
Genomic and Multi-Omics Analysis of Phlebopus portentosus: Effects of Cultivation on Secondary Metabolites


Abstract
1. Introduction
2. Materials and Methods
2.1. Source of Strains and Culture Media
2.2. Genome Sequencing
2.3. Gene Prediction and Annotation
2.4. Additional Annotation
2.5. Secondary Metabolite Biosynthesis Gene Cluster Analysis
2.6. Cluster Analysis
2.7. Prediction of TPS Proteins
2.8. Transcription Factor Identification Analysis
2.9. Differentially Expressed Metabolites and Enrichment Analysis
2.10. Transcriptome Sequencing and Differential Gene Expression Analysis
2.11. Prediction of Transcription Factor Binding Sites
2.12. Quantitative Real-Time PCR Analysis
3. Results
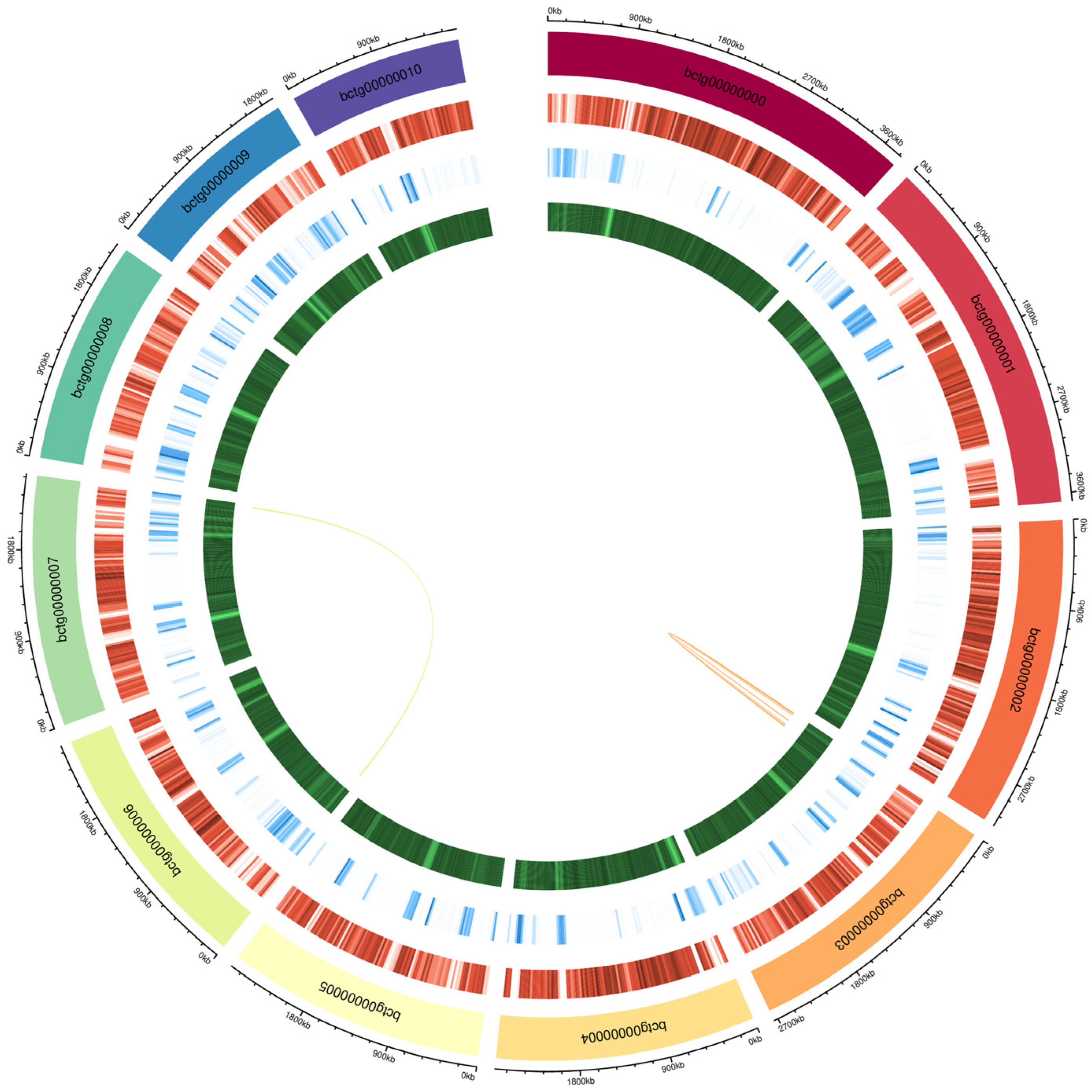
3.1. Genome Assembly
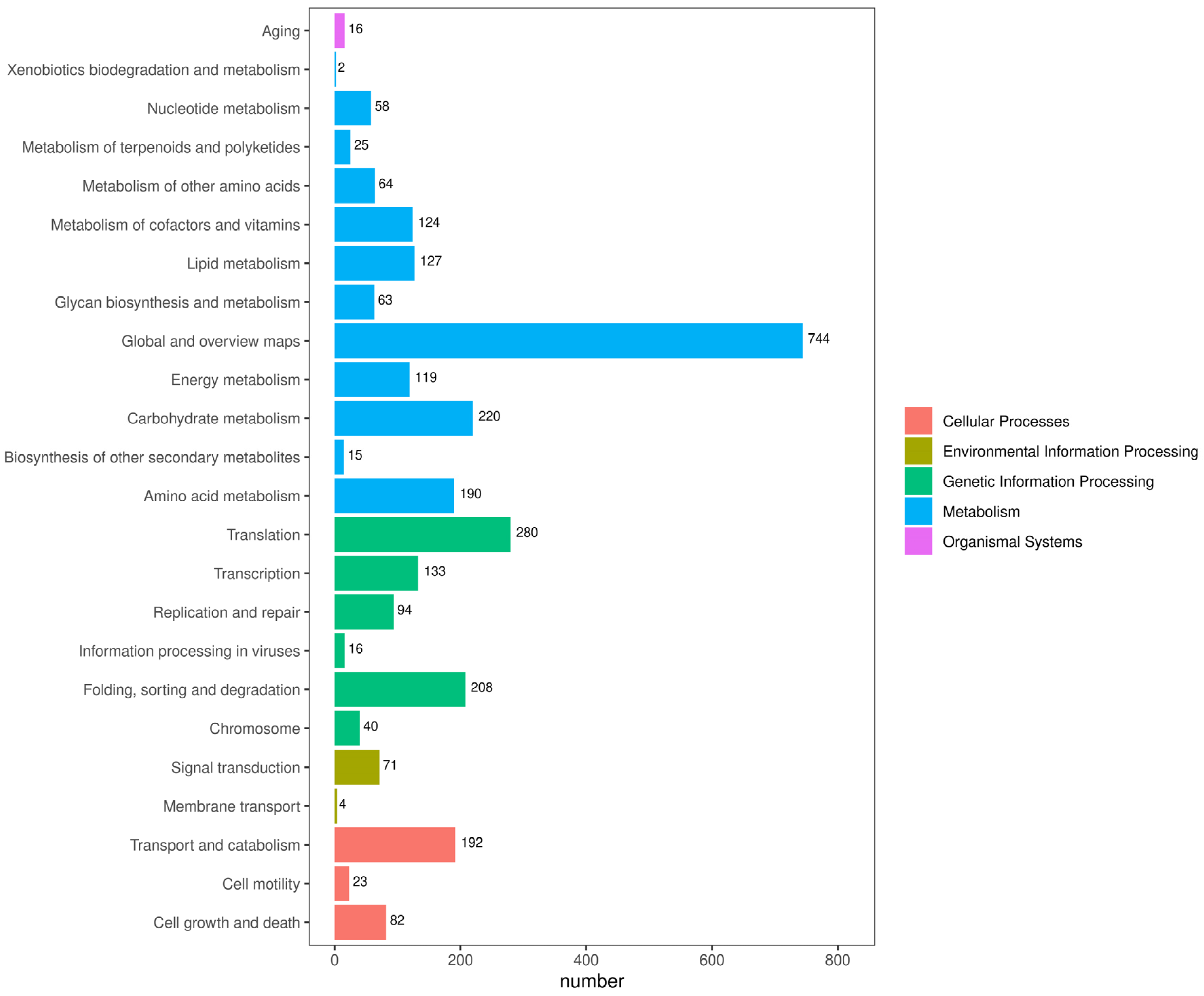
3.2. Genome Annotation
3.3. Extended Annotation
3.3.1. Carbohydrate Enzyme Annotation
3.3.2. Virulence Factor (VFDB) Analysis
3.3.3. Pathogen–Host Interaction (PHI) Analysis
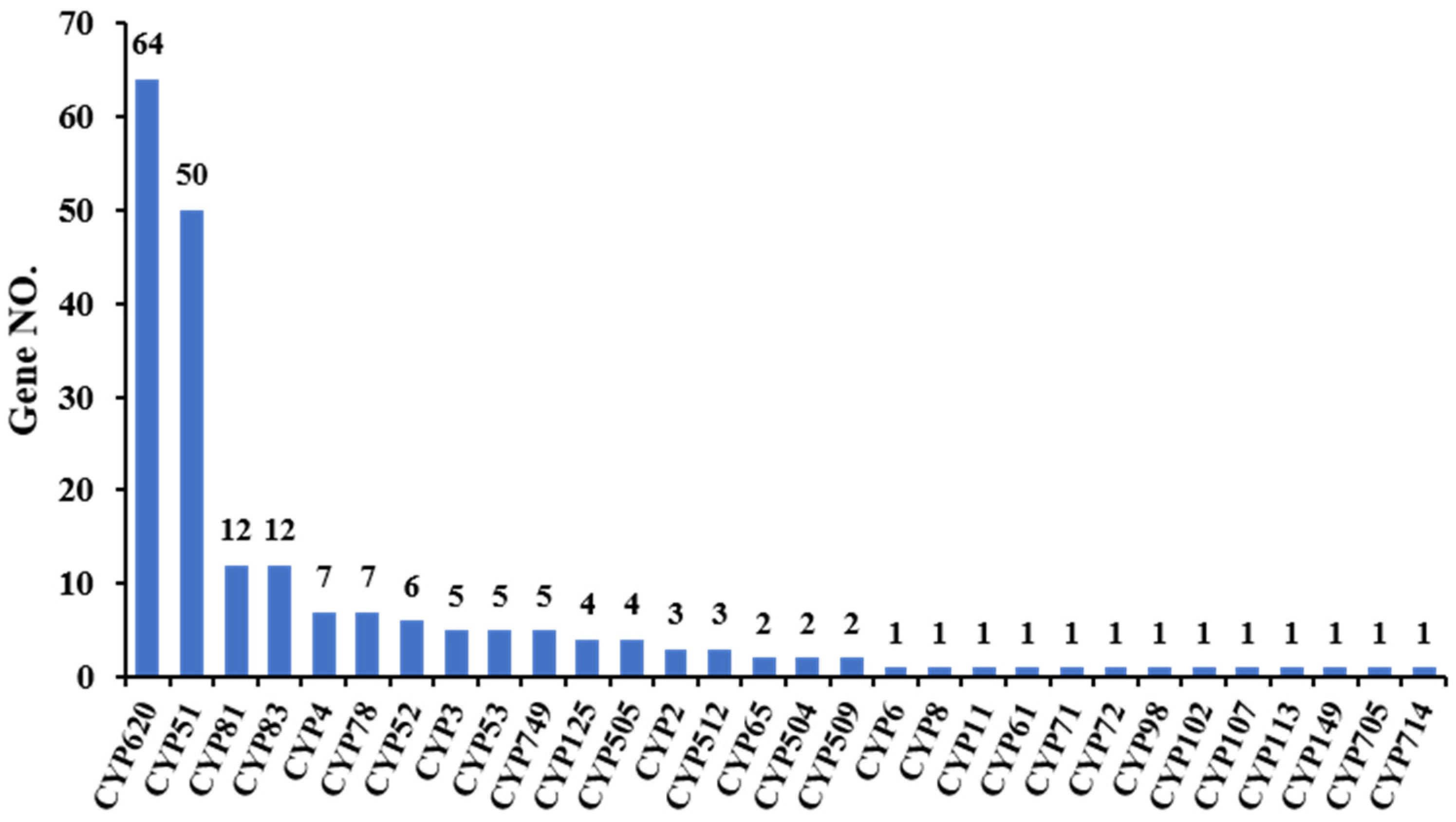
3.3.4. Cytochrome P450 Annotation
3.4. Metabolome Analysis of Different Cultivation Conditions
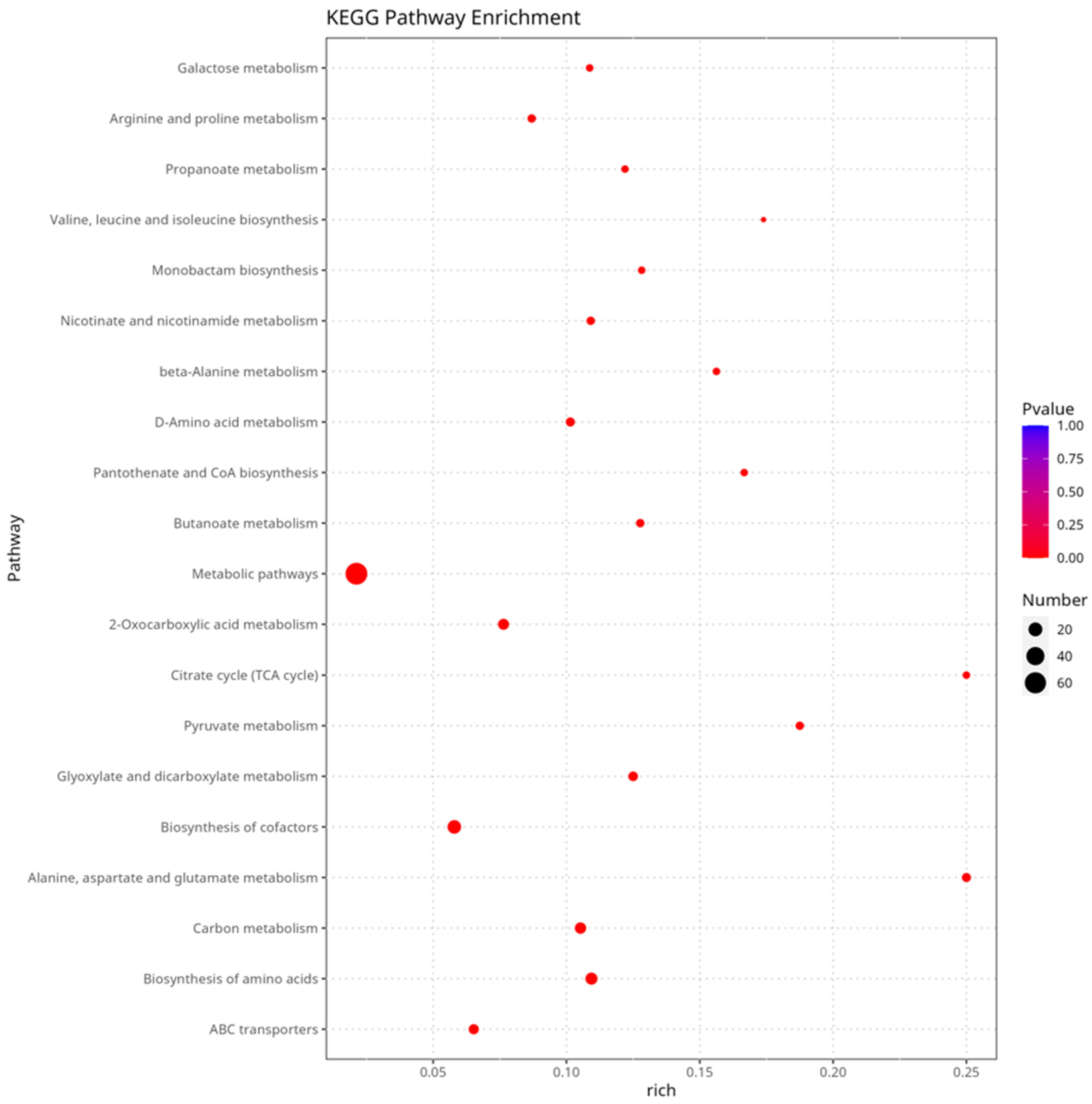
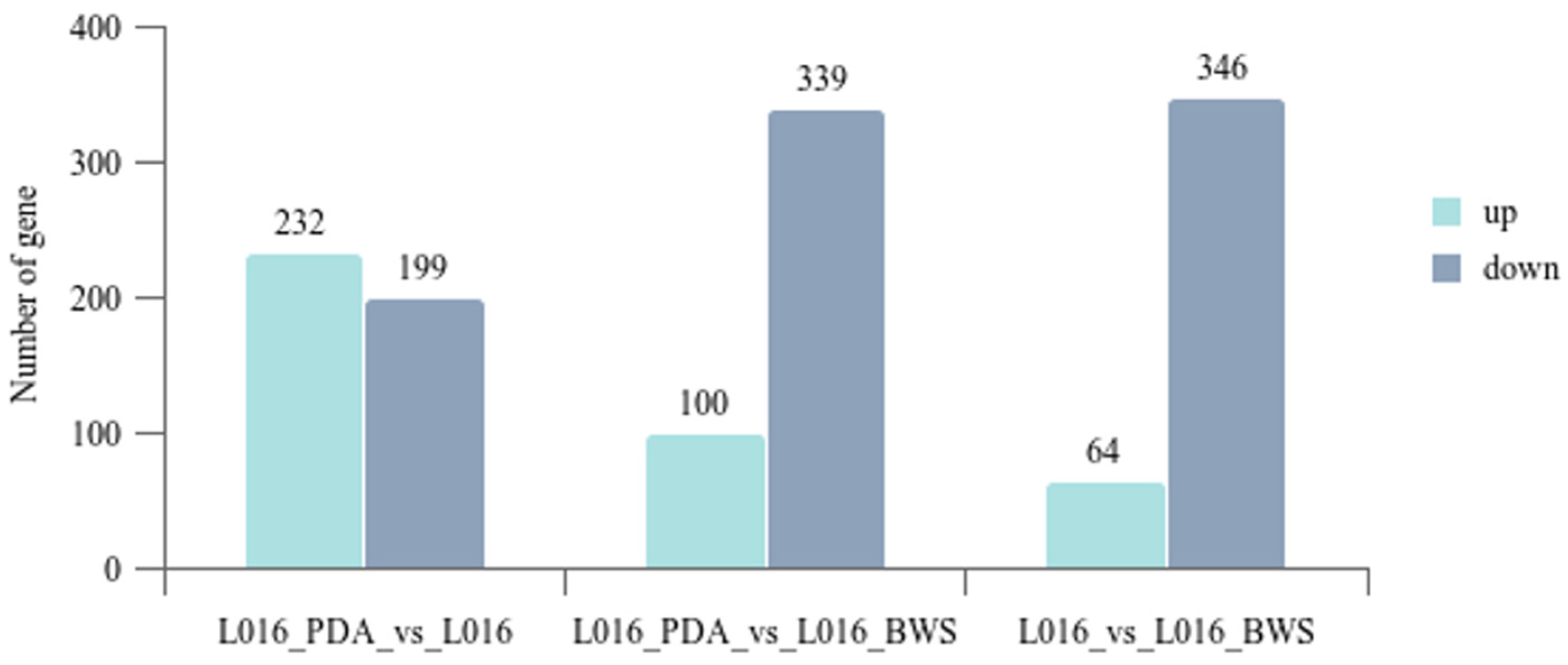
3.5. Transcriptome Analysis of Different Cultivation Conditions
3.6. Analysis of Biosynthetic Gene Clusters
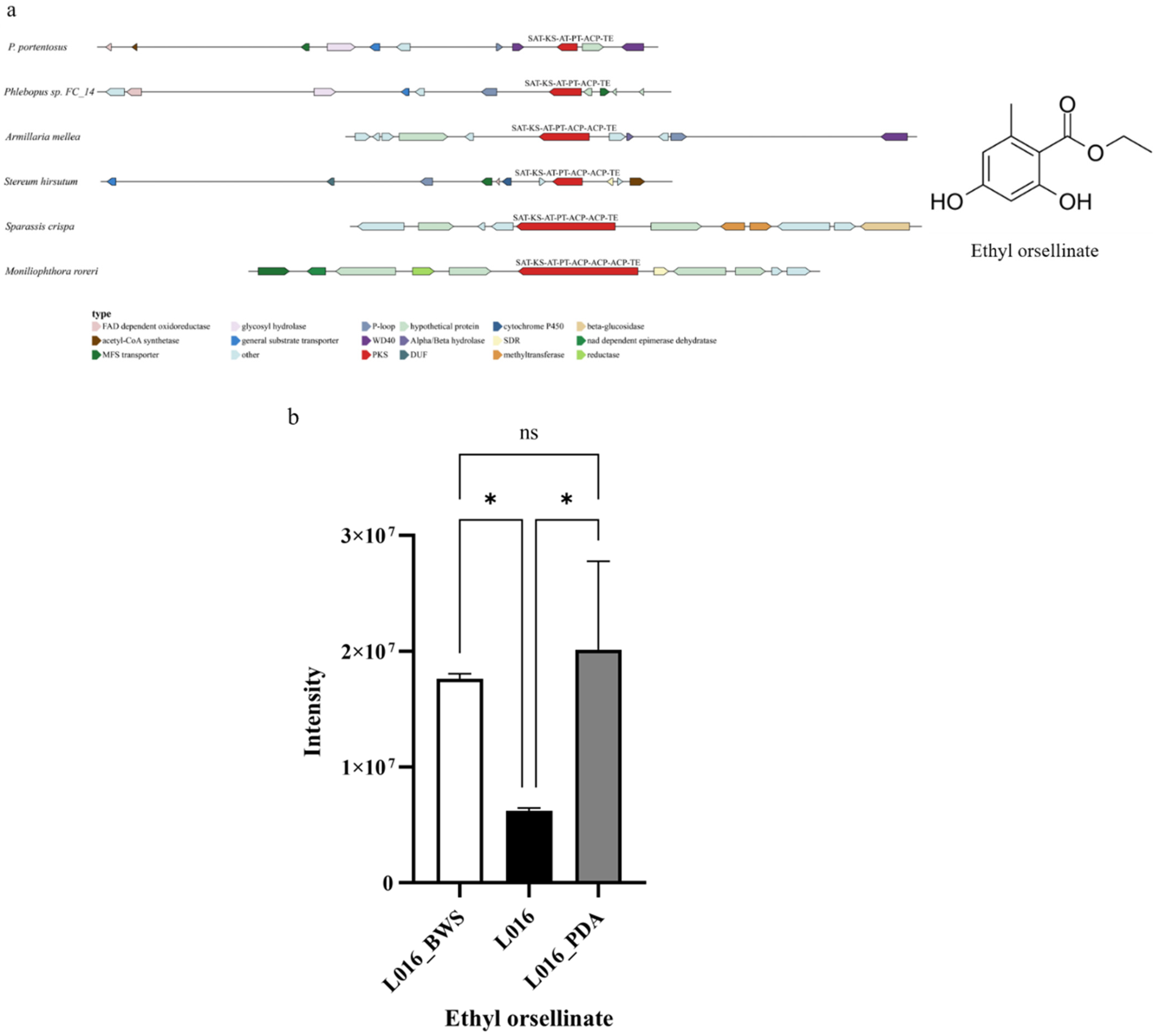
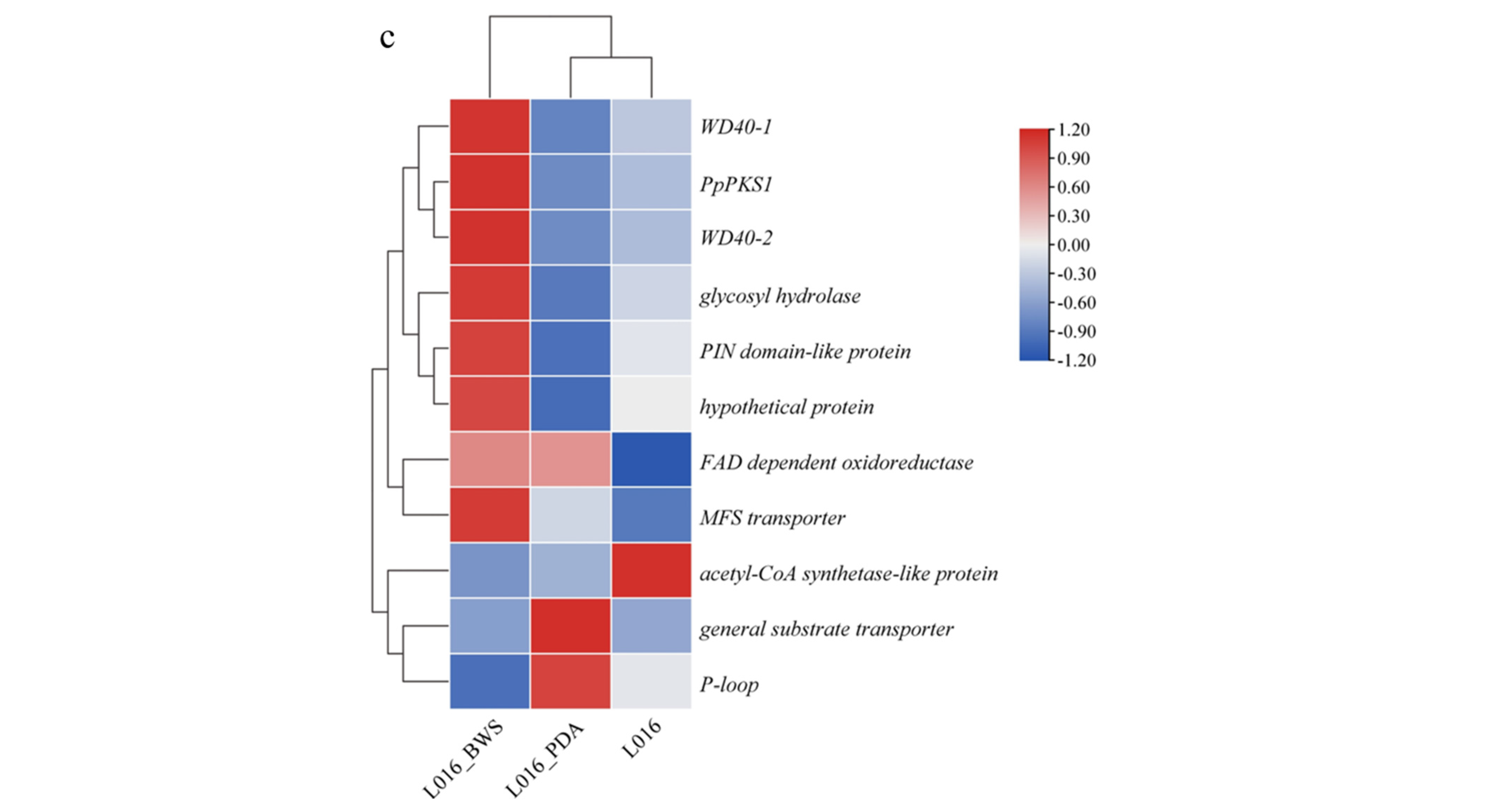
3.6.1. Ethyl Orsellinate Biosynthesis Gene in P. portentosus
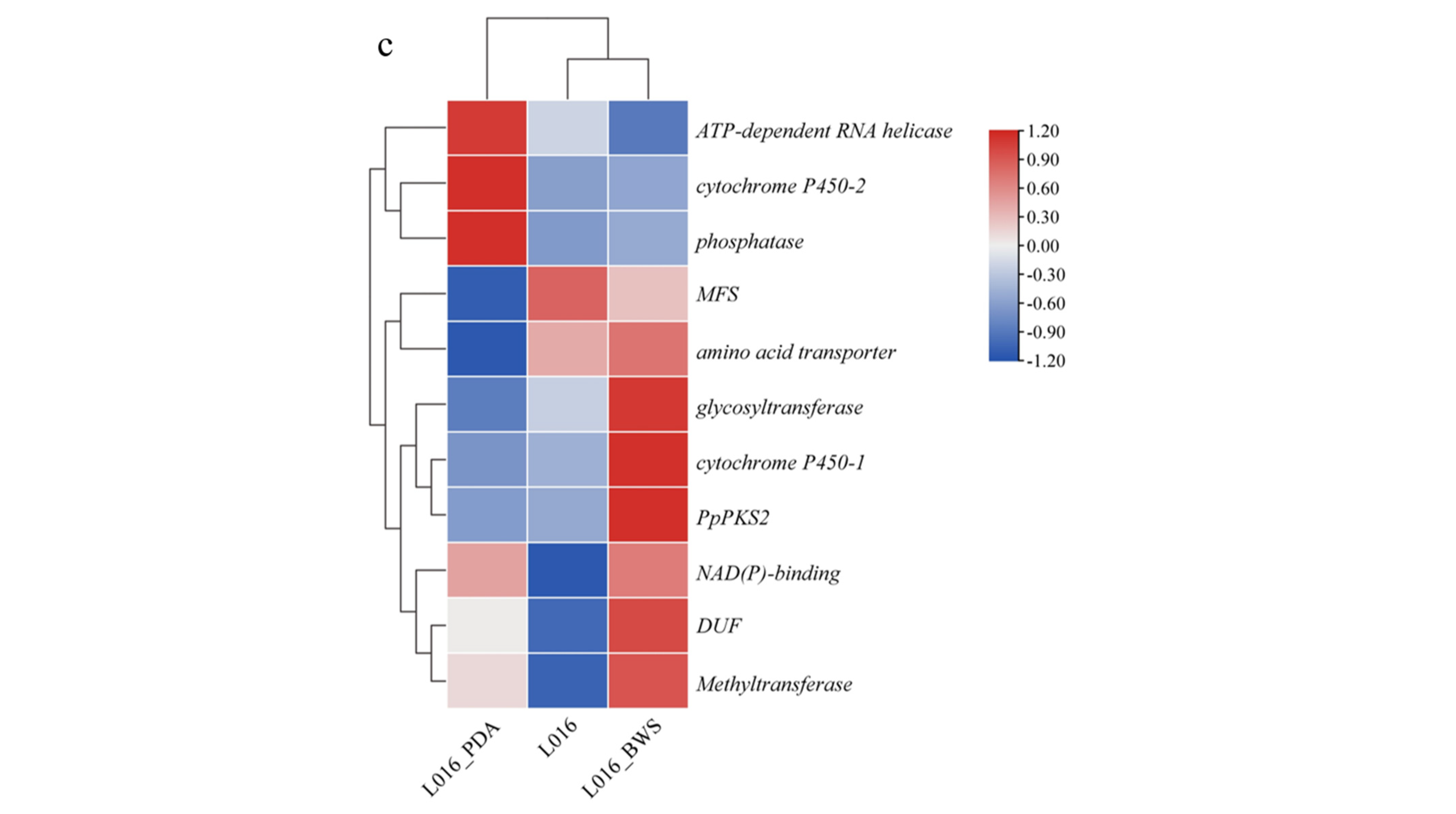
3.6.2. 6-Methylsalicylic Acid Biosynthesis Gene in P. portentosus
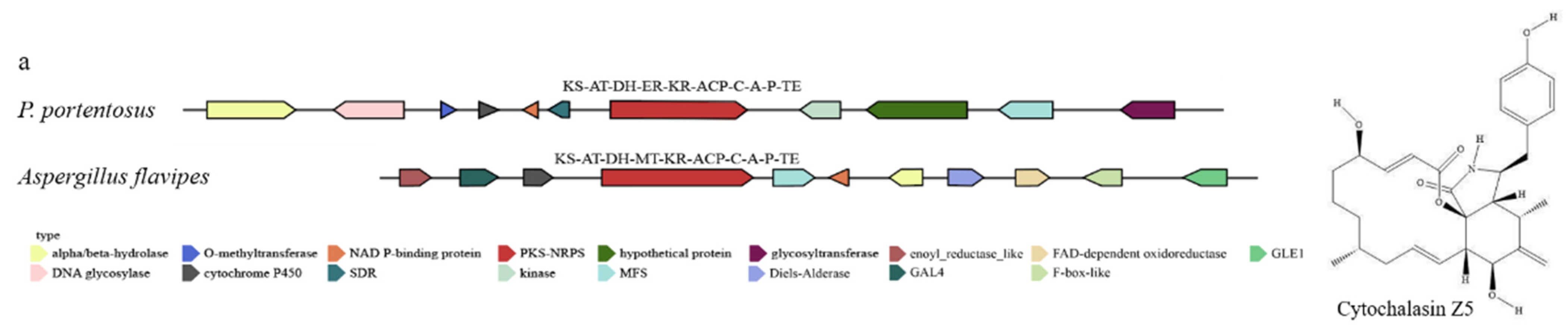
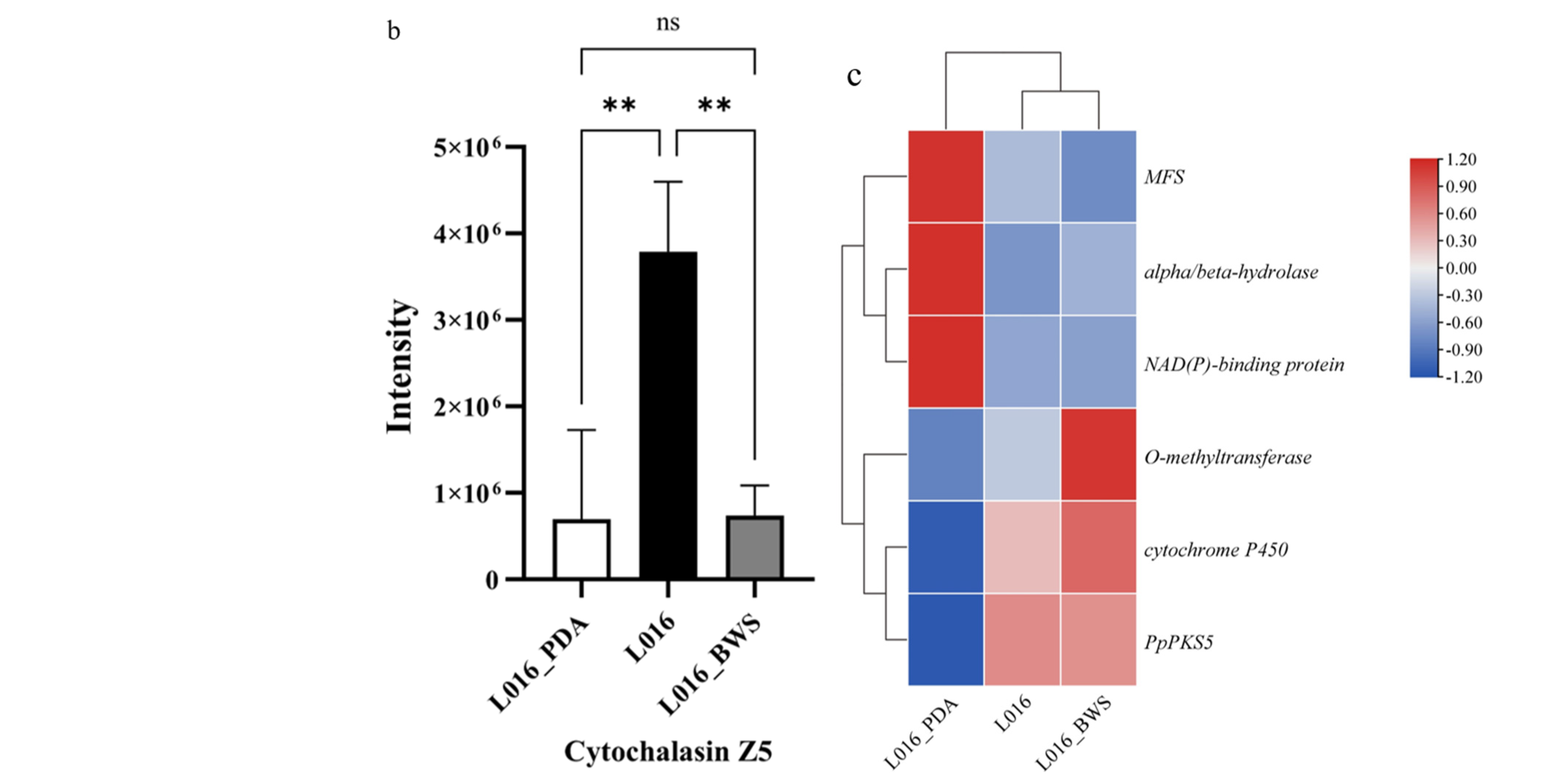
3.6.3. Cytochalasin Z5 Biosythesis Gene in P. portentosus
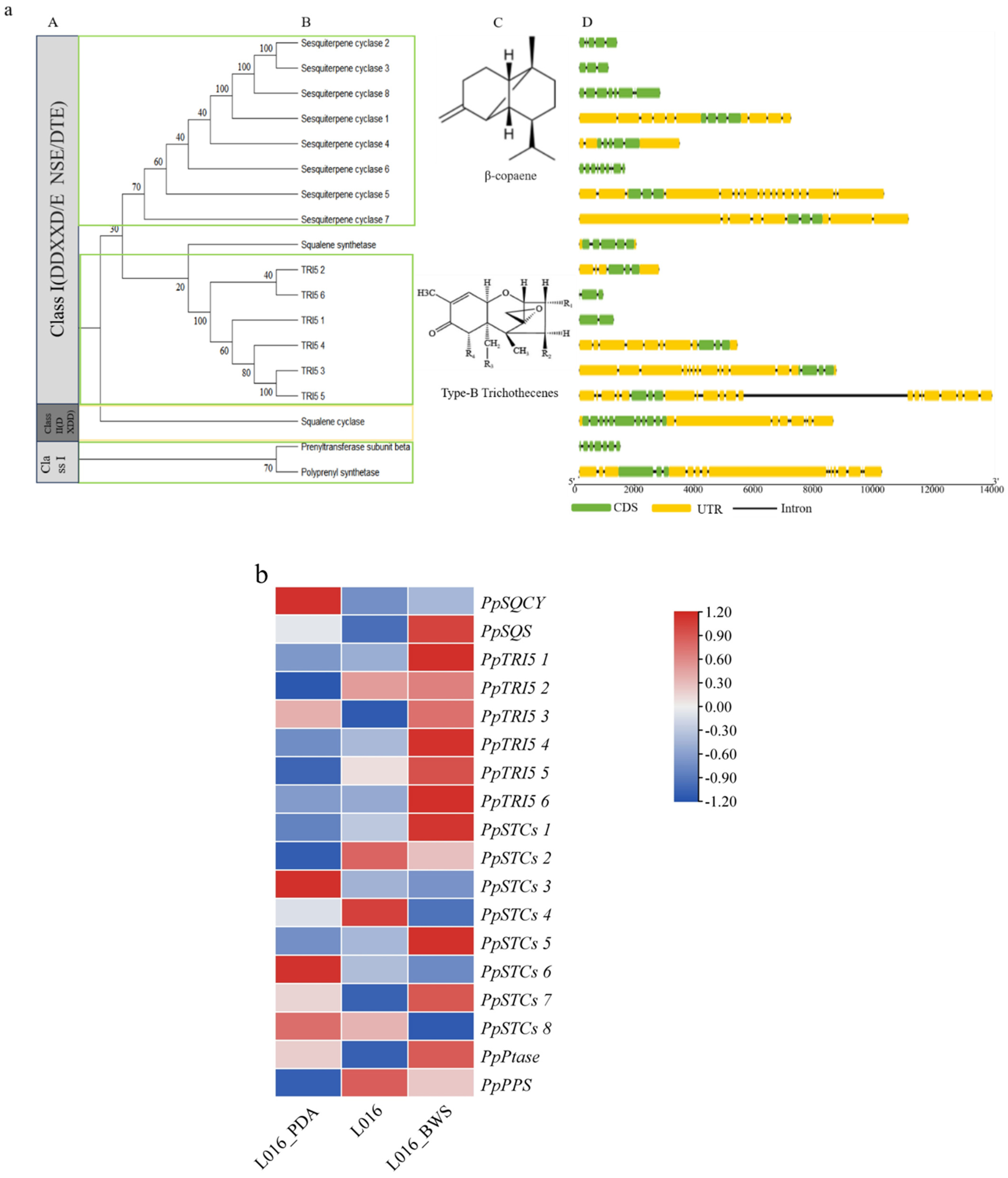
3.7. Characterization of PpTPS Proteins
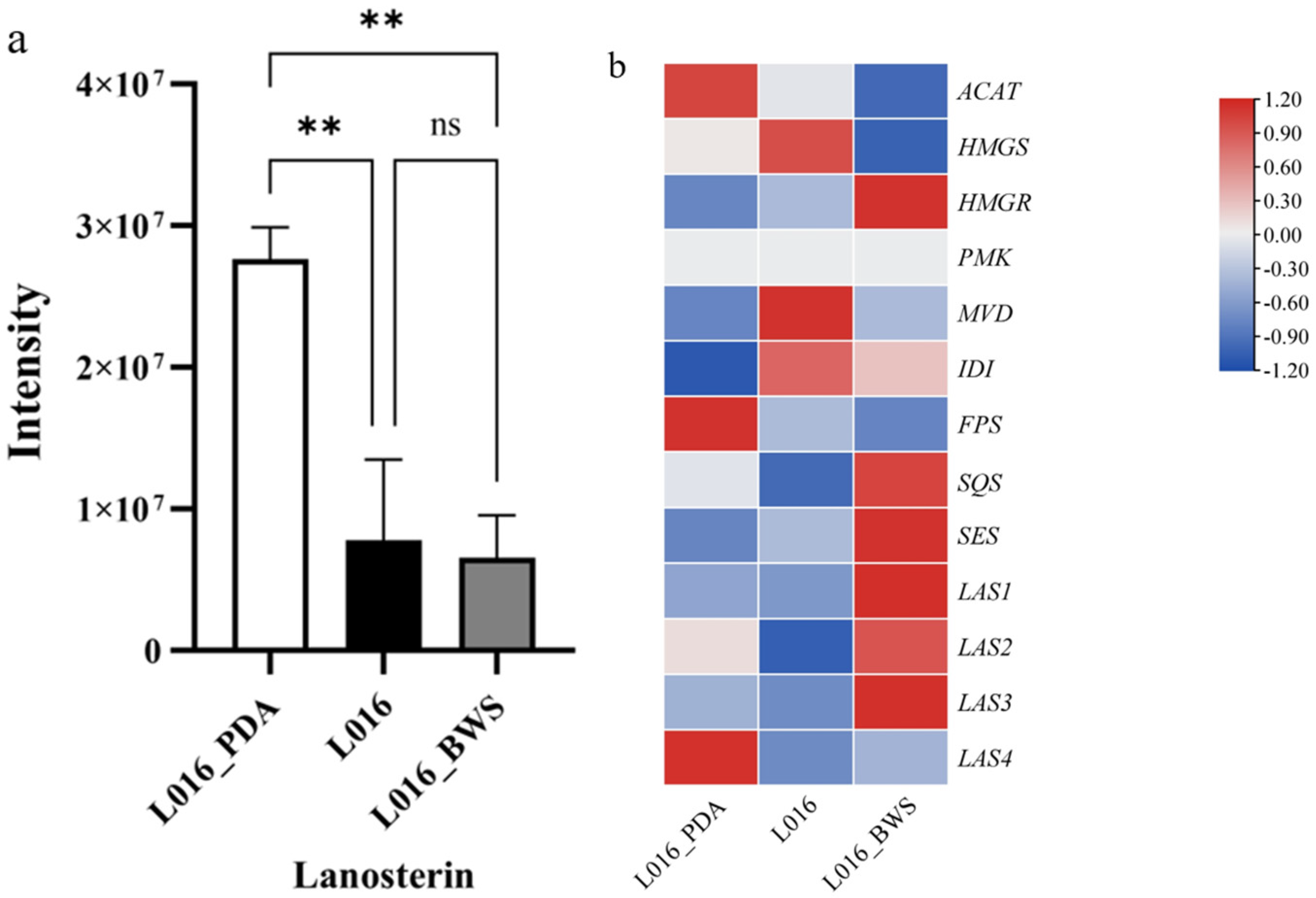
3.8. Analysis of Lanosterin Transcriptome and Metabolome Expression Under Different Cultivation Conditions
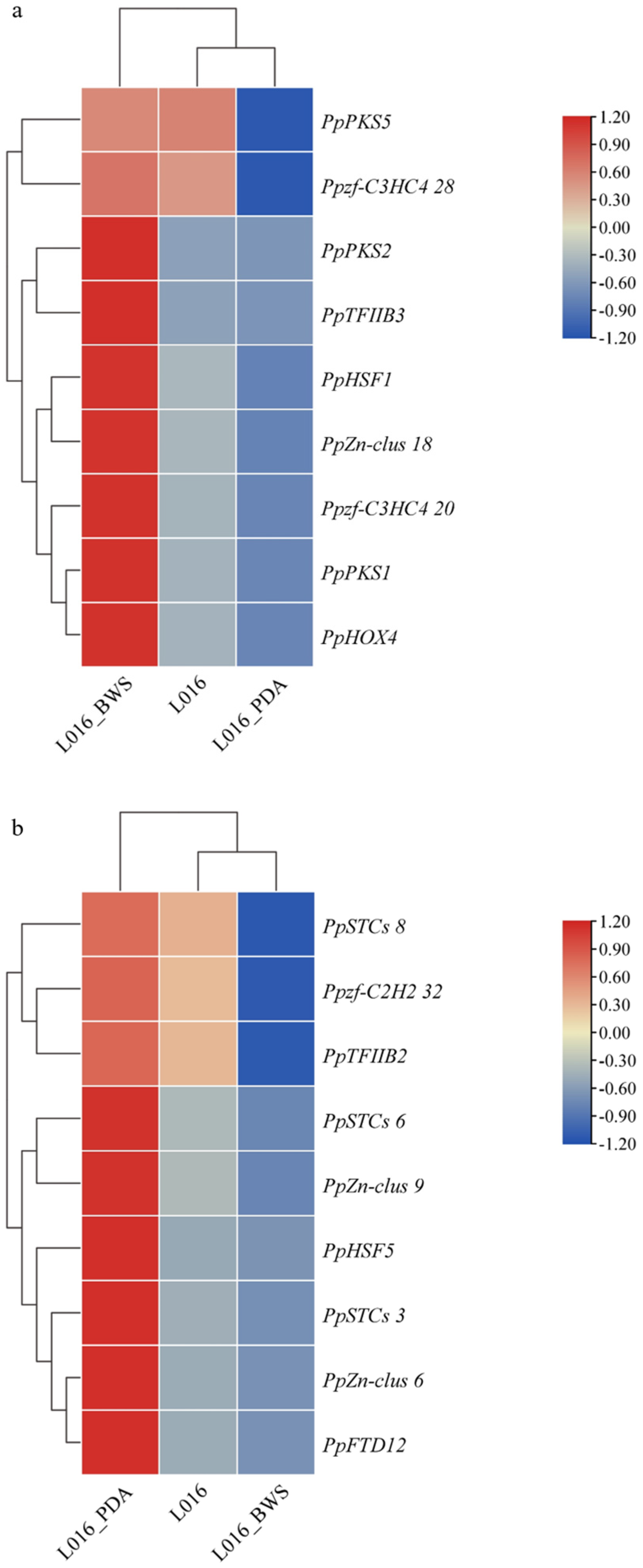
3.9. Analysis of Gene Expression and Prediction of Binding Sites
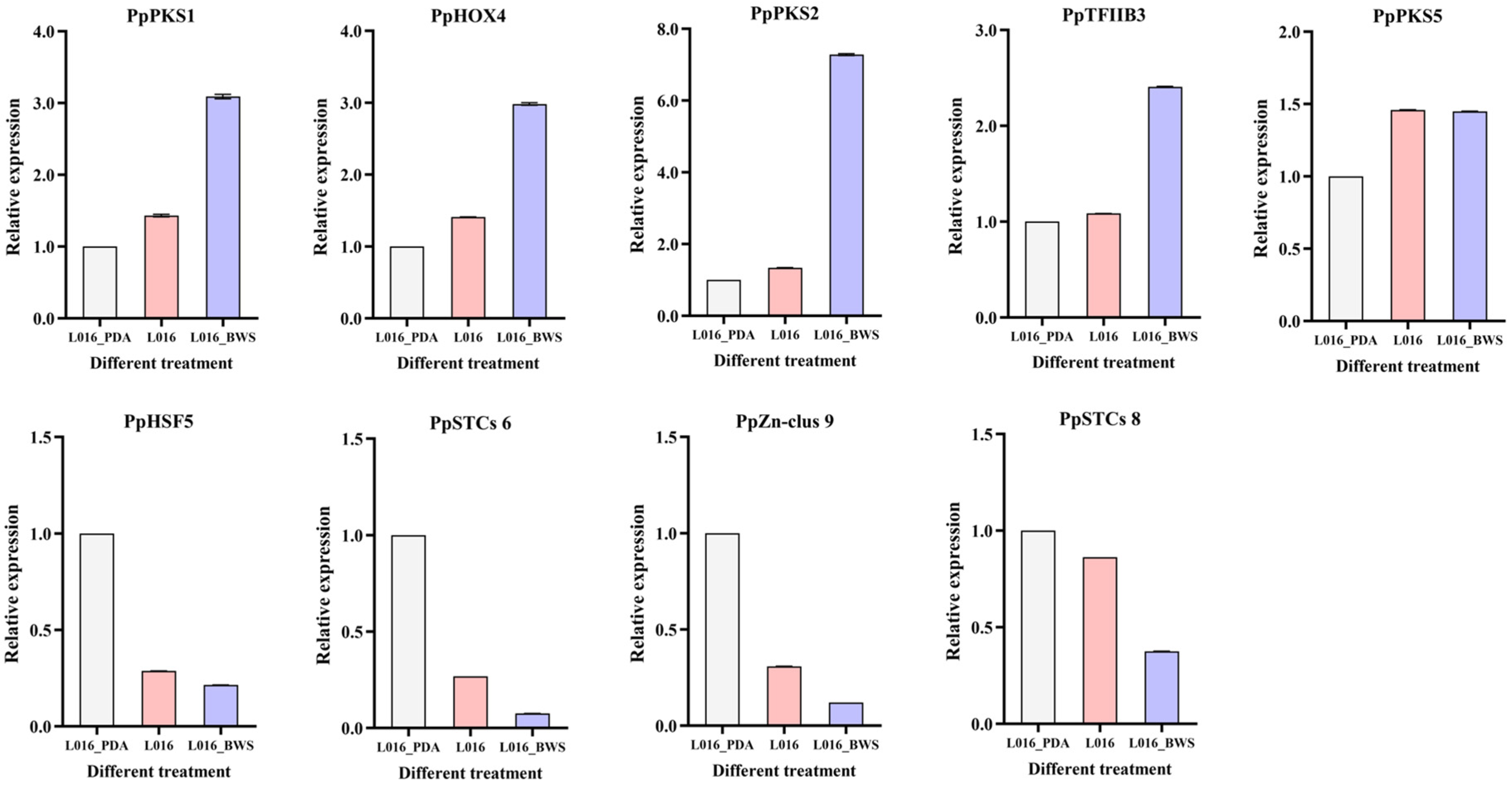
3.10. Gene Expression Analysis by qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jaworska, G.; Bernaś, E. Amino acid content of frozen Agaricus bisporus and Boletus edulis mushrooms: Effects of pretreatments. Int. J. Food Prop. 2013, 16, 139–153. [Google Scholar] [CrossRef]
- Yuwa-Amornpitak, T.; Butkhup, L.; Yeunyaw, P.N. Amino acids and antioxidant activities of extracts from wild edible mushrooms from a community forest in the Nasrinual District, Maha Sarakham, Thailand. Food Sci. Technol. 2020, 40, 712–720. [Google Scholar] [CrossRef]
- Zhang, C.X.; Ji, K.P.; He, M.X.; Cao, Y.; Liu, J.; Wang, W.B. Analysis on nutrient components of Phlebopus portentosus fruit bodies. J. Yunnan Univ. 2010, 32, 702–704. [Google Scholar]
- Ji, K.P.; Cao, Y.; Zhang, C.X.; He, M.X.; Liu, J.; Wang, W.B.; Wang, Y. Cultivation of Phlebopus portentosus in southern China. Mycol. Prog. 2011, 10, 293–300. [Google Scholar] [CrossRef]
- Zhang, C.; He, M.; Liu, J.; Xu, X.; Cao, Y.; Gao, F.; Fang, Y.W.; Wang, W.B.; Wang, Y. Brief introduction to a unique edible Bolete-Phlebopus portentosus in Southern China. J. Agric. Sci. Technol. B 2017, 7, 386–394. [Google Scholar]
- Raghoonundon, B.; Raspé, O.; Thongklang, N.; Hyde, K.D. Phlebopus (boletales, boletinellaceae), a peculiar bolete genus with widely consumed edible species and potential for economic development in tropical countries. Food Biosci. 2021, 41, 100962. [Google Scholar] [CrossRef]
- Yang, T.; Liu, J.; Xu, X.; He, M.; Gao, F.; Fang, Y.; Wang, W.; Dai, L.; Wang, Y.; Zhang, C. Sclerotia formation of Phlebopus portentosus under natural and artificial conditions. Forests 2023, 14, 1096. [Google Scholar] [CrossRef]
- Yuwa-Amornpitak, T.; Butkhup, L.; Chookietwattana, P.-N. Cultivation of edible ectomycorrhizal mushrooms (Phelobopus portentosus) associated with Sesbania javanica Miq. Jordan J. Biol. Sci. 2023, 16, 125–130. [Google Scholar]
- Ji, K.P.; Zhang, C.X.; Zeng, Y.; Liu, C.F.; He, M.X.; Wang, W.B. Artificial fungal colony and its fruiting of Phlebopus portentosus (Boletaceae) in pot. Acta. Bot. Yunnan 2007, 5, 554–558. [Google Scholar]
- Ji, K.P.; He, M.X.; Zhang, C.X.; Liu, J.; Wang, W.B.; Hou, J.Y. Semi-artificial simulate cultivation of Phlebopus portentosus and the durability of hyphae on host roots. Microbiol. China 2009, 36, 377–382. [Google Scholar]
- Li, X.B.; Hu, C.M.; Li, C.H.; Ji, G.Y.; Luo, S.Z.; Cao, Y.; Ji, K.P.; Tan, Q.; Bao, D.P.; Shang, J.J.; et al. LC/MS-and GC/MS-based metabolomic profiling to determine changes in flavor quality and bioactive components of Phlebopus portentosus under low-temperature storage. Front. Nutr. 2023, 10, 1168025. [Google Scholar] [CrossRef]
- Pan, Z.C.; Li, A.Z.; Zeng, N.K.; Yang, X.Q.; Xie, H.J.; Chen, J.; Zhao, K.; Wang, Y. Structural characteristics and hypoglycemic activity of a polysaccharide from an edible bolete Phlebopus portentosus. Int. J. Biol. Macromol. 2025, 142587. [Google Scholar] [CrossRef]
- Sun, Z.C.; Hu, M.G.; Sun, Z.H.; Zhu, N.L.; Yang, J.S.; Ma, G.X.; Xu, X.D. Pyrrole alkaloids from the edible mushroom Phlebopus portentosus with their bioactive activities. Molecules 2018, 23, 1198. [Google Scholar] [CrossRef] [PubMed]
- Chuankid, B.; Schrey, H.; Thongbai, B.; Raspé, O.; Arnold, N.; Hyde, K.D.; Stadler, M. Secondary metabolites of Phlebopus species from Northern Thailand. Mycol. Prog. 2020, 19, 1525–1536. [Google Scholar] [CrossRef]
- Wang, Z.X.; Zhou, W.; He, Y.H.; Zhao, Z.Y.; Cao, Y.; Luo, S.Z.; Ji, G.Y.; Ji, K.P.; Chen, J.; Li, J.Y.; et al. Bioactive secondary metabolites of two Chinese edible Boletes, Phlebopus portentosus and Butyriboletus roseoflavus. Molecules 2025, 30, 1197. [Google Scholar] [CrossRef]
- Xiang, T.; Ge, F.; Zhu, L.; Shao, G.; Wu, T.; Fan, H.; Liu, Y.; Wang, L.; Wang, J.; Li, P. Integrated metabolomic and transcriptomic analysis reveals the regulatory effects of IAA on polyphenol biosynthesis and antioxidant activity in Phlebopus portentosus. Food Biosci. 2025, 65, 106116. [Google Scholar] [CrossRef]
- Feng, J.F.; Zhou, R.C.; Guo, X.T.; Zhang, Y. Polyketide and its application. Modern Agric. Sci. Technol. 2011, 39, 24–26. [Google Scholar]
- Lin, Z.; Phadke, S.; Lu, Z.; Beyhan, S.; Abdel Aziz, M.H.; Reilly, C.; Schmidt, E.W. Onydecalins, fungal polyketides with anti-Histoplasma and anti-TRP activity. J. Nat. Prod. 2018, 81, 2605–2611. [Google Scholar] [CrossRef]
- Dujovne, C.A. Red yeast rice preparations: Are they suitable substitutions for statins? Am. J. Med. 2017, 130, 1148–1150. [Google Scholar] [CrossRef]
- Tao, H.; Abe, I. Enzymology and biosynthesis of the orsellinic acid derived medicinal meroterpenoids. Curr. Opin. Biotechnol. 2021, 69, 52–59. [Google Scholar] [CrossRef]
- Wei, G.; Tan, D.; Chen, C.; Tong, Q.; Li, X.N.; Huang, J.; Liu, J.; Xue, Y.; Wang, J.; Luo, Z. Flavichalasines A–M, cytochalasan alkaloids from Aspergillus flavipes. Sci. Rep. 2017, 7, 42434. [Google Scholar] [CrossRef]
- Ismail, A.; Gonçalves, B.L.; de Neeff, D.V.; Ponzilacqua, B.; Coppa, C.F.; Hintzsche, H.; Sajid, M.; Cruz, A.G.; Corassin, C.H.; Oliveira, C.A. Aflatoxin in foodstuffs: Occurrence and recent advances in decontamination. Food Res. Int. 2018, 113, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.J.; Mao, X.M.; Chen, X.A.; Li, Y.Q. Recent advances in acyltransferase domain of type I polyktide synthases. Chin. J. Organ. Chem. 2018, 38, 2377. [Google Scholar] [CrossRef]
- Herbst, D.A.; Townsend, C.A.; Maier, T. The architectures of iterative type I PKS and FAS. Nat. Prod. Rep. 2018, 35, 1046–1069. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.K. Secondary metabolites from higher fungi in China and their biological activity. Drug Discov. Ther. 2007, 1, 94–103. [Google Scholar]
- Velíšek, J.; Cejpek, K. Pigments of higher fungi—A review. Czech J. Food Sci. 2011, 29, 87–102. [Google Scholar] [CrossRef]
- De Silva, D.D.; Rapior, S.; Sudarman, E.; Stadler, M.; Xu, J.; Aisyah Alias, S.; Hyde, K.D. Bioactive metabolites from macrofungi: Ethnopharmacology, biological activities and chemistry. Fungal Divers. 2013, 62, 1–40. [Google Scholar] [CrossRef]
- Duru, M.E.; Çayan, G.T. Biologically active terpenoids from mushroom origin: A review. Rec. Nat. Prod. 2015, 9, 456. [Google Scholar]
- Sun, X.; Shi, Y.; Shi, D.; Tu, Y.; Liu, L. Biological activities of secondary metabolites from the edible-medicinal macrofungi. J. Fungi 2024, 10, 144. [Google Scholar] [CrossRef]
- Zhao, P.; Guan, M.; Tang, W.; Walayat, N.; Ding, Y.; Liu, J. Structural diversity, fermentation production, bioactivities and applications of triterpenoids from several common medicinal fungi: Recent advances and future perspectives. Fitoterapia 2023, 166, 105470. [Google Scholar] [CrossRef]
- Nieto, I.J.; Chegwin, A.C. The effect of different substrates on triterpenoids and fatty acids in fungi of the genus Pleurotus. J. Chil. Chem. Soc. 2013, 58, 1580–1583. [Google Scholar] [CrossRef]
- He, Y.C.; He, K.Z.; Pu, Q.; Li, J.; Zhao, Z.J. Optimization of cultivating conditions for triterpenoids production from Antrodia cinnmomea. Indian J. Microbiol. 2012, 52, 648–653. [Google Scholar] [CrossRef]
- Luo, P.; Huang, J.H.; Lv, J.M.; Wang, G.Q.; Hu, D.; Gao, H. Biosynthesis of fungal terpenoids. Nat. Prod. Rep. 2024, 41, 748–783. [Google Scholar] [CrossRef] [PubMed]
- Christianson, D.W. Structural and chemical biology of terpenoid cyclases. Chem. Rev. 2017, 117, 11570–11648. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, E.; Lin, F.Y. Terpene biosynthesis: Modularity rules. Angew. Chem. Int. Ed. 2012, 51, 1124–1137. [Google Scholar] [CrossRef]
- Wan, J.N.; Li, Y.; Guo, T.; Ji, G.Y.; Luo, S.Z.; Ji, K.P.; Cao, Y.; Tan, Q.; Bao, D.P.; Yang, R.H. Whole-genome and transcriptome sequencing of Phlebopus portentosus reveals its associated ectomycorrhizal niche and conserved pathways involved in fruiting body development. Front. Microbiol. 2021, 12, 732458. [Google Scholar] [CrossRef]
- Cao, Y.; Zhang, Y.; Yu, Z.; Wang, P.; Tang, X.; He, X.; Mi, F.; Liu, C.; Yang, D.; Xu, J. Genome sequence of Phlebopus portentosus strain PP33, a cultivated bolete. Genome Announc. 2015, 3, e00326-15. [Google Scholar] [CrossRef]
- Yang, M.; Yuan, X.; Kong, L.; Gao, Y.; Liu, C.; Wang, Y. Draft genome sequence of ectomycorrhizal edible mushroom Phlebopus portentosus strain YAF023. Microbiol. Resour. Announc. 2024, 13, e00588-24. [Google Scholar] [CrossRef]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Lepesheva, G.I.; Hargrove, T.Y.; Kleshchenko, Y.; Nes, W.D.; Villalta, F.; Waterman, M.R. CYP51: A major drug target in the cytochrome P450 superfamily. Lipids 2008, 43, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Lah, L.; Podobnik, B.; Novak, M.; Korošec, B.; Berne, S.; Vogelsang, M.; Kraševec, N.; Zupanec, N.; Stojan, J.; Bohlmann, J. The versatility of the fungal cytochrome P450 monooxygenase system is instrumental in xenobiotic detoxification. Mol. Microbiol. 2011, 81, 1374–1389. [Google Scholar] [CrossRef] [PubMed]
- Mischko, W.; Hirte, M.; Fuchs, M.; Mehlmer, N.; Brück, T.B. Identification of sesquiterpene synthases from the Basidiomycota Coniophora puteana for the efficient and highly selective β-copaene and cubebol production in E. coli. Microb. Cell Fact. 2018, 17, 164. [Google Scholar] [CrossRef]
- Zhang, Y.; Mao, L.; Wang, H.; Brocker, C.; Yin, X.; Vasiliou, V.; Fei, Z.; Wang, X. Genome-wide identification and analysis of grape aldehyde dehydrogenase (ALDH) gene superfamily. PLoS ONE 2012, 7, e32153. [Google Scholar] [CrossRef]
- Kumla, J.; Hobbie, E.A.; Suwannarach, N.; Lumyong, S. The ectomycorrhizal status of a tropical black bolete, Phlebopus portentosus, assessed using mycorrhizal synthesis and isotopic analysis. Mycorrhiza 2016, 26, 333–343. [Google Scholar] [CrossRef]
- Kumla, J.; Bussaban, B.; Suwannarach, N.; Lumyong, S.; Danell, E. Basidiome formation of an edible wild, putatively ectomycorrhizal fungus, Phlebopus portentosus without host plant. Mycologia 2012, 104, 597–603. [Google Scholar] [CrossRef]
- Martin, F.; Aerts, A.; Ahrén, D.; Brun, A.; Danchin, E.; Duchaussoy, F.; Gibon, J.; Kohler, A.; Lindquist, E.; Pereda, V. The genome of Laccaria bicolor provides insights into mycorrhizal symbiosis. Nature 2008, 452, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, B.E.; Tulloss, R.E.; Pringle, A. The irreversible loss of a decomposition pathway marks the single origin of an ectomycorrhizal symbiosis. PLoS ONE 2012, 7, e39597. [Google Scholar] [CrossRef]
- Yang, R.H.; Li, Y.; Tang, L.H.; Li, C.H.; Bao, D.P. Genome-wide comparison of lignocellulose degradation enzymes in Agaricales. Mycosystema 2017, 36, 705–717. [Google Scholar]
- Zhao, L.; Zhang, J.; Zhao, D.; Jia, L.; Qin, B.; Cao, X.; Zang, L.; Lu, F.; Liu, F. Biological degradation of lignin: A critical review on progress and perspectives. Ind. Crop. Prod. 2022, 188, 115715. [Google Scholar] [CrossRef]
- Zhou, C.L.; Guo, T.; Ji, G.Y.; Ji, K.P.; Bao, D.P.; Yang, R.H. Genomic and transcriptomic analysis to uncover transcription factors and laccase expression regulators in Phlebopus portentosus. Northern Hort. 2023, 46, 108–116. [Google Scholar]
- Busk, P.K.; Lange, M.; Pilgaard, B.; Lange, L. Several genes encoding enzymes with the same activity are necessary for aerobic fungal degradation of cellulose in nature. PLoS ONE 2014, 9, e114138. [Google Scholar] [CrossRef] [PubMed]
- Baldrian, P.; Valášková, V. Degradation of cellulose by basidiomycetous fungi. FEMS Microbiol. Rev. 2008, 32, 501–521. [Google Scholar] [CrossRef] [PubMed]
- Pfister, B.; Zeeman, S.C. Formation of starch in plant cells. Cell. Mol. Life Sci. 2016, 73, 2781–2807. [Google Scholar] [CrossRef] [PubMed]
- Southall, S.M.; Simpson, P.J.; Gilbert, H.J.; Williamson, G.; Williamson, M.P. The starch-binding domain from glucoamylase disrupts the structure of starch. FEBS Lett. 1999, 447, 58–60. [Google Scholar] [CrossRef]
- He, M.; Wang, T.; Tang, C.; Xiao, M.; Pu, X.; Qi, J.; Li, Y.; Li, X. Metabolomics and transcriptomics reveal the effects of different fermentation times on antioxidant activities of Ophiocordyceps sinensis. J. Fungi 2025, 11, 51. [Google Scholar] [CrossRef]
- Chao, K.; Qi, T.; Wan, Q.; Li, T. Insights into the flavor differentiation between two wild edible boletus species through metabolomic and transcriptomic analyses. Foods 2023, 12, 2728. [Google Scholar] [CrossRef]
- Xiao, M.; Wang, T.; Tang, C.; He, M.; Li, Y.; Li, X. Effects of different drying methods on amino acid metabolite content and quality of Ophiocordyceps sinensis by LC-MS/MS combined with multivariate statistical methods. Metabolites 2024, 14, 459. [Google Scholar] [CrossRef]
- Sun, L.B.; Zhang, Z.Y.; Xin, G.; Sun, B.X.; Bao, X.J.; Wei, Y.Y.; Zhao, X.M.; Xu, H.R. Advances in umami taste and aroma of edible mushrooms. Trends Food Sci. Technol. 2020, 96, 176–187. [Google Scholar] [CrossRef]
- Salyha, N.; Salyha, Y. Protective role of l-glutamic acid and l-cysteine in mitigation the chlorpyrifos-induced oxidative stress in rats. Environ. Toxicol. Pharmacol. 2018, 64, 155–163. [Google Scholar] [CrossRef]
- Ishkaeva, R.A.; Nizamov, I.S.; Blokhin, D.S.; Urakova, E.A.; Klochkov, V.V.; Nizamov, I.D.; Gareev, B.I.; Salakhieva, D.V.; Abdullin, T.I. Dithiophosphate-induced redox conversions of reduced and oxidized glutathione. Molecules 2021, 26, 2973. [Google Scholar] [CrossRef] [PubMed]
- Nayak, B.N.; Singh, R.B.; Buttar, H.S. Biochemical and dietary functions of tryptophan and its metabolites in human health. In Functional Foods and Nutraceuticals in Metabolic and Non-Communicable Diseases; Elsevier: Amsterdam, The Netherlands, 2022; pp. 783–798. [Google Scholar]
- Lackner, G.; Bohnert, M.; Wick, J.; Hoffmeister, D. Assembly of melleolide antibiotics involves a polyketide synthase with cross-coupling activity. Chem. Biol. 2013, 20, 1101–1106. [Google Scholar] [CrossRef] [PubMed]
- Abugrain, M.E.; Brumsted, C.J.; Osborn, A.R.; Philmus, B.; Mahmud, T. A highly promiscuous ß-ketoacyl-acp synthase (kas) III-like protein is involved in pactamycin biosynthesis. ACS Chem. Biol. 2017, 12, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.; Tang, M.; Lin, S.; Chen, G.; Feng, Q.; Wang, Y.; Hua, H.; Bai, J.; Wang, H.; Pei, Y. Cytotoxic cytochalasans from Aspergillus flavipes PJ03-11 by OSMAC method. Tetrahedron Lett. 2018, 59, 1767–1771. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Yuan, X.; Zhang, H.; Zheng, Y. Genomic features of Taiwanofungus gaoligongensis and the transcriptional regulation of secondary metabolite biosynthesis. J. Fungi 2024, 10, 826. [Google Scholar] [CrossRef]
- Hautbergue, T.; Jamin, E.L.; Debrauwer, L.; Puel, O.; Oswald, I.P. From genomics to metabolomics, moving toward an integrated strategy for the discovery of fungal secondary metabolites. Nat. Prod. Rep. 2018, 35, 147–173. [Google Scholar] [CrossRef]
- Caceres, I.; Al Khoury, A.; El Khoury, R.; Lorber, S.; Oswald, I.P.; El Khoury, A.; Atoui, A.; Puel, O.; Bailly, J.D. Aflatoxin biosynthesis and genetic regulation: A review. Toxins 2020, 12, 150. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Number | Item | Count | Percentage (%) |
|---|---|---|---|---|
| Total_length (bp) | 31,421,828 | All | 7928 | 100.00 |
| Scaffolds | 15 | Annotation | 7749 | 97.74 |
| GC_content (%) | 48.91 | KEGG | 3112 | 39.25 |
| N50 (bp) | 2,638,669 | Pathway | 1702 | 21.47 |
| N90 (bp) | 1,761,355 | Nr | 7638 | 96.34 |
| Average (bp) | 2,094,788.53 | Uniprot | 7554 | 95.28 |
| Median (bp) | 2,550,121 | GO | 4981 | 62.83 |
| Min (bp) | 93,900 | KOG | 1625 | 20.50 |
| Max (bp) | 38,374,347 | Pfam | 5385 | 67.92 |
| Total number of genes | 7928 | Interproscan | 7305 | 92.14 |
| The average cds_length of per gene (bp) | 1500.19 | Refseq | 7398 | 93.31 |
| The average exon_number of per gene | 8.39 | Tigerfam | 1764 | 22.25 |
| Ecology Inches | Species | Genome Size (Mb) | No. of Genes | Accession Numbers | Total | AA | CBM | CE | GH | GT | PL |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ectomycorrhiza | P. portentosus YAF023 | 29.97 | 7928 | JAYXKI000000000 | 201 | 42 | 4 | 8 | 89 | 53 | 5 |
| P. portentosus PP17026 | 32.74 | 9464 | JAHRGP000000000 | 301 | 57 | 19 | 47 | 110 | 62 | 6 | |
| Paxillus ubicundulus | 53.01 | 22,354 | JMDR00000000 | 273 | 47 | 12 | 39 | 108 | 61 | 6 | |
| Paxillus involutus | 58.30 | 17,984 | JOMD00000000 | 404 | 62 | 14 | 64 | 171 | 84 | 9 | |
| Suillus luteus | 37.01 | 18,419 | JMSM00000000 | 309 | 57 | 7 | 42 | 131 | 66 | 6 | |
| Pisolithus tinctorius | 71.01 | 22,845 | JMDO00000000 | 241 | 42 | 2 | 38 | 87 | 69 | 3 | |
| Saprophytic | Schizophyll umcommune | 38.48 | 13,189 | ADMJ00000000 | 525 | 85 | 22 | 82 | 241 | 77 | 18 |
| Lentinula edodes | 41.82 | 14,889 | LDAT00000000 | 547 | 90 | 40 | 80 | 249 | 78 | 10 | |
| Volvariella volvacea | 36.45 | 11,084 | AMXZ00000000 | 561 | 121 | 70 | 63 | 215 | 63 | 29 | |
| Coprinus cinereus | 38.70 | 16,862 | JAAGWA000000000 | 567 | 132 | 67 | 86 | 192 | 74 | 16 |
| Classification of Metabolites | Positive Ion Mode | Negative Ion Mode |
|---|---|---|
| Organoheterocyclic compounds | 164 | 103 |
| Organic acids and derivatives | 148 | 123 |
| Lipids and lipid-like molecules | 130 | 129 |
| Benzenoids | 85 | 71 |
| Organic oxygen compounds | 40 | 54 |
| Phenylpropanoids and polyketides | 36 | 48 |
| Organic nitrogen compounds | 31 | 2 |
| Nucleosides, nucleotides, and analogs | 24 | 14 |
| Alkaloids and derivatives | 16 | 1 |
| Other | 7 | 6 |
| Unclassified | 252 | 98 |
| Total | 933 | 649 |
| TPS ID | Score | Relative Score | Sequence ID | Start | End | Strand | Predicted Sequence |
|---|---|---|---|---|---|---|---|
| PpHOX4 | 6.9166865 | 1 | PpPKS1 | 573 | 576 | + | TAAT |
| PpHSF1 | 11.563199 | 1 | PpPKS1 | 2003 | 2009 | + | ATGGAAC |
| Ppzf-C2H2 32 | 9.598243 | 1 | PpSTCs 8 | 608 | 613 | + | CCCCAC |
| PpHSF5 | 8.239277 | 1 | PpSTCs 3 | 213 | 217 | + | GGCC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, Z.; Yuan, X.; Zhang, C.; Wang, Y.; Li, L.; Zheng, Y. Genomic and Multi-Omics Analysis of Phlebopus portentosus: Effects of Cultivation on Secondary Metabolites. J. Fungi 2025, 11, 323. https://doi.org/10.3390/jof11040323
Kang Z, Yuan X, Zhang C, Wang Y, Li L, Zheng Y. Genomic and Multi-Omics Analysis of Phlebopus portentosus: Effects of Cultivation on Secondary Metabolites. Journal of Fungi. 2025; 11(4):323. https://doi.org/10.3390/jof11040323
Chicago/Turabian StyleKang, Zujiang, Xiaolong Yuan, Chuanguang Zhang, Yi Wang, Lu Li, and Yuan Zheng. 2025. "Genomic and Multi-Omics Analysis of Phlebopus portentosus: Effects of Cultivation on Secondary Metabolites" Journal of Fungi 11, no. 4: 323. https://doi.org/10.3390/jof11040323
APA StyleKang, Z., Yuan, X., Zhang, C., Wang, Y., Li, L., & Zheng, Y. (2025). Genomic and Multi-Omics Analysis of Phlebopus portentosus: Effects of Cultivation on Secondary Metabolites. Journal of Fungi, 11(4), 323. https://doi.org/10.3390/jof11040323