 vs. T28, and T28 vs. T18, we identified 103, 162, and 153 DAMs, respectively. Notably, the number of DAMs in the low-temperature and warm-temperature comparison groups was lower than that in the high-temperature comparison group. The identified DAMs across the three comparison groups predominantly included Threonine, Catechol, L-Prolinamide, Taurine, Creatine, and Acetylphosphate. The change in temperature can affect the activity of enzymes related to threonine anabolism in Cordyceps sinensis and thereby influence the content of acid [
41]. L-Prolinamide has been shown to inhibit the release of melanocyte-stimulating hormone (MSH), improve pharmacological mechanisms within the central nervous system, and exhibit antioxidant effects that enhance sperm quality [
42]. Taurine has potential mechanisms for regulating the tumor microenvironment and tumor cell metabolism, contributing to enhanced immunotherapy through the modulation of tumor microenvironment metabolism and relevant molecules, thereby exerting anti-tumor effects [
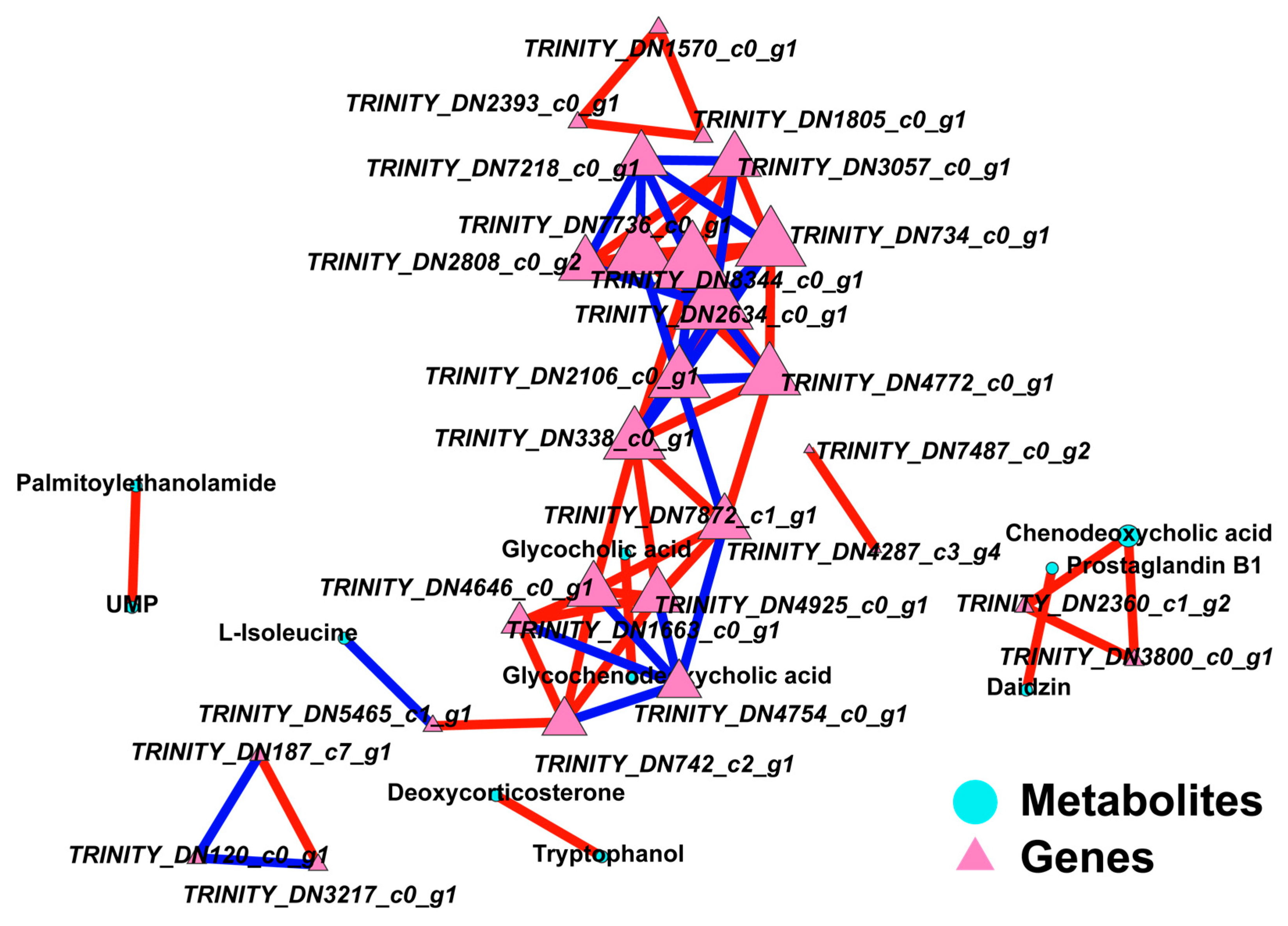
43]. The varying levels of these active secondary metabolites in strains at different temperatures suggest that the nutrient composition and medicinal value of the fungi are altered in response to changes in growth environment temperature, indicating that temperature fluctuations may lead to inconsistencies in the metabolite synthesis patterns among strains.
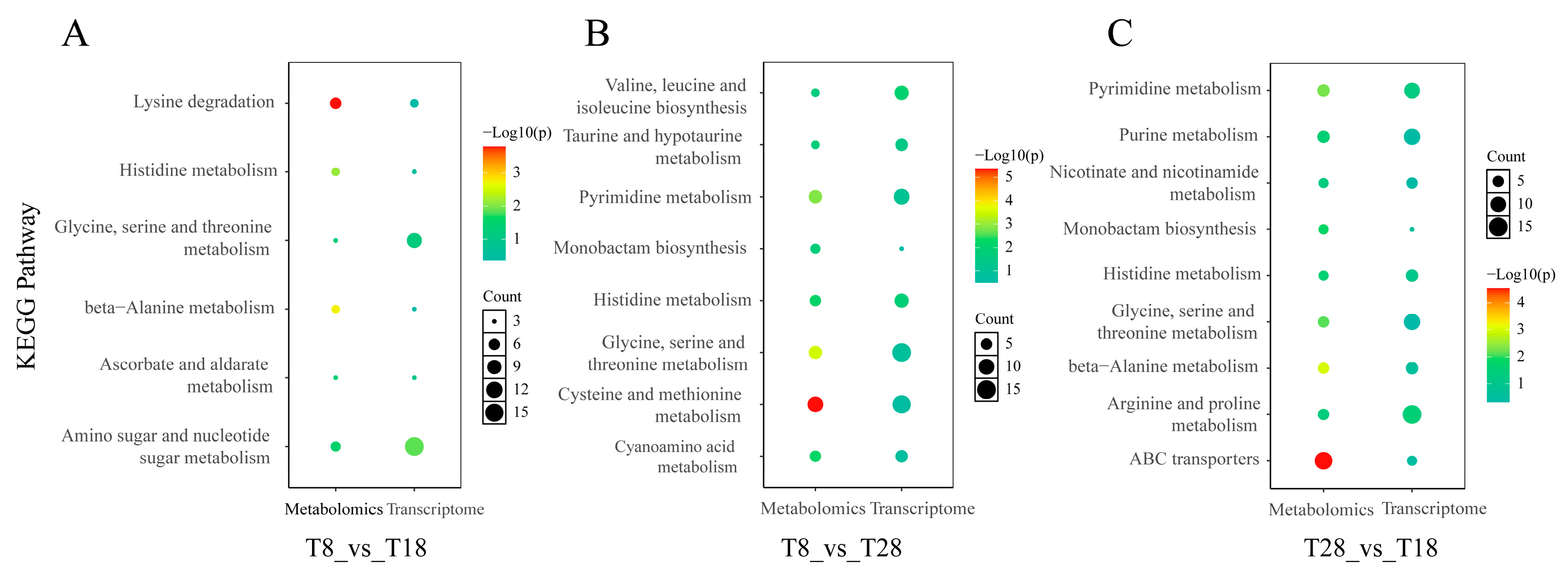
The KEGG databases serve as essential tools for elucidating the advanced functions and applications of cells, organisms, and ecosystems through molecular-level information. Consequently, the KEGG pathway analysis is crucial for comprehending the roles of metabolites and their interrelationships. The KEGG enrichment analysis of differentially accumulated metabolites (DAMs) across three distinct groups revealed that, in the comparisons between low-temperature strains, warm-temperature strains, and high-temperature strains (T8 vs. T18 and T8 vs. T28), the metabolic pathways significantly enriched included lysine degradation, lysine biosynthesis, and protein digestion and absorption. In the comparison between high-temperature and temperate strains (T28 vs. T18), significant enrichment was observed in the metabolism of linoleic acid and cysteine-methionine. Amino acids, recognized for their beneficial effects on human health, play a crucial role in enhancing immunity and promoting wound healing [
44]. However, in filamentous fungi, the type of amino acids is a critical factor influencing the growth rate of mycelia [
45]. Cysteine functions as an antioxidant and plays a crucial role in the synthesis of antioxidant substances. It effectively scavenges reactive oxygen species generated during metabolism, reduces oxidative damage, enhances the organism’s resistance to oxidative stress, and maintains the stability of cellular structure and function under adverse conditions such as high temperatures and high salinity [
46].
With the continuous advancement of transcriptomics, its application has expanded to various fields, including animals, plants, microorganisms, and medicine [
47,
48]. By investigating organisms through transcriptomic analysis, researchers can identify changes in gene expression across different species [
49]. In this study, transcriptomics were employed to examine the differentially expressed genes of
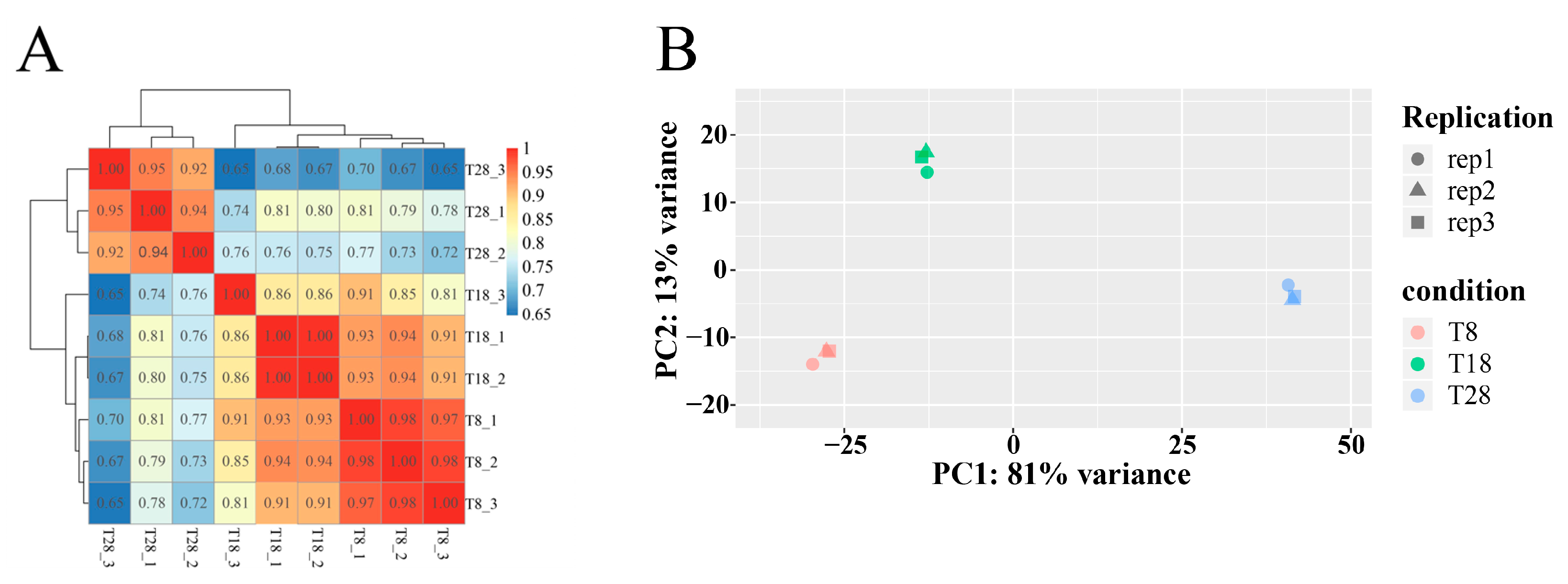
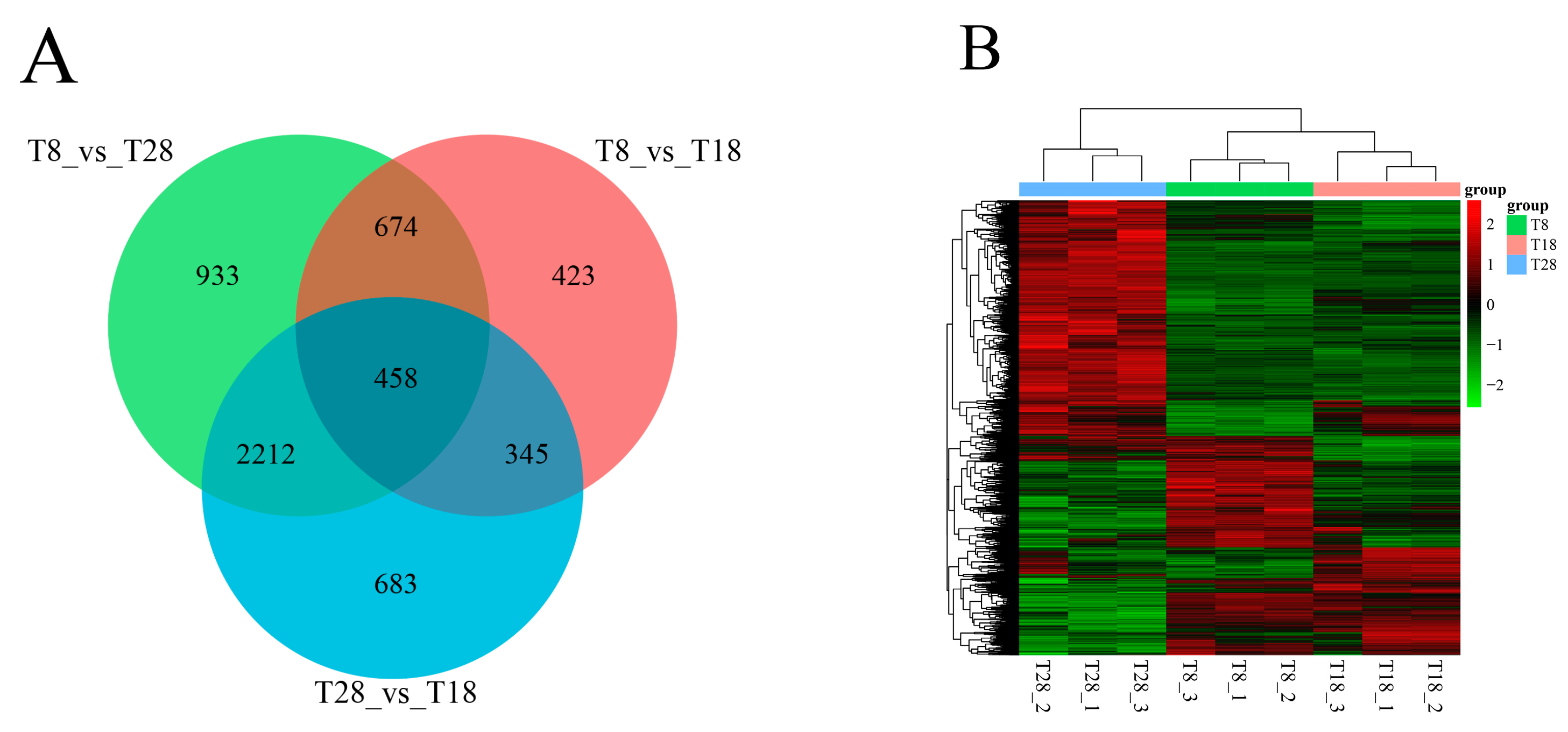
O. sinensis under varying temperature treatments, leading to the identification of these genes. In the T8 vs. T18 comparison group, the majority of genes were down-regulated. Conversely, in the T8 vs. T28 comparison group, up-regulated genes predominated, while in the T28 vs. T18 comparison group, down-regulated genes outnumbered up-regulated ones. As temperature increased, the number of up-regulated genes gradually decreased, whereas down-regulated genes increased. Additionally, the clustering analysis of the three strains at different temperatures revealed that the strains at T8 and T18 clustered together, whereas those at T28 formed a distinct cluster. Integrating these findings with a comprehensive analysis of morphology, physiological indicators, and metabolomics suggests that the strains at 28 °C may experience high-temperature stress. Ultimately, significant changes were observed in growth rates, physiological and biochemical indices, differential metabolites, and the number of differentially expressed genes. These results indicate that the antioxidant oxidase system of the strain was compromised under different temperature stresses, leading to reduced spore production and adversely affecting normal growth and development. The variability in the number of differentially expressed genes among the strain highlights differing trends in expression. Furthermore, gene expression profiles under varying temperature stresses differed, reflecting the strain’s adaptive mechanisms to temperature fluctuations.
In recent years, the integration of high-throughput sequencing platforms with RNA-Seq technology has significantly advanced molecular biology research [
50]. Consequently, this study conducted Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses of differentially expressed genes (DEGs) across various comparison groups using Illumina RNA-Seq technology. The GO enrichment analysis revealed that DEGs were primarily categorized into three functional domains: molecular function (MF), biological process (BP), and cellular component (CC). Further analysis indicated that the most prominent GO terms in the T8 vs. T18 comparison group included nucleolus, pre-ribosome, 90s ribosome, RNA binding, biological origin of ribosomes, ribosome processing, and metabolic processes. In the T8 vs. T28 comparison group, GO terms were predominantly enriched in extracellular regions and intrinsic components of the plasma membrane. The main GO terms enriched in the T28 vs. T18 comparison group included membrane composition, isotropic transport activity, and solute cation symporter activity. Temperature stress induces the peroxidation of the
O. sinensis plasma membrane, resulting in compositional changes to adapt to elevated external temperatures [
51]. This suggests that temperature stress significantly impacts the metabolic substances of strains, leading to continuous alterations in their physiological, biochemical, and metabolic processes. Additionally, the KEGG pathways associated with DEGs across different comparison groups were analyzed. The results indicated that in the T8 vs. T18 comparison group, DEGs were highly enriched in KEGG pathways related to ribosome biogenesis, as well as the biosynthesis of ubiquinone and other terpenoid quinones in eukaryotes. In the T8 vs. T28 comparison group, DEGs showed high enrichment in KEGG pathways related to protein efflux, tryptophan metabolism, arginine and proline metabolism, protein processing in the endoplasmic reticulum, and phenylalanine metabolism. The KEGG pathways associated with DEGs in the T28 vs. T18 comparison group were significantly enriched in ribosome and eukaryotic ribosome biogenesis pathways. This evidence supports the notion that temperature variations influence the original gene expression patterns of
O. sinensis, and that the strain’s stress response mechanisms to temperature changes are distinct.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}