
Induction of Antifungal Tolerance Reveals Genetic and Phenotypic Changes in Candida glabrata

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Candida glabrata Isolates
2.2. MIC Determination
2.3. DNA Extraction, Sequencing and Variant Calling
2.4. Comparative Chromosomal Abnormality Analyses
2.5. Adhesion Assay
2.6. Quantification of Rhodamine 6G Efflux
2.7. Virulence Assay
2.8. Adhesion to Human Epithelial Cells
2.9. Ergosterol Quantification
2.10. Chitin Quantification
2.11. Biofilm Quantification
2.12. Statistical Analysis
3. Results
3.1. MICs After Drug Exposure
3.2. Amino Acid Substitutions
3.3. Comparative SNP and Chromosomal Abnormality Analyses
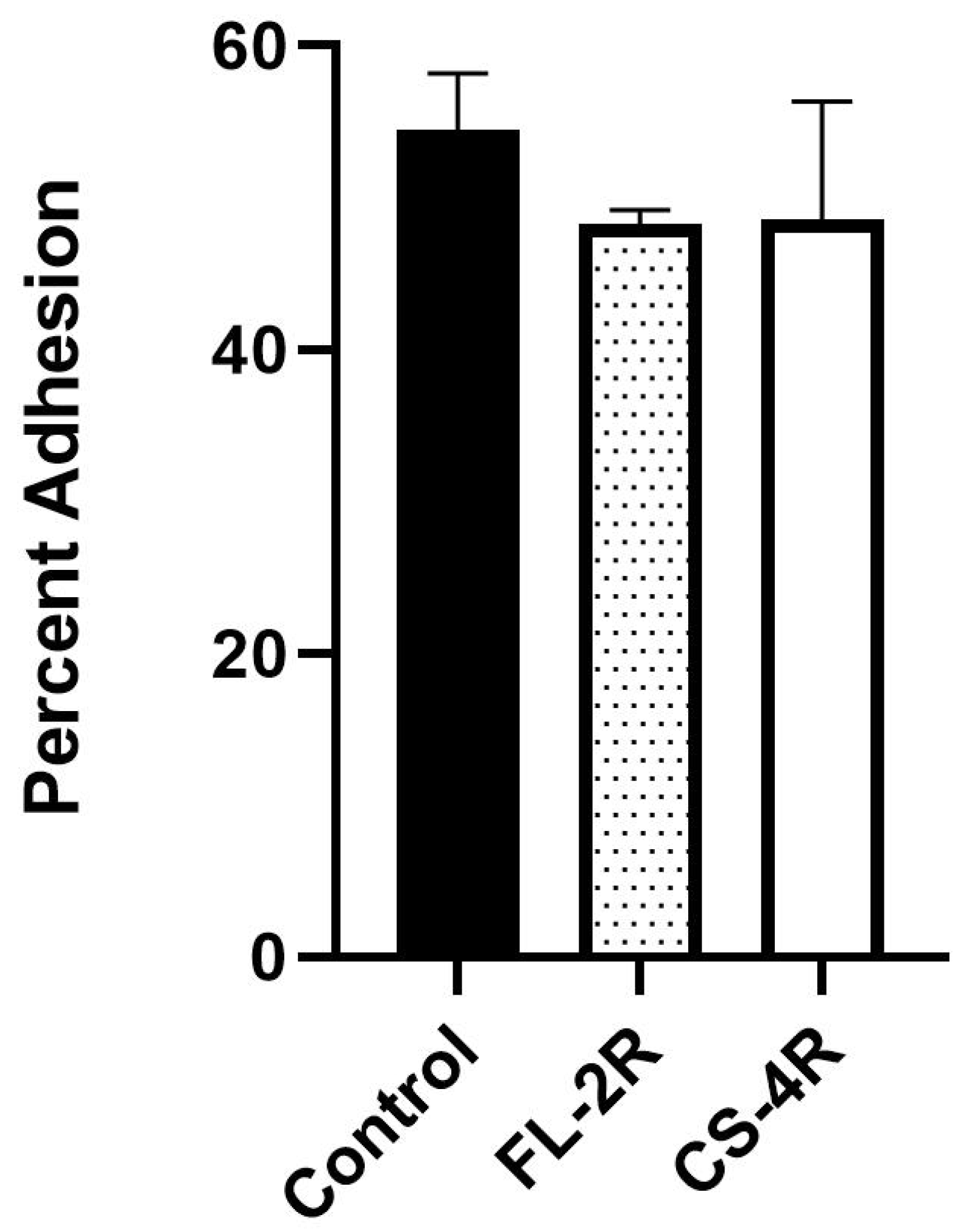
3.4. Decreased Adhesion in Drug-Tolerant Isolates
3.5. Decreased Virulence in Drug-Tolerant Isolates
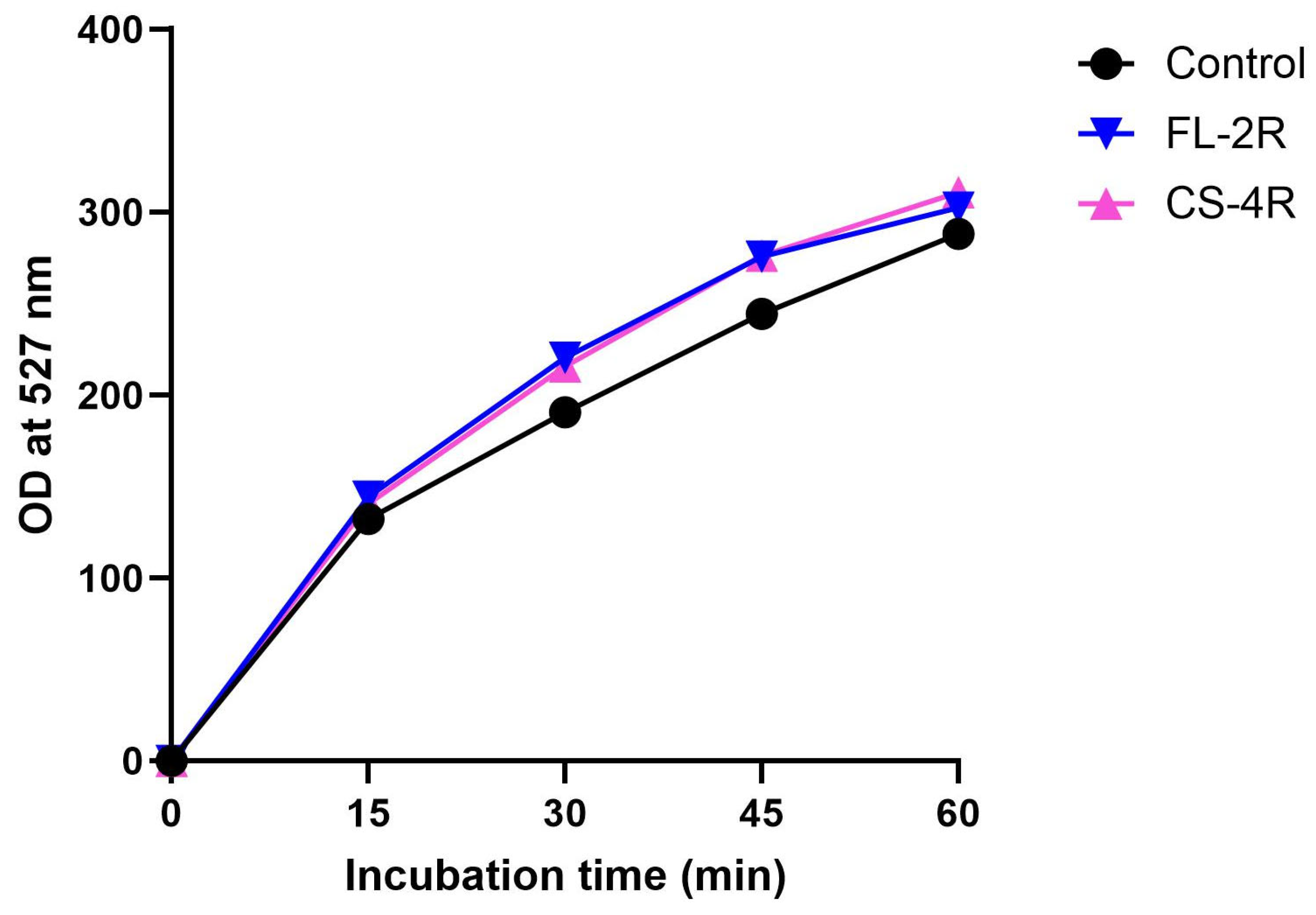
3.6. Upregulation of Efflux Pumps in Drug-Tolerant Isolates
3.7. Slightly Decreased Adhesion of Drug-Tolerant Isolates to Human Cells
3.8. No Changes in Ergosterol and Chitin Contents Upon Development of Drug Tolerance
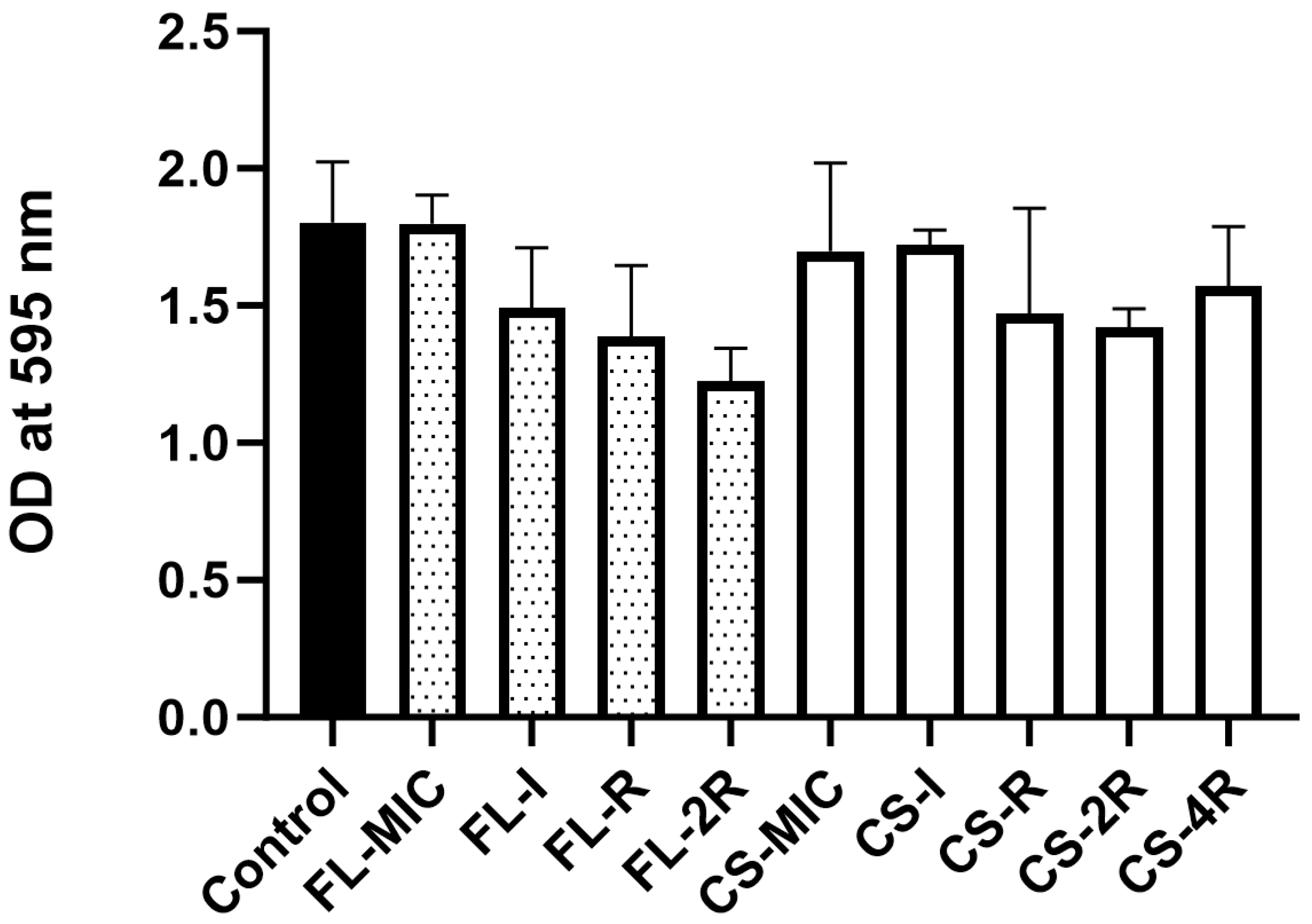
3.9. Absence of Significant Biofilm Formation in Drug-Tolerant Isolates
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Silva, S.; Negri, M.; Henriques, M.; Oliveira, R.; Williams, D.W.; Azeredo, J. Candida Glabrata, Candida Parapsilosis and Candida Tropicalis: Biology, Epidemiology, Pathogenicity and Antifungal Resistance. FEMS Microbiol. Rev. 2012, 36, 288–305. [Google Scholar] [CrossRef] [PubMed]
- Charlet, R.; Pruvost, Y.; Tumba, G.; Istel, F.; Poulain, D.; Kuchler, K.; Sendid, B.; Jawhara, S. Remodeling of the Candida Glabrata Cell Wall in the Gastrointestinal Tract Affects the Gut Microbiota and the Immune Response. Sci. Rep. 2018, 8, 3316. [Google Scholar] [CrossRef] [PubMed]
- Pérez, J.C. The Interplay between Gut Bacteria and the Yeast Candida Albicans. Gut Microbes 2021, 13, 1979877. [Google Scholar] [CrossRef] [PubMed]
- Kullberg, B.J.; Arendrup, M.C. Invasive Candidiasis. N. Engl. J. Med. 2015, 373, 1445–1456. [Google Scholar] [CrossRef] [PubMed]
- Hirano, R.; Sakamoto, Y.; Kudo, K.; Ohnishi, M. Retrospective Analysis of Mortality and Candida Isolates of 75 Patients with Candidemia: A Single Hospital Experience. Infect. Drug Resist. 2015, 8, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.; Meltzer, M.I.; Plikaytis, B.D.; Sofair, A.N.; Huie-White, S.; Wilcox, S.; Harrison, L.H.; Seaberg, E.C.; Hajjeh, R.A.; Teutsch, S.M. Excess Mortality, Hospital Stay, and Cost Due to Candidemia: A Case-Control Study Using Data from Population-Based Candidemia Surveillance. Infect. Control Hosp. Epidemiol. 2005, 26, 540–547. [Google Scholar] [CrossRef]
- Gabaldón, T.; Fairhead, C. Genomes Shed Light on the Secret Life of Candida Glabrata: Not so Asexual, Not so Commensal. Curr. Genet. 2019, 65, 93–98. [Google Scholar] [CrossRef]
- Kumar, K.; Askari, F.; Sahu, M.S.; Kaur, R. Candida Glabrata: A Lot More Than Meets the Eye. Microorganisms 2019, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Barada, G.; Basma, R.; Khalaf, R.A. Microsatellite DNA Identification and Genotyping of Candida Albicans from Lebanese Clinical Isolates. Mycopathologia 2008, 165, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Gabaldón, T.; Gómez-Molero, E.; Bader, O. Molecular Typing of Candida Glabrata. Mycopathologia 2020, 185, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Csank, C.; Haynes, K. Candida Glabrata Displays Pseudohyphal Growth. FEMS Microbiol. Lett. 2000, 189, 115–120. [Google Scholar] [CrossRef]
- Sasani, E.; Khodavaisy, S.; Agha Kuchak Afshari, S.; Darabian, S.; Aala, F.; Rezaie, S. Pseudohyphae Formation in Candida Glabrata Due to CO2 Exposure. Curr. Med. Mycol. 2016, 2, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Girois, S.B.; Chapuis, F.; Decullier, E.; Revol, B.G.P. Adverse Effects of Antifungal Therapies in Invasive Fungal Infections: Review and Meta-Analysis. Eur. J. Clin. Microbiol. Infect. Dis. 2006, 25, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Farmakiotis, D.; Kontoyiannis, D.P. Epidemiology of Antifungal Resistance in Human Pathogenic Yeasts: Current Viewpoint and Practical Recommendations for Management. Int. J. Antimicrob. Agents 2017, 50, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Sae-Tia, S.; Fries, B.C. Candidiasis and Mechanisms of Antifungal Resistance. Antibiotics 2020, 9, 312. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Berman, J. Beyond Resistance: Antifungal Heteroresistance and Antifungal Tolerance in Fungal Pathogens. Curr. Opin. Microbiol. 2024, 78, 102439. [Google Scholar] [CrossRef]
- Suchodolski, J.; Muraszko, J.; Bernat, P.; Krasowska, A. A Crucial Role for Ergosterol in Plasma Membrane Composition, Localisation, and Activity of Cdr1p and H+-ATPase in Candida Albicans. Microorganisms 2019, 7, 378. [Google Scholar] [CrossRef]
- Sanglard, D.; Odds, F.C. Resistance of Candida Species to Antifungal Agents: Molecular Mechanisms and Clinical Consequences. Lancet Infect. Dis. 2002, 2, 73–85. [Google Scholar] [CrossRef]
- Arendrup, M.C.; Perlin, D.S. Echinocandin Resistance: An Emerging Clinical Problem? Curr. Opin. Infect. Dis. 2014, 27, 484–492. [Google Scholar] [CrossRef]
- Yang, F.; Teoh, F.; Tan, A.S.M.; Cao, Y.; Pavelka, N.; Berman, J. Aneuploidy Enables Cross-Adaptation to Unrelated Drugs. Mol. Biol. Evol. 2019, 36, 1768–1782. [Google Scholar] [CrossRef] [PubMed]
- Berman, J.; Krysan, D.J. Drug Resistance and Tolerance in Fungi. Nat. Rev. Microbiol. 2020, 18, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Schwarzmüller, T.; Ma, B.; Hiller, E.; Istel, F.; Tscherner, M.; Brunke, S.; Ames, L.; Firon, A.; Green, B.; Cabral, V.; et al. Systematic Phenotyping of a Large-Scale Candida Glabrata Deletion Collection Reveals Novel Antifungal Tolerance Genes. PLoS Pathog. 2014, 10, e1004211. [Google Scholar] [CrossRef]
- Iyer, K.R.; Robbins, N.; Cowen, L.E. The Role of Candida Albicans Stress Response Pathways in Antifungal Tolerance and Resistance. iScience 2022, 25, 103953. [Google Scholar] [CrossRef]
- Prasad, R.; Rawal, M.K. Efflux Pump Proteins in Antifungal Resistance. Front. Pharmacol. 2014, 5, 202. [Google Scholar] [CrossRef] [PubMed]
- Bonhomme, J.; d’Enfert, C. Candida Albicans Biofilms: Building a Heterogeneous, Drug-Tolerant Environment. Curr. Opin. Microbiol. 2013, 16, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Cowen, L.E.; Sanglard, D.; Howard, S.J.; Rogers, P.D.; Perlin, D.S. Mechanisms of Antifungal Drug Resistance. Cold Spring Harb. Perspect. Med. 2015, 5, a019752. [Google Scholar] [CrossRef]
- Prasad, R.; Nair, R.; Banerjee, A. Multidrug Transporters of Candida Species in Clinical Azole Resistance. Fungal Genet. Biol. 2019, 132, 103252. [Google Scholar] [CrossRef] [PubMed]
- Campoy, S.; Adrio, J.L. Antifungals. Biochem. Pharmacol. 2017, 133, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, M.; Posteraro, B.; Lass-Flörl, C. Antifungal Drug Resistance among Candida Species: Mechanisms and Clinical Impact. Mycoses 2015, 58 (Suppl. S2), 2–13. [Google Scholar] [CrossRef] [PubMed]
- Ueno, K.; Namiki, Y.; Mitani, H.; Yamaguchi, M.; Chibana, H. Differential Cell Wall Remodeling of Two Chitin Synthase Deletants Δchs3A and Δchs3B in the Pathogenic Yeast Candida Glabrata. FEMS Yeast Res. 2011, 11, 398–407. [Google Scholar] [CrossRef]
- Rai, M.N.; Sharma, V.; Balusu, S.; Kaur, R. An Essential Role for Phosphatidylinositol 3-Kinase in the Inhibition of Phagosomal Maturation, Intracellular Survival and Virulence in Candida Glabrata. Cell Microbiol. 2015, 17, 269–287. [Google Scholar] [CrossRef] [PubMed]
- Timmermans, B.; De Las Peñas, A.; Castaño, I.; Van Dijck, P. Adhesins in Candida Glabrata. J. Fungi 2018, 4, 60. [Google Scholar] [CrossRef] [PubMed]
- d’Enfert, C.; Janbon, G. Biofilm Formation in Candida Glabrata: What Have We Learnt from Functional Genomics Approaches? FEMS Yeast Res. 2016, 16, fov111. [Google Scholar] [CrossRef] [PubMed]
- Valotteau, C.; Prystopiuk, V.; Cormack, B.P.; Dufrêne, Y.F. Atomic Force Microscopy Demonstrates That Candida Glabrata Uses Three Epa Proteins to Mediate Adhesion to Abiotic Surfaces. mSphere 2019, 4, e00277-19. [Google Scholar] [CrossRef] [PubMed]
- El Hachem, S.; Fattouh, N.; Chedraoui, C.; Finianos, M.; Bitar, I.; Khalaf, R.A. Sequential Induction of Drug Resistance and Characterization of an Initial Candida Albicans Drug-Sensitive Isolate. J. Fungi 2024, 10, 347. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute. Performance Standards for Antifungal Susceptibility Testing of Yeasts; Alexander, B.D., Procop, G.W., Dufresne, P., Clinical and Laboratory Standards Institute, Eds.; CLSI: Wayne, PA, USA, 2017; ISBN 978-1-56238-828-7. [Google Scholar]
- Subcommittee on Antifungal Susceptibility Testing (AFST) of the ESCMID European Committee for Antimicrobial Susceptibility Testing (EUCAST); Rodríguez-Tudela, J.L.; Barchiesi, F.; Bille, J.; Chryssanthou, E.; Cuenca-Estrella, M.; Denning, D.; Donnelly, J.P.; Dupont, B.; Fegeler, W.; et al. Method for the Determination of Minimum Inhibitory Concentration (MIC) by Broth Dilution of Fermentative Yeasts. Clin. Microbiol. Infect. 2003, 9, i–viii. [Google Scholar] [CrossRef]
- Fattouh, N.; Husni, R.; Finianos, M.; Bitar, I.; Khalaf, R.A. Adhesive and Biofilm-Forming Candida Glabrata Lebanese Hospital Isolates Harbour Mutations in Subtelomeric Silencers and Adhesins. Mycoses 2024, 67, e13750. [Google Scholar] [CrossRef] [PubMed]
- Fattouh, N.; Khalaf, R.A.; Husni, R. Candida Glabrata Hospital Isolate from Lebanon Reveals Micafungin Resistance Associated with Increased Chitin and Resistance to a Cell-Surface-Disrupting Agent. J. Glob. Antimicrob. Resist. 2024, 37, 62–68. [Google Scholar] [CrossRef]
- Abbey, D.A.; Funt, J.; Lurie-Weinberger, M.N.; Thompson, D.A.; Regev, A.; Myers, C.L.; Berman, J. YMAP: A Pipeline for Visualization of Copy Number Variation and Loss of Heterozygosity in Eukaryotic Pathogens. Genome Med. 2014, 6, 100. [Google Scholar] [CrossRef] [PubMed]
- Toutounji, M.; Tokajian, S.; Khalaf, R.A. Genotypic and Phenotypic Characterization of Candida Albicans Lebanese Hospital Isolates Resistant and Sensitive to Caspofungin. Fungal Genet. Biol. 2019, 127, 12–22. [Google Scholar] [CrossRef]
- Maesaki, S.; Marichal, P.; Bossche, H.V.; Sanglard, D.; Kohno, S. Rhodamine 6G Efflux for the Detection of CDR1-Overexpressing Azole-Resistant Candidaalbicans Strains. J. Antimicrob. Chemother. 1999, 44, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Fattouh, N.; Hdayed, D.; Geukgeuzian, G.; Tokajian, S.; Khalaf, R.A. Molecular Mechanism of Fluconazole Resistance and Pathogenicity Attributes of Lebanese Candida Albicans Hospital Isolates. Fungal Genet. Biol. 2021, 153, 103575. [Google Scholar] [CrossRef]
- Younes, S.; Bahnan, W.; Dimassi, H.I.; Khalaf, R.A. The Candida Albicans Hwp2 Is Necessary for Proper Adhesion, Biofilm Formation and Oxidative Stress Tolerance. Microbiol. Res. 2011, 166, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Arthington-Skaggs, B.A.; Jradi, H.; Desai, T.; Morrison, C.J. Quantitation of Ergosterol Content: Novel Method for Determination of Fluconazole Susceptibility of Candida Albicans. J. Clin. Microbiol. 1999, 37, 3332–3337. [Google Scholar] [CrossRef]
- El Khoury, P.; Awad, A.; Wex, B.; Khalaf, R.A. Proteomic Analysis of a Candida Albicans Pir32 Null Strain Reveals Proteins Involved in Adhesion, Filamentation and Virulence. PLoS ONE 2018, 13, e0194403. [Google Scholar] [CrossRef] [PubMed]
- Kapteyn, J.C.; Hoyer, L.L.; Hecht, J.E.; Müller, W.H.; Andel, A.; Verkleij, A.J.; Makarow, M.; Van Den Ende, H.; Klis, F.M. The Cell Wall Architecture of Candida Albicans Wild-Type Cells and Cell Wall-Defective Mutants. Mol. Microbiol. 2000, 35, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.; Henriques, M.; Martins, A.; Oliveira, R.; Williams, D.; Azeredo, J. Biofilms of Non-Candida Albicans Candida Species: Quantification, Structure and Matrix Composition. Med. Mycol. 2009, 47, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Vale-Silva, L.A.; Sanglard, D. Tipping the Balance Both Ways: Drug Resistance and Virulence in Candida Glabrata. FEMS Yeast Res. 2015, 15, fov025. [Google Scholar] [CrossRef]
- Al-Baqsami, Z.F.; Ahmad, S.; Khan, Z. Antifungal Drug Susceptibility, Molecular Basis of Resistance to Echinocandins and Molecular Epidemiology of Fluconazole Resistance among Clinical Candida Glabrata Isolates in Kuwait. Sci. Rep. 2020, 10, 6238. [Google Scholar] [CrossRef]
- Sanguinetti, M.; Posteraro, B.; Fiori, B.; Ranno, S.; Torelli, R.; Fadda, G. Mechanisms of Azole Resistance in Clinical Isolates of Candida Glabrata Collected during a Hospital Survey of Antifungal Resistance. Antimicrob. Agents Chemother. 2005, 49, 668–679. [Google Scholar] [CrossRef]
- Robbins, N.; Cowen, L.E. Antifungal Drug Resistance: Deciphering the Mechanisms Governing Multidrug Resistance in the Fungal Pathogen Candida Glabrata. Curr. Biol. 2021, 31, R1520–R1523. [Google Scholar] [CrossRef]
- Healey, K.R.; Perlin, D.S. Fungal Resistance to Echinocandins and the MDR Phenomenon in Candida Glabrata. J. Fungi 2018, 4, 105. [Google Scholar] [CrossRef]
- Walker, L.A.; Munro, C.A.; de Bruijn, I.; Lenardon, M.D.; McKinnon, A.; Gow, N.A.R. Stimulation of Chitin Synthesis Rescues Candida Albicans from Echinocandins. PLoS Pathog. 2008, 4, e1000040. [Google Scholar] [CrossRef]
- Reithofer, V.; Fernández-Pereira, J.; Alvarado, M.; de Groot, P.; Essen, L.-O. A Novel Class of Candida Glabrata Cell Wall Proteins with β-Helix Fold Mediates Adhesion in Clinical Isolates. PLoS Pathog. 2021, 17, e1009980. [Google Scholar] [CrossRef]
- Gómez-Molero, E.; de Boer, A.D.; Dekker, H.L.; Moreno-Martínez, A.; Kraneveld, E.A.; Ichsan; Chauhan, N.; Weig, M.; de Soet, J.J.; de Koster, C.G.; et al. Proteomic Analysis of Hyperadhesive Candida Glabrata Clinical Isolates Reveals a Core Wall Proteome and Differential Incorporation of Adhesins. FEM Yeast Res. 2015, 15, fov098. [Google Scholar] [CrossRef]
- Rodrigues, C.F.; Silva, S.; Henriques, M. Candida Glabrata: A Review of Its Features and Resistance. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Maestre-Reyna, M.; Diderrich, R.; Veelders, M.S.; Eulenburg, G.; Kalugin, V.; Brückner, S.; Keller, P.; Rupp, S.; Mösch, H.-U.; Essen, L.-O. Structural Basis for Promiscuity and Specificity during Candida Glabrata Invasion of Host Epithelia. Proc. Natl. Acad. Sci. USA 2012, 109, 16864–16869. [Google Scholar] [CrossRef] [PubMed]
- Daneshnia, F.; de Almeida Júnior, J.N.; Ilkit, M.; Lombardi, L.; Perry, A.M.; Gao, M.; Nobile, C.J.; Egger, M.; Perlin, D.S.; Zhai, B.; et al. Worldwide Emergence of Fluconazole-Resistant Candida Parapsilosis: Current Framework and Future Research Roadmap. Lancet Microbe 2023, 4, e470–e480. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Ischer, F.; Calabrese, D.; Posteraro, B.; Sanguinetti, M.; Fadda, G.; Rohde, B.; Bauser, C.; Bader, O.; Sanglard, D. Gain of Function Mutations in CgPDR1 of Candida Glabrata Not Only Mediate Antifungal Resistance but Also Enhance Virulence. PLoS Pathog. 2009, 5, e1000268. [Google Scholar] [CrossRef] [PubMed]
- Bohner, F.; Papp, C.; Gácser, A. The Effect of Antifungal Resistance Development on the Virulence of Candida Species. FEMS Yeast Res. 2022, 22, foac019. [Google Scholar] [CrossRef]
- Alexander, B.D.; Johnson, M.D.; Pfeiffer, C.D.; Jiménez-Ortigosa, C.; Catania, J.; Booker, R.; Castanheira, M.; Messer, S.A.; Perlin, D.S.; Pfaller, M.A. Increasing Echinocandin Resistance in Candida Glabrata: Clinical Failure Correlates with Presence of FKS Mutations and Elevated Minimum Inhibitory Concentrations. Clin. Infect. Dis. 2013, 56, 1724–1732. [Google Scholar] [CrossRef] [PubMed]
- Helmstetter, N.; Chybowska, A.D.; Delaney, C.; Da Silva Dantas, A.; Gifford, H.; Wacker, T.; Munro, C.; Warris, A.; Jones, B.; Cuomo, C.A.; et al. Population Genetics and Microevolution of Clinical Candida Glabrata Reveals Recombinant Sequence Types and Hyper-Variation within Mitochondrial Genomes, Virulence Genes, and Drug Targets. Genetics 2022, 221, iyac031. [Google Scholar] [CrossRef] [PubMed]
- Flowers, S.A.; Colón, B.; Whaley, S.G.; Schuler, M.A.; Rogers, P.D. Contribution of Clinically Derived Mutations in ERG11 to Azole Resistance in Candida Albicans. Antimicrob. Agents Chemother. 2015, 59, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.A.; Gow, N.A.R.; Munro, C.A. Elevated Chitin Content Reduces the Susceptibility of Candida Species to Caspofungin. Antimicrob. Agents Chemother. 2013, 57, 146–154. [Google Scholar] [CrossRef]
- Caudle, K.E.; Barker, K.S.; Wiederhold, N.P.; Xu, L.; Homayouni, R.; Rogers, P.D. Genomewide Expression Profile Analysis of the Candida Glabrata Pdr1 Regulon. Eukaryot. Cell 2010, 10, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Shields, R.K.; Kline, E.G.; Healey, K.R.; Kordalewska, M.; Perlin, D.S.; Nguyen, M.H.; Clancy, C.J. Spontaneous Mutational Frequency and FKS Mutation Rates Vary by Echinocandin Agent against Candida Glabrata. Antimicrob. Agents Chemother. 2019, 63, e01692-18. [Google Scholar] [CrossRef]
- Cavalheiro, M.; Costa, C.; Silva-Dias, A.; Miranda, I.M.; Wang, C.; Pais, P.; Pinto, S.N.; Mil-Homens, D.; Sato-Okamoto, M.; Takahashi-Nakaguchi, A.; et al. A Transcriptomics Approach To Unveiling the Mechanisms of In Vitro Evolution towards Fluconazole Resistance of a Candida Glabrata Clinical Isolate. Antimicrob. Agents Chemother. 2019, 63, e00995-18. [Google Scholar] [CrossRef] [PubMed]
- Ksiezopolska, E.; Schikora-Tamarit, M.À.; Beyer, R.; Nunez-Rodriguez, J.C.; Schüller, C.; Gabaldón, T. Narrow Mutational Signatures Drive Acquisition of Multidrug Resistance in the Fungal Pathogen Candida Glabrata. Curr. Biol. 2021, 31, 5314–5326.e10. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.C.; Alastruey-Izquierdo, A.; Berman, J.; Bicanic, T.; Bignell, E.M.; Bowyer, P.; Bromley, M.; Brüggemann, R.; Garber, G.; Cornely, O.A.; et al. Tackling the Emerging Threat of Antifungal Resistance to Human Health. Nat. Rev. Microbiol. 2022, 20, 557–571. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (a) | |
| Isolate Name | Concentration of Fluconazole (µg/mL) |
| FL-MIC | 2 |
| FL-I | 33 |
| FL-R | 64 |
| FL-2R | 128 |
| (b) | |
| Isolate Name | Concentration of Caspofungin (µg/mL) |
| CS-MIC | 0.19 |
| CS-I | 0.345 |
| CS-R | 0.5 |
| CS-2R | 1 |
| CS-4R | 2 |
| (a) | |
| MIC (μg/mL) | |
| FL-MIC | 4 |
| FL-I | 1.5 |
| FL-R | 7 |
| FL-2R | 6 |
| (b) | |
| MIC (μg/mL) | |
| CS-MIC | <0.125 |
| CS-I | <0.125 |
| CS-R | <0.125 |
| CS-2R | <0.125 |
| CS-4R | <0.125 |
| (a) | ||||||
| Amino Acid Substitution | ||||||
| Function | Gene | FL-MIC | FL-I | FL-R | FL-2R | |
| Adhesion | AWP2 | TT633II | TT633II | TT633II | TT633II | |
| AWP3a | x | V64A | V64A | V64A | ||
| AWP8 | x | x | N654K | N654K | ||
| AWP9 | Y31D | Y31D | Y31D | Y31D | ||
| x | x | C312G | C312G | |||
| x | x | X | C267G | |||
| x | x | X | C132G | |||
| C124G | C124G | C124G | C124G | |||
| AWP10 | x | x | X | C2069S | ||
| x | x | X | C3127S | |||
| PWP1 | S6604T | S6604T | S6604T | S6604T | ||
| N4199K | N4199K | N4199K | N4199K | |||
| (b) | ||||||
| Amino Acid Substitution | ||||||
| Function | Gene | CS-MIC | CS-I | CS-R | CS-2R | CS-4R |
| Adhesion | EPA16 | x | x | R1356G | R1356G | R1356G |
| EPA15 | x | x | x | X | P137Q | |
| EPA22 | T1460P | T1460P | T1460P | T1460P | T1460P | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chedraoui, C.; Fattouh, N.; El Hachem, S.; Younes, M.; Khalaf, R.A. Induction of Antifungal Tolerance Reveals Genetic and Phenotypic Changes in Candida glabrata. J. Fungi 2025, 11, 284. https://doi.org/10.3390/jof11040284
Chedraoui C, Fattouh N, El Hachem S, Younes M, Khalaf RA. Induction of Antifungal Tolerance Reveals Genetic and Phenotypic Changes in Candida glabrata. Journal of Fungi. 2025; 11(4):284. https://doi.org/10.3390/jof11040284
Chicago/Turabian StyleChedraoui, Christy, Nour Fattouh, Setrida El Hachem, Maria Younes, and Roy A. Khalaf. 2025. "Induction of Antifungal Tolerance Reveals Genetic and Phenotypic Changes in Candida glabrata" Journal of Fungi 11, no. 4: 284. https://doi.org/10.3390/jof11040284
APA StyleChedraoui, C., Fattouh, N., El Hachem, S., Younes, M., & Khalaf, R. A. (2025). Induction of Antifungal Tolerance Reveals Genetic and Phenotypic Changes in Candida glabrata. Journal of Fungi, 11(4), 284. https://doi.org/10.3390/jof11040284