
Whole-Genome Sequencing and Comparative Genomic Analysis of Three Clinical Bloodstream Infection Isolates of Trichosporon austroamericanum

 , , , , , , and
, , , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Fungal Isolates
2.2. DNA Extraction and Genome Sequencing
2.3. Genome Assembly
2.4. Gene Prediction and Functional Annotation
2.5. Phylogenetic Analysis
2.6. Ortholog Analysis
2.7. Galleria mellonella Infection Model
3. Results
3.1. Genome Assembly and Gene Annotation
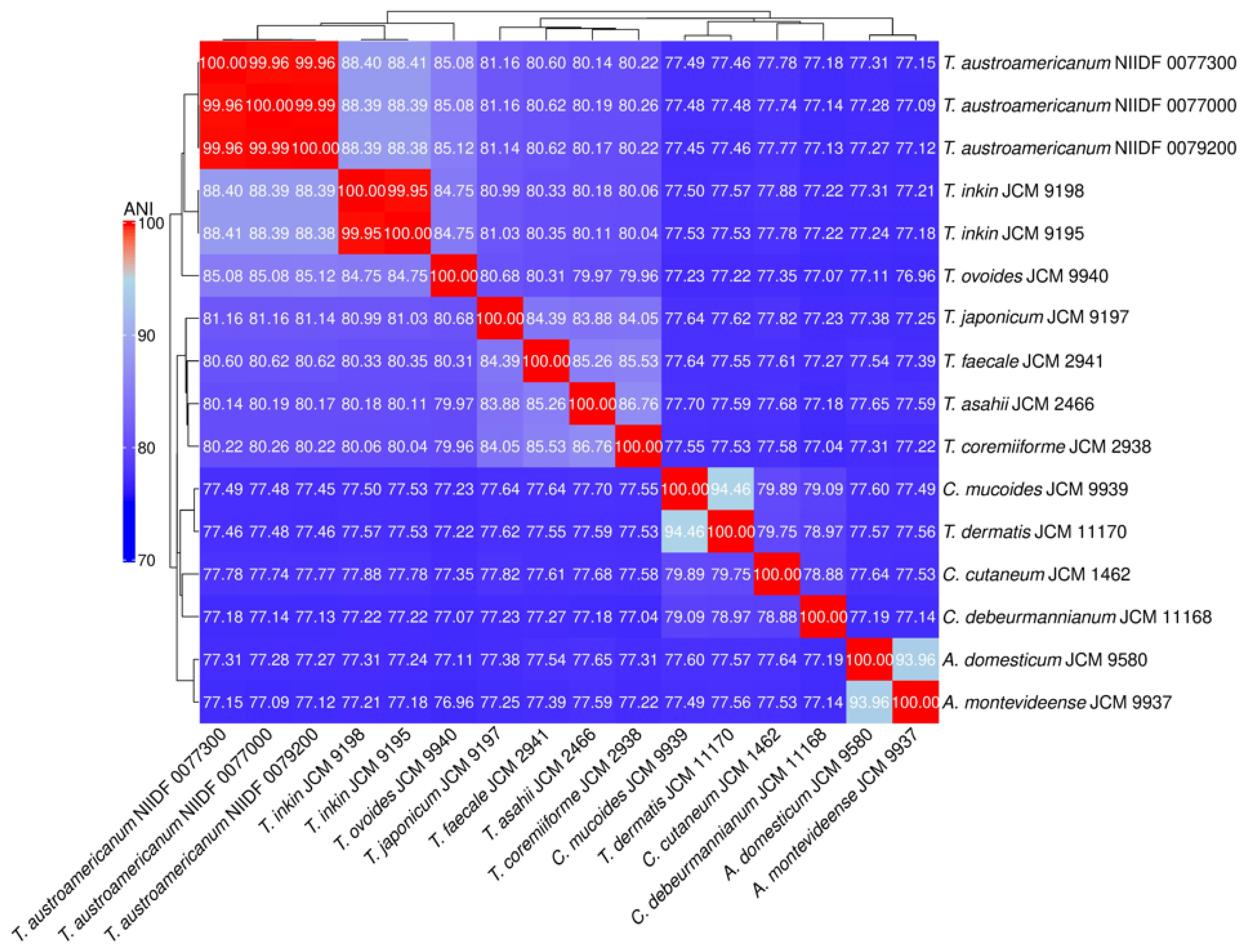
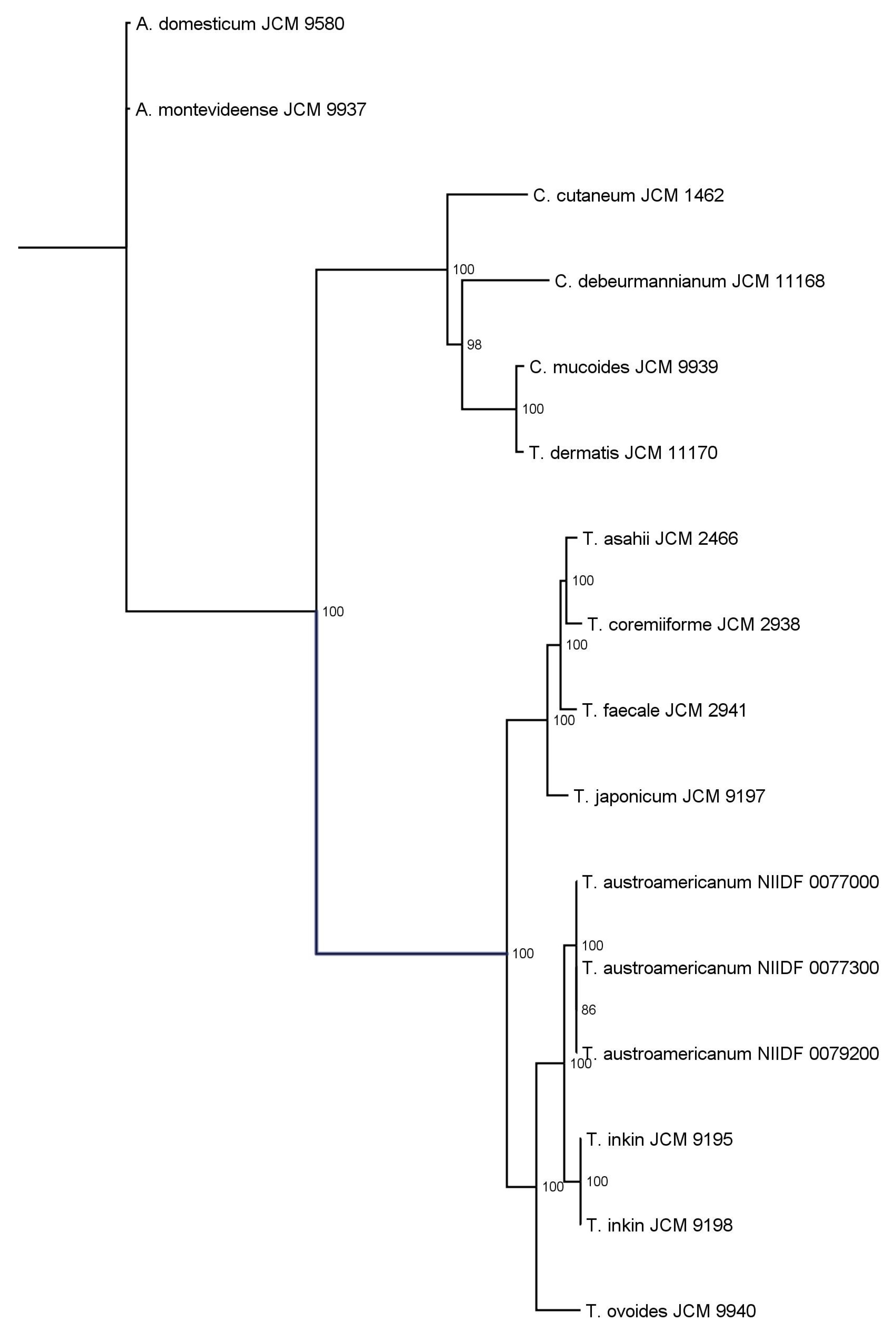
3.2. Phylogenetic Analysis
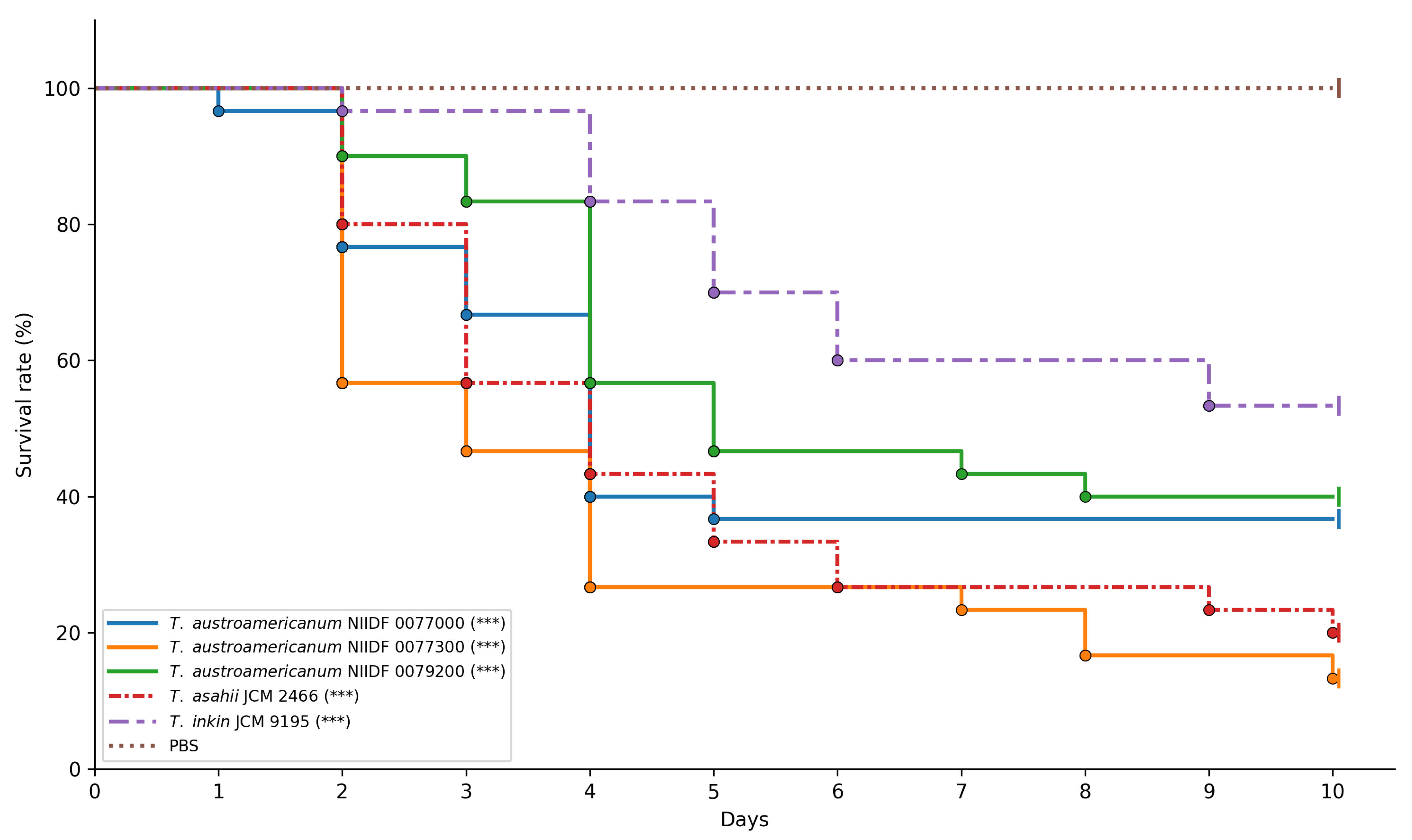
3.3. Virulence Assessment Using Galleria mellonella Infection Model
3.4. Ortholog Analysis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Colombo, A.L.; Padovan, A.C.B.; Chaves, G.M. Current knowledge of Trichosporon spp. and Trichosporonosis. Clin. Microbiol. Rev. 2011, 24, 682–700. [Google Scholar] [CrossRef]
- de Almeida, J.N.; Francisco, E.C.; Ruiz, A.H.; Cuéllar, L.E.; Aquino, V.R.; Mendes, A.V.; Queiroz-Telles, F.; Santos, D.W.; Guimarães, T.; Chaves, G.M.; et al. Epidemiology, Clinical Aspects, Outcomes and Prognostic Factors Associated with Trichosporon Fungaemia: Results of an International Multicentre Study Carried Out at 23 Medical Centres. J. Antimicrob. Chemother. 2021, 76, 1907–1915. [Google Scholar] [CrossRef]
- de Almeida Júnior, J.N.; Hennequin, C. Invasive Trichosporon Infection: A Systematic Review on a Re-emerging Fungal Pathogen. Front. Microbiol. 2016, 7, 1629. [Google Scholar] [CrossRef]
- Francisco, E.C.; de Almeida Junior, J.N.; de Queiroz Telles, F.; Aquino, V.R.; Mendes, A.V.A.; de Andrade Barberino, M.G.M.; de Tarso O Castro, P.; Guimarães, T.; Hahn, R.C.; Padovan, A.C.B.; et al. Species distribution and antifungal susceptibility of 358 Trichosporon clinical isolates collected in 24 medical centres. Clin. Microbiol. Infect. 2019, 25, 909.e1–909.e5. [Google Scholar] [CrossRef]
- Guo, L.N.; Yu, S.Y.; Hsueh, P.R.; Al-Hatmi, A.M.S.; Meis, J.F.; Hagen, F.; Xiao, M.; Wang, H.; Barresi, C.; Zhou, M.L.; et al. Invasive Infections Due to Trichosporon: Species Distribution, Genotyping, and Antifungal Susceptibilities from a Multicenter Study in China. J. Clin. Microbiol. 2019, 57, e01505-18. [Google Scholar] [CrossRef]
- Dabas, Y.; Xess, I.; Kale, P. Molecular and antifungal susceptibility study on trichosporonemia and emergence of Trichosporon mycotoxinivorans as a bloodstream pathogen. Med. Mycol. 2017, 55, 518–527. [Google Scholar] [CrossRef]
- Santos, F.A.; Leite-Andrade, M.C.; Vasconcelos, M.A.; Alves, A.I.; Buonafina-Paz, M.D.; Araújo-Neto, L.N.; Macêdo, D.P.; Neves, R.P. Trichosporon inkin fungemia case report: Clinical and laboratory management. Future Microbiol. 2022, 17, 81–87. [Google Scholar] [CrossRef]
- Rodriguez-Tudela, J.L.; Diaz-Guerra, T.M.; Mellado, E.; Cano, V.; Tapia, C.; Perkins, A.; Gomez-Lopez, A.; Rodero, L.; Cuenca-Estrella, M. Susceptibility patterns and molecular identification of Trichosporon species. Antimicrob. Agents Chemother. 2005, 49, 4026–4034. [Google Scholar] [CrossRef]
- Francisco, E.C.; Desnos-Ollivier, M.; Dieleman, C.; Boekhout, T.; Santos, D.W.C.L.; Medina-Pestana, J.O.; Colombo, A.L.; Hagen, F. Unveiling Trichosporon austroamericanum sp. nov.: A Novel Emerging Opportunistic Basidiomycetous Yeast Species. Mycopathologia 2024, 189, 43. [Google Scholar] [CrossRef]
- Libkind, D.; Čadež, N.; Opulente, D.A.; Langdon, Q.K.; Rosa, C.A.; Sampaio, J.P.; Gonçalves, P.; Hittinger, C.T.; Lachance, M.A. Towards yeast taxogenomics: Lessons from novel species descriptions based on complete genome sequences. FEMS Yeast Res. 2020, 20, foaa042. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Kayamori, A.; Aoki, K.; Shiwa, Y.; Matsutani, M.; Fujita, N.; Sugita, T.; Iwasaki, W.; Tanaka, N.; Takashima, M. Chromosome-level genome assemblies of Cutaneotrichosporon spp. (Trichosporonales, Basidiomycota) reveal imbalanced evolution between nucleotide sequences and chromosome synteny. BMC Genom. 2023, 24, 609. [Google Scholar] [CrossRef]
- Oxford Nanopore Technologies. Dorado: A High-Performance Basecaller for Nanopore Reads. Available online: https://github.com/nanoporetech/dorado (accessed on 5 March 2025).
- Wick, R. Filtlong: Quality Filtering Tool for Nanopore Reads. Available online: https://github.com/rrwick/Filtlong (accessed on 5 March 2025).
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef]
- Oxford Nanopore Technologies. Medaka: Sequence Consensus Polishing Tool. Available online: https://github.com/nanoporetech/medaka (accessed on 5 March 2025).
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Funannotate. Funannotate: Fungal Genome Annotation Pipeline. Available online: https://github.com/nextgenusfs/funannotate (accessed on 5 March 2025).
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: Two open-source ab initio eukaryotic gene finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef]
- Korf, I. Gene finding in novel genomes. Bioinformatics 2004, 5, 59. [Google Scholar] [CrossRef]
- Borodovsky, M.; Lomsadze, A. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 2011, 39, e147. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 7.0: New and improved predictions for detection, regulation, chemical structures, and visualisation. Nucleic Acids Res. 2023, 51, W46–W50. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Käll, L.; Krogh, A.; Sonnhammer, E.L. A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 2004, 338, 1027–1036. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez, R.L.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. Corrigendum to: IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 2461. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods. 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree v1.4.4. Institute of Evolutionary Biology, University of Edinburgh. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 13 May 2025).
- Urban, M.; Cuzick, A.; Seager, J.; Wood, V.; Rutherford, K.; Venkatesh, S.Y.; De Silva, N.; Martinez, M.C.; Pedro, H.; Yates, A.D.; et al. PHI-base: The pathogen–host interactions database. Nucleic Acids Res. 2020, 48, D613–D620. [Google Scholar] [CrossRef] [PubMed]
- Mariné, M.; Bom, V.L.P.; de Castro, P.A.; Winkelstroter, L.K.; Ramalho, L.N.; Brown, N.A.; Goldman, G.H. The development of animal infection models and antifungal efficacy assays against clinical isolates of Trichosporon asahii, T. asteroides and T. inkin. Virulence 2015, 6, 476–486. [Google Scholar] [CrossRef]
- Davidson-Pilon, C. Lifelines: Survival Analysis in Python. Available online: https://lifelines.readthedocs.io (accessed on 24 March 2025).
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- MacPherson, S.; Larochelle, M.; Turcotte, B. A fungal family of transcriptional regulators: The zinc cluster proteins. Microbiol. Mol. Biol. Rev. 2006, 70, 583–604. [Google Scholar] [CrossRef]
- Pla, J.; Gil, C.; Monteoliva, L.; Navarro-García, F.; Sánchez, M.; Nombela, C. Understanding Candida albicans at the molecular level. Yeast 1996, 12, 1677–1702. [Google Scholar] [CrossRef]
- Leberer, E.; Ziegelbauer, K.; Schmidt, A.; Harcus, D.; Dignard, D.; Ash, J.; Johnson, L.; Thomas, D.Y. Virulence and hyphal formation of Candida albicans require the Ste20p-like protein kinase CaCla4p. Curr. Biol. 1997, 7, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.J.; Köhler, J.R.; DiDomenico, B.; Loebenberg, D.; Cacciapuoti, A.; Fink, G.R. Nonfilamentous Candida albicans mutants are avirulent. Cell 1997, 90, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Giammarino, A.; Bellucci, N.; Angiolella, L. Galleria mellonella as a Model for the Study of Fungal Pathogens: Advantages and Disadvantages. Pathogens 2024, 13, 233. [Google Scholar] [CrossRef]
- Serrano, I.; Verdial, C.; Tavares, L.; Oliveira, M. The Virtuous Galleria mellonella Model for Scientific Experimentation. Antibiotics 2023, 12, 505. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Strains | Strain ID | Source |
|---|---|---|
| Trichosporon austroamericanum | NIIDF 0077000 | human blood |
| Trichosporon austroamericanum | NIIDF 0077300 | human blood |
| Trichosporon austroamericanum | NIIDF 0079200 | human blood |
| Trichosporon inkin | JCM 9195 | human skin |
| Trichosporon inkin | JCM 9198 | surgical wound |
| Trichosporon ovoides | JCM 9940 | scalp |
| Trichosporon asahii | JCM 2466 | human skin |
| Trichosporon coremiiforme | JCM 2938 | lesion on farmer head |
| Trichosporon dermatis | JCM 11170 | human skin |
| Trichosporon faecale | JCM 2941 | human feces |
| Trichosporon japonicum | JCM 9197 | unknown |
| Apiotrichum domesticum | JCM 9580 | damp and rotten wooden sideboard |
| Apiotrichum montevideense | JCM 9937 | water purification tank |
| Cutaneotrichosporon cutaneum | JCM 1462 | unknown |
| Cutaneotrichosporon debeurmannianum | JCM 11168 | human bronchial secretion |
| Cutaneotrichosporon mucoides | JCM 9939 | case of meningitis |
| Strains | Scaffolds | Total Length (Mb) | Largest Scaffold (Mb) | GC (%) | N50 (Mb) | Repeat Sequences (%) |
|---|---|---|---|---|---|---|
| T. austroamericanum NIIDF 0077000 | 9 | 21.02 | 3.93 | 50.03 | 3.19 | 19.37 |
| T. austroamericanum NIIDF 0077300 | 10 | 21.05 | 5.69 | 50.01 | 3.88 | 19.39 |
| T. austroamericanum NIIDF 0079200 | 9 | 21.06 | 3.92 | 50.04 | 3.41 | 19.32 |
| T. inkin JCM 9195 | 36 | 20.36 | 3.66 | 50.01 | 2.71 | 18.62 |
| T. inkin JCM 9198 | 36 | 20.44 | 4.50 | 50.02 | 2.74 | 18.68 |
| T. ovoides JCM 9940 | 55 | 40.40 | 3.19 | 50.01 | 1.87 | 19.89 |
| T. asahii JCM 2466 | 40 | 24.82 | 4.04 | 49.99 | 2.29 | 20.28 |
| T. coremiiforme JCM 2938 | 153 | 43.00 | 1.53 | 50.00 | 0.91 | 20.13 |
| T. dermatis JCM 11170 | 24 | 23.42 | 3.89 | 49.90 | 2.78 | 20.01 |
| T. faecale JCM 2941 | 58 | 24.81 | 2.90 | 50.06 | 1.37 | 19.84 |
| T. japonicum JCM 9197 | 28 | 23.55 | 4.60 | 50.07 | 3.28 | 18.57 |
| A. domesticum JCM 9580 | 20 | 24.65 | 7.92 | 49.98 | 3.53 | 20.77 |
| A. montevideense JCM 9937 | 24 | 25.18 | 3.76 | 49.96 | 2.17 | 20.87 |
| C. cutaneum JCM 1462 | 122 | 24.22 | 1.65 | 50.06 | 0.37 | 18.73 |
| C. debeurmannianum JCM 11168 | 28 | 19.15 | 3.80 | 49.92 | 2.97 | 20.34 |
| C. mucoides JCM 9939 | 36 | 42.75 | 4.21 | 49.96 | 3.35 | 19.90 |
| Strains | Complete (%) | Single-Copy (%) | Duplicated (%) | Fragmented (%) | Missing (%) |
|---|---|---|---|---|---|
| T. austroamericanum NIIDF 0077000 | 91.4 | 91.3 | 0.1 | 2.8 | 5.8 |
| T. austroamericanum NIIDF 0077300 | 91.3 | 91.2 | 0.1 | 2.9 | 5.8 |
| T. austroamericanum NIIDF 0079200 | 90.3 | 90.2 | 0.1 | 2.8 | 6.5 |
| T. inkin JCM 9195 | 88.3 | 87.9 | 0.4 | 4.1 | 7.6 |
| T. inkin JCM 9198 | 88.3 | 87.9 | 0.4 | 4.1 | 7.6 |
| T. ovoides JCM 9940 | 95.3 | 24.7 | 70.6 | 1.1 | 3.6 |
| T. asahii JCM 2466 | 88.4 | 87.7 | 0.7 | 4.8 | 6.8 |
| T. coremiiforme JCM 2938 | 91.0 | 23.4 | 67.6 | 3.4 | 5.6 |
| T. dermatis JCM 11170 | 92.1 | 91.2 | 0.9 | 2.8 | 5.1 |
| T. faecale JCM 2941 | 88.3 | 87.7 | 0.6 | 4.9 | 6.8 |
| T. japonicum JCM 9197 | 88.2 | 87.7 | 0.5 | 4.8 | 7.0 |
| A. domesticum JCM 9580 | 89.0 | 88.2 | 0.8 | 2.7 | 8.3 |
| A. montevideense JCM 9937 | 88.7 | 88.0 | 0.7 | 2.8 | 8.5 |
| C. cutaneum JCM 1462 | 93.2 | 92.5 | 0.7 | 1.7 | 5.1 |
| C. debeurmannianum JCM 11168 | 91.0 | 90.4 | 0.6 | 2.9 | 6.1 |
| C. mucoides JCM 9939 | 93.9 | 30.0 | 63.9 | 1.9 | 4.2 |
| Strains | Genes | InterProScan (%) | eggNOG (%) | Pfam (%) | CAZyme (%) | MEROPS (%) | Phobius (%) |
|---|---|---|---|---|---|---|---|
| T. austroamericanum NIIDF 0077000 | 7427 | 5674 (76.4%) | 6206 (83.6%) | 4974 (67.0%) | 185 (2.5%) | 232 (3.1%) | 517 (7.0%) |
| T. austroamericanum NIIDF 0077300 | 7442 | 5653 (76.0%) | 6202 (83.3%) | 4959 (66.6%) | 185 (2.5%) | 232 (3.1%) | 511 (6.9%) |
| T. austroamericanum NIIDF 0079200 | 7574 | 5736 (75.7%) | 6291 (83.1%) | 5037 (66.5%) | 187 (2.5%) | 231 (3.0%) | 528 (7.0%) |
| T. inkin JCM 9195 | 7325 | 5715 (78.0%) | 6267 (85.6%) | 5031 (68.7%) | 184 (2.5%) | 230 (3.1%) | 494 (6.7%) |
| T. inkin JCM 9198 | 7178 | 5612 (78.2%) | 6149 (85.7%) | 4913 (68.4%) | 182 (2.5%) | 228 (3.2%) | 485 (6.8%) |
| T. ovoides JCM 9940 | 14321 | 10864 (75.9%) | 11193 (78.2%) | 9494 (66.3%) | 348 (2.4%) | 440 (3.1%) | 943 (6.6%) |
| T. asahii JCM 2466 | 8530 | 5932 (69.5%) | 6530 (76.6%) | 5143 (60.3%) | 194 (2.3%) | 245 (2.9%) | 639 (7.5%) |
| T. coremiiforme JCM 2938 | 15235 | 10972 (72.0%) | 12053 (79.1%) | 9533 (62.6%) | 350 (2.3%) | 459 (3.0%) | 1063 (7.0%) |
| T. dermatis JCM 11170 | 8285 | 6246 (75.4%) | 6816 (82.3%) | 5469 (66.0%) | 233 (2.8%) | 270 (3.3%) | 540 (6.5%) |
| T. faecale JCM 2941 | 8774 | 5950 (67.8%) | 6539 (74.5%) | 5147 (58.7%) | 191 (2.2%) | 242 (2.8%) | 681 (7.8%) |
| T. japonicum JCM 9197 | 8199 | 5874 (71.6%) | 6405 (78.1%) | 5096 (62.2%) | 196 (2.4%) | 248 (3.0%) | 652 (8.0%) |
| A. domesticum JCM 9580 | 7975 | 5871 (73.6%) | 6430 (80.6%) | 5148 (64.6%) | 208 (2.6%) | 244 (3.1%) | 613 (7.7%) |
| A. montevideense JCM 9937 | 14315 | 10887 (76.1%) | 11874 (82.9%) | 9560 (66.8%) | 403 (2.8%) | 466 (3.3%) | 959 (6.7%) |
| C. cutaneum JCM 1462 | 8982 | 6315 (70.3%) | 6903 (76.9%) | 5524 (61.5%) | 202 (2.2%) | 296 (3.3%) | 597 (6.6%) |
| C. debeurmannianum JCM 11168 | 6946 | 5553 (79.9%) | 6069 (87.4%) | 4897 (70.5%) | 171 (2.5%) | 233 (3.4%) | 380 (5.5%) |
| C. mucoides JCM 9939 | 14315 | 10,887 (76.1%) | 11,874 (82.9%) | 9560 (66.8%) | 403 (2.8%) | 466 (3.3%) | 959 (6.7%) |
| Comparison | Adjusted p-Value |
|---|---|
| T. asahii JCM 2466 vs. T. austroamericanum NIIDF 0077000 | 1.0000 |
| T. asahii JCM 2466 vs. T. austroamericanum NIIDF 0077300 | 1.0000 |
| T. asahii JCM 2466 vs. T. austroamericanum NIIDF 0079200 | 0.9312 |
| T. asahii JCM 2466 vs. T. inkin JCM 9195 | 0.0154 |
| T. inkin JCM 9195 vs. T. austroamericanum NIIDF 0077000 | 0.5687 |
| T. inkin JCM 9195 vs. T. austroamericanum NIIDF 0077300 | 0.0005 |
| T. inkin JCM 9195 vs. T. austroamericanum NIIDF 0079200 | 1.0000 |
| T. austroamericanum NIIDF 0077000 vs. T. austroamericanum NIIDF 0077300 | 0.7089 |
| T. austroamericanum NIIDF 0077000 vs. T. austroamericanum NIIDF 0079200 | 1.0000 |
| T. austroamericanum NIIDF 0077300 vs. T. austroamericanum NIIDF 0079200 | 0.0707 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horiguchi, T.; Umeyama, T.; Tomuro, H.; Otani, A.; Shinohara, T.; Abe, M.; Takatsuka, S.; Miyazawa, K.; Nagi, M.; Muraosa, Y.; et al. Whole-Genome Sequencing and Comparative Genomic Analysis of Three Clinical Bloodstream Infection Isolates of Trichosporon austroamericanum. J. Fungi 2025, 11, 401. https://doi.org/10.3390/jof11050401
Horiguchi T, Umeyama T, Tomuro H, Otani A, Shinohara T, Abe M, Takatsuka S, Miyazawa K, Nagi M, Muraosa Y, et al. Whole-Genome Sequencing and Comparative Genomic Analysis of Three Clinical Bloodstream Infection Isolates of Trichosporon austroamericanum. Journal of Fungi. 2025; 11(5):401. https://doi.org/10.3390/jof11050401
Chicago/Turabian StyleHoriguchi, Takanori, Takashi Umeyama, Hiroko Tomuro, Amato Otani, Takayuki Shinohara, Masahiro Abe, Shogo Takatsuka, Ken Miyazawa, Minoru Nagi, Yasunori Muraosa, and et al. 2025. "Whole-Genome Sequencing and Comparative Genomic Analysis of Three Clinical Bloodstream Infection Isolates of Trichosporon austroamericanum" Journal of Fungi 11, no. 5: 401. https://doi.org/10.3390/jof11050401
APA StyleHoriguchi, T., Umeyama, T., Tomuro, H., Otani, A., Shinohara, T., Abe, M., Takatsuka, S., Miyazawa, K., Nagi, M., Muraosa, Y., Hoshino, Y., Sakoh, T., Araoka, H., Uchida, N., Kaneko, T., Nagano, Y., Tsukada, H., Miyazaki, T., & Miyazaki, Y. (2025). Whole-Genome Sequencing and Comparative Genomic Analysis of Three Clinical Bloodstream Infection Isolates of Trichosporon austroamericanum. Journal of Fungi, 11(5), 401. https://doi.org/10.3390/jof11050401