
Candida glabrata, Friend and Foe


Abstract
:
{kind=link}
{kind=link}
{kind=link}
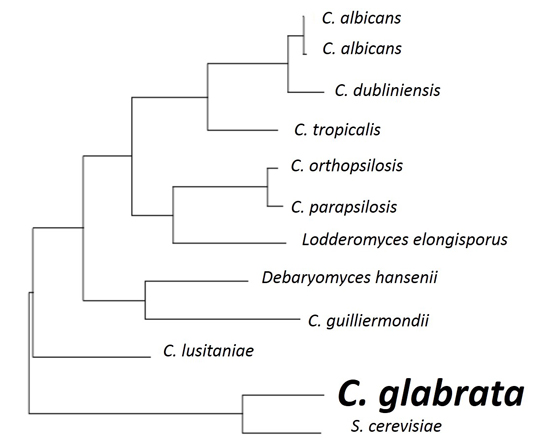
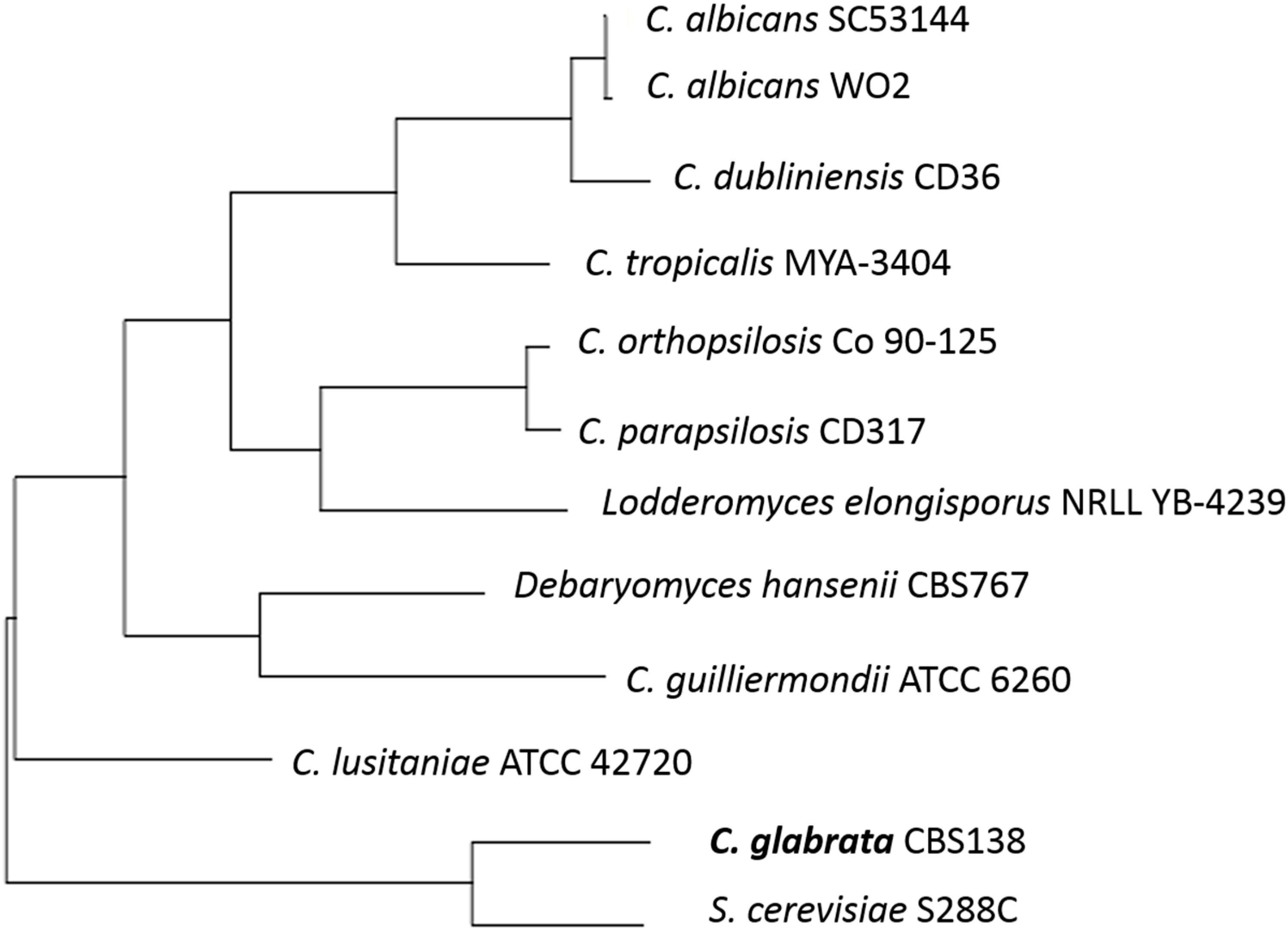
1. Introduction
2. The Opportunistic Pathogen
2.1. Adherence and Cell Wall
2.2. Biofilm Production
2.3. Extracellular Phospholipases and Enzyme Production
3. Drug Resistance
3.1. Azole Resistance
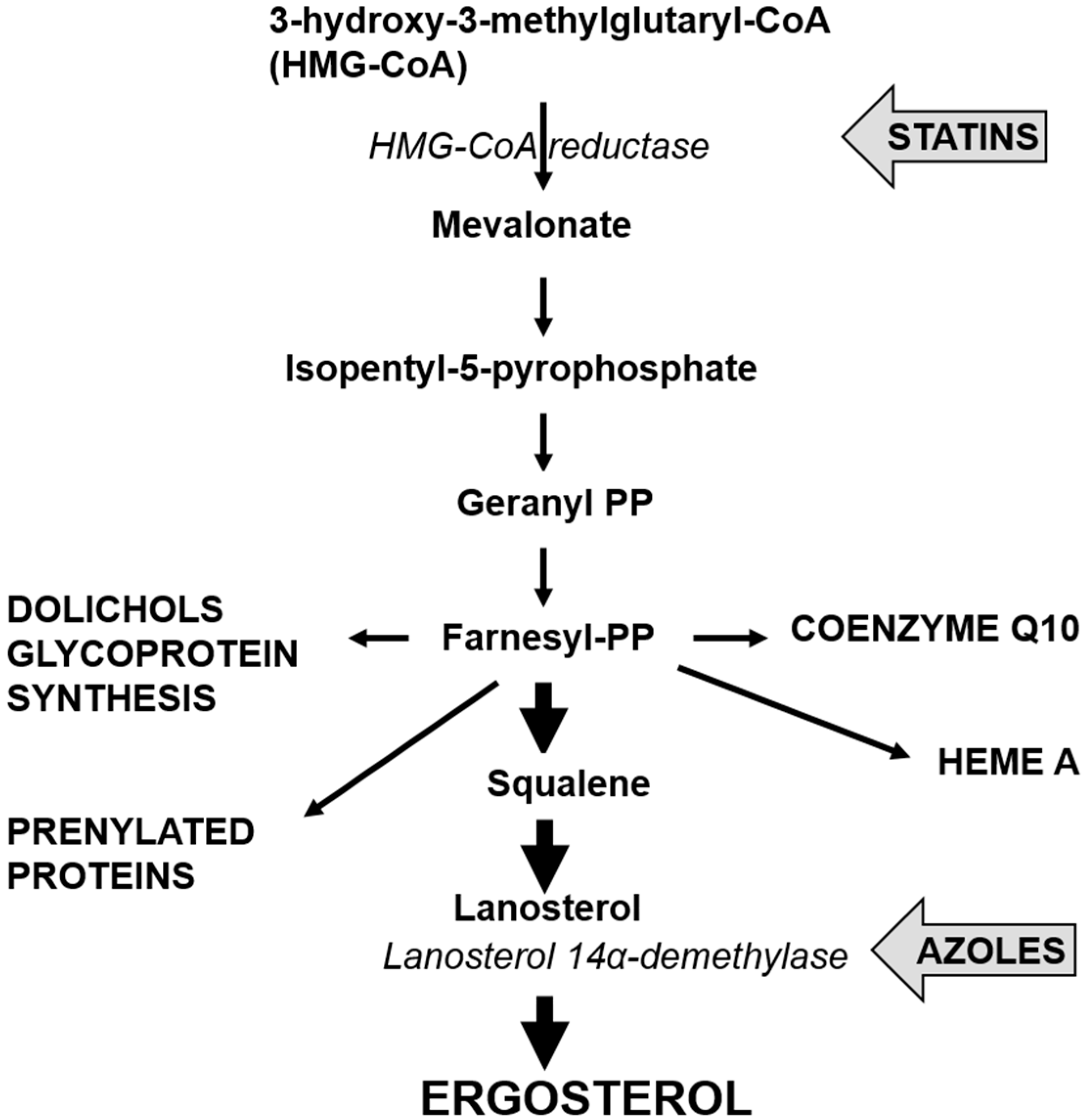
3.2. Echinocandin Resistance
3.3. Polyenes Resistance
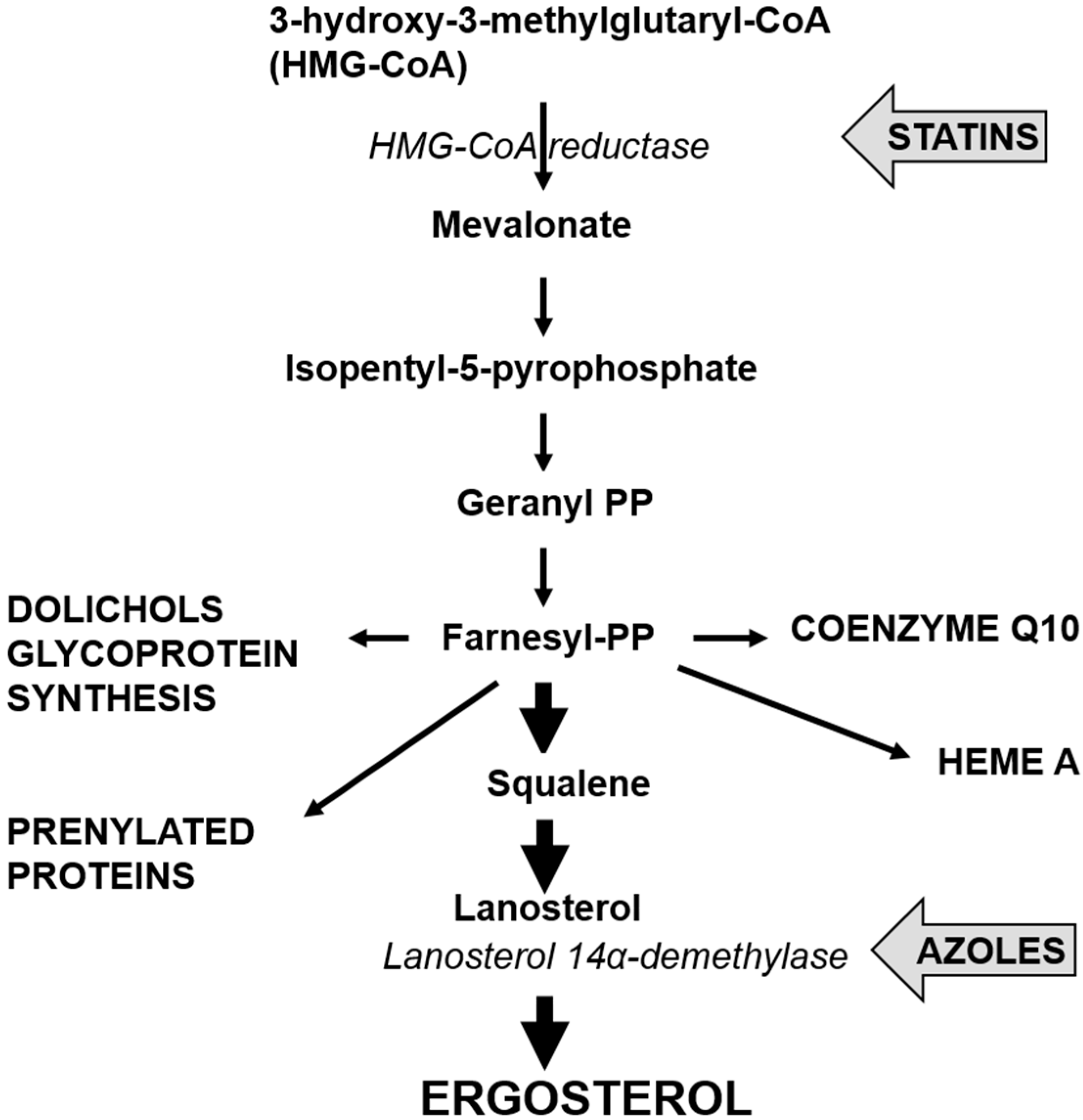
4. Cholesterol Lowering Statins and Candida glabrata
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Hommel, R.K. Torulopsis. In Encyclopedia of Food Microbiology, 2nd ed.; Academic Press: San Diego, CA, USA, 2014; pp. 596–602. [Google Scholar]
- Tsuyoshi, N.; Fudou, R.; Yamanaka, S.; Kozaki, M.; Tamang, N.; Thapa, S.; Tamang, J.P. Identification of yeast strains isolated from marcha in Sikkim, a microbial starter for amylolytic fermentation. Int. J. Food Microbiol. 2005, 99, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Fidel, P.L.; Vazquez, J.A.; Sobel, J.D. Candida glabrata: Review of epidemiology, pathogenesis, and clinical disease with comparison to C. albicans. Clin. Microbiol. Rev. 1999, 12, 80–96. [Google Scholar] [PubMed]
- Sinnott, J.T.; Cullison, J.P.; Sweeney, M.P. Candida (Torulopsis) glabrata. Infect. Control. 1987, 8, 334–336. [Google Scholar] [PubMed]
- Bialkova, A.; Šubík, J. Biology of the pathogenic yeast Candida glabrata. Folia Microbiol. 2006, 51, 3–20. [Google Scholar] [CrossRef]
- Brunke, S.; Hube, B. Two unlike cousins: Candida albicans and C. glabrata infection strategies. Cell Microbiol. 2013, 15, 701–708. [Google Scholar] [CrossRef] [PubMed]
- White, C.; Zainasheff, J. Yeast: The practical guide to beer fermentation; Brewers Publications: Boulder, CO, USA, 2010; pp. 26–29. [Google Scholar]
- Hajar, S.; Noorhisham, T.; Nurina, A. Short Technical Communication Yeast identification from domestic ragi for food fermentation by PCR method. Int. Food Res. J. 2012, 19, 775–777. [Google Scholar]
- Greppi, A.; Krych, L.; Costantini, A.; Rantsiou, K.; Hounhouigan, D.J.; Arneborg, N.; Cocolin, L.; Jespersen, L. Phytase-producing capacity of yeasts isolated from traditional African fermented food products and PHYPk gene expression of Pichia kudriavzevii strains. Int. J. Food Microbiol. 2015, 205, 81–89. [Google Scholar] [CrossRef] [PubMed]
- De Melo Pereira, G.V.; Soccol, V.T.; Pandey, A.; Medeiros, A.B.; Andrade Lara, J.M.; Gollo, A.L.; Soccol, C.R. Isolation, selection and evaluation of yeasts for use in fermentation of coffee beans by the wet process. Int. J. Food Microbiol. 2014, 188, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Steensels, J.; Verstrepen, K.J. Taming wild yeast: Potential of conventional and nonconventional yeasts in industrial fermentations. Annu. Rev. Microbiol. 2014, 68, 61–80. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.F.; Silva, S.; Henriques, M. Candida glabrata: A review of its features and resistance. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Riera, M.; Mogensen, E.; d’Enfert, C.; Janbon, G. New regulators of biofilm development in Candida glabrata. Res. Microbiol. 2012, 163, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Redding, S.; Dongari-Bagtzoglou, A. Candida glabrata, an emerging oral opportunistic pathogen. J. Dent. Res. 2007, 86, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Lachke, S.A.; Srikantha, T.; Tsai, L.K.; Daniels, K.; Soll, D.R. Phenotypic switching in Candida glabrata involves phase-specific regulation of the metallothionein gene MT-II and the newly discovered hemolysin gene HLP. Infect. Immun. 2000, 68, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Desai, C.; Mavrianos, J.; Chauhan, N. Candida glabrata Pwp7p and Aed1p are required for adherence to human endothelial cells. FEMS Yeast Res. 2011, 11, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Kramer, A.; Schwebke, I.; Kampf, G. How long do nosocomial pathogens persist on inanimate surfaces? A systematic review. BMC Infect. Dis. 2006, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- West, L.; Lowman, D.W.; Mora-Montes, H.M.; Grubb, S.; Murdoch, C.; Thornhill, M.H.; Gow, N.A.; Williams, D.; Haynes, K. Differential virulence of Candida glabrata glycosylation mutants. J. Biol. Chem. 2013, 288, 22006–22018. [Google Scholar] [CrossRef] [PubMed]
- Berila, N.; Hyroššová, P.; Šubík, J. Oxidative stress response and virulence factors in Candida glabrata clinical isolates. Folia Microbiol. 2011, 56, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Cormack, B.P.; Ghori, N.; Falkow, S. An adhesin of the yeast pathogen Candida glabrata mediating adherence to human epithelial cells. Science 1999, 285, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Castaño, I.; Pan, S.J.; Zupancic, M.; Hennequin, C.; Dujon, B.; Cormack, B.P. Telomere length control and transcriptional regulation of subtelomeric adhesins in Candida glabrata. Mol. Microbiol. 2005, 55, 1246–1258. [Google Scholar] [CrossRef] [PubMed]
- Almshawit, H.; Pouniotis, D.; Macreadie, I. Cell density impacts on Candida glabrata survival in hypo-osmotic stress. FEMS Yeast Res. 2014, 14, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Izumida, F.E.; Moffa, E.B.; Vergani, C.E.; Machado, A.L.; Jorge, J.H.; Giampaolo, E.T. In vitro evaluation of adherence of Candida albicans, Candida glabrata, and Streptococcus mutans to an acrylic resin modified by experimental coatings. Biofouling 2014, 30, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Coco, B.; Bagg, J.; Cross, L.; Jose, A.; Cross, J.; Ramage, G. Mixed Candida albicans and Candida glabrata populations associated with the pathogenesis of denture stomatitis. Oral Microbiol. Immunol. 2008, 23, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Jayatilake, J.; Samaranayake, Y.; Cheung, L.; Samaranayake, L. Quantitative evaluation of tissue invasion by wild type, hyphal and sap mutants of Candida albicans, and non-albicans Candida species in reconstituted human oral epithelium. J. Oral Pathol. Med. 2006, 35, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Calcagno, A.M.; Bignell, E.; Warn, P.; Jones, M.D.; Denning, D.W.; Mühlschlegel, F.A.; Rogers, T.R.; Haynes, K. Candida glabrata STE12 is required for wild-type levels of virulence and nitrogen starvation induced filamentation. Mol. Microbiol. 2003, 50, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Ghannoum, M.A. Potential role of phospholipases in virulence and fungal pathogenesis. Clin. Microbiol. Rev. 2000, 13, 122–143. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Sanguinetti, M.; Torelli, R.; Posteraro, B.; Sanglard, D. Contribution of CgPDR1-regulated genes in enhanced virulence of azole-resistant Candida glabrata. PLoS ONE 2011, 6, e17589. [Google Scholar] [CrossRef] [PubMed]
- Pfaller, M.; Castanheira, M.; Lockhart, S.; Ahlquist, A.; Messer, S.; Jones, R. Frequency of decreased susceptibility and resistance to echinocandins among fluconazole-resistant bloodstream isolates of Candida glabrata. J. Clin. Microbiol. 2012, 50, 1199–1203. [Google Scholar] [CrossRef] [PubMed]
- Dixon, D.M.; Walsh, T.J. Chapter 76: Antifungal Agents. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Silva, S.; Negri, M.; Henriques, M.; Oliveira, R.; Williams, D.W.; Azeredo, J. Candida glabrata, Candida parapsilosis and Candida tropicalis: Biology, epidemiology, pathogenicity and antifungal resistance. FEMS Microbiol. Rev. 2012, 36, 288–305. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Ischer, F.; Calabrese, D.; Posteraro, B.; Sanguinetti, M.; Fadda, G.; Rohde, B.; Bauser, C.; Bader, O.; Sanglard, D. Gain of function mutations in CgPDR1 of Candida glabrata not only mediate antifungal resistance but also enhance virulence. PLoS Pathog. 2009, 5, e1000268. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, M.; Posteraro, B.; Fiori, B.; Ranno, S.; Torelli, R.; Fadda, G. Mechanisms of azole resistance in clinical isolates of Candida glabrata collected during a hospital survey of antifungal resistance. Antimicrob. Agents Chemother. 2005, 49, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Marichal, P.; Bossche, H.V.; Odds, F.C.; Nobels, G.; Warnock, D.W.; Timmerman, V.; van Broeckhoven, C.; Fay, S.; Mose-Larsen, P. Molecular biological characterization of an azole-resistant Candida glabrata isolate. Antimicrob. Agents Chemother. 1997, 41, 2229–2237. [Google Scholar] [PubMed]
- Redding, S.W.; Kirkpatrick, W.R.; Saville, S.; Coco, B.J.; White, W.; Fothergill, A.; Rinaldi, M.; Eng, T.; Patterson, T.F.; Lopez-Ribot, J. Multiple patterns of resistance to fluconazole in Candida glabrata isolates from a patient with oropharyngeal candidiasis receiving head and neck radiation. J. Clin. Microbiol. 2003, 41, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Berila, N.; Borecka, S.; Dzugasova, V.; Bojnansky, J.; Subik, J. Mutations in the CgPDR1 and CgERG11 genes in azole-resistant Candida glabrata clinical isolates from Slovakia. Int. J. Antimicrob. Agents 2009, 33, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Alexander, B.D.; Johnson, M.D.; Pfeiffer, C.D.; Jiménez-Ortigosa, C.; Catania, J.; Booker, R.; Castanheira, M.; Messer, S.A.; Perlin, D.S.; Pfaller, M.A. Increasing echinocandin resistance in Candida glabrata: Clinical failure correlates with presence of FKS mutations and elevated minimum inhibitory concentrations. Clin. Infect. Dis. 2013, 56, 1724–1732. [Google Scholar] [CrossRef] [PubMed]
- Perlin, D.S. Resistance to echinocandin-class antifungal drugs. Drug Resist. Updates 2007, 10, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Effron, G.; Lee, S.; Park, S.; Cleary, J.D.; Perlin, D.S. Effect of Candida glabrata FKS1 and FKS2 mutations on echinocandin sensitivity and kinetics of 1,3-β-d-glucan synthase: Implication for the existing susceptibility breakpoint. Antimicrob. Agents Chemother. 2009, 53, 3690–3699. [Google Scholar] [CrossRef] [PubMed]
- Katiyar, S.K.; Alastruey-Izquierdo, A.; Healey, K.R.; Johnson, M.E.; Perlin, D.S.; Edlind, T.D. FKS1 and FKS2 are functionally redundant but differentially regulated in Candida glabrata: Implications for echinocandin resistance. Antimicrob. Agents Chemother. 2012, 56, 6304–6309. [Google Scholar] [CrossRef] [PubMed]
- Vandeputte, P.; Tronchin, G.; Bergès, T.; Hennequin, C.; Chabasse, D.; Bouchara, J.-P. Reduced susceptibility to polyenes associated with a missense mutation in the ERG6 gene in a clinical isolate of Candida glabrata with pseudohyphal growth. Antimicrob. Agents Chemother. 2007, 51, 982–990. [Google Scholar] [CrossRef] [PubMed]
- Vandeputte, P.; Tronchin, G.; Larcher, G.; Ernoult, E.; Bergès, T.; Chabasse, D.; Bouchara, J.-P. A nonsense mutation in the ERG6 gene leads to reduced susceptibility to polyenes in a clinical isolate of Candida glabrata. Antimicrob. Agents Chemother. 2008, 52, 3701–3709. [Google Scholar] [CrossRef] [PubMed]
- Macreadie, I.G.; Johnson, G.; Schlosser, T.; Macreadie, P.I. Growth inhibition of Candida species and Aspergillus fumigatus by statins. FEMS Microbiol. Lett. 2006, 262, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Westermeyer, C.; Macreadie, I.G. Simvastatin reduces ergosterol levels, inhibits growth and causes loss of mtDNA in Candida glabrata. FEMS Yeast Res. 2007, 7, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Forrest, G.N.; Kopack, A.M.; Perencevich, E.N. Statins in candidemia: Clinical outcomes from a matched cohort study. BMC Infect. Dis. 2010, 10, 152. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, G.; Garcia-Vidal, C.; Nucci, M.; Puchades, F.; Fernández-Ruiz, M.; Mykietiuk, A.; Manzur, A.; Gudiol, C.; Pemán, J.; Viasus, D. Effect of statin use on outcomes of adults with candidemia. PLoS ONE 2013, 8, e77317. [Google Scholar] [CrossRef] [PubMed]
- Welch, M.L.; Liappis, A.P.; Kan, V.L. Candidemia outcomes not improved with statin use. Med. Mycol. 2013, 51, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Wikhe, K.; Westermeyer, C.; Macreadie, I. Biological consequences of statins in Candida species and possible implications for human health. Biochem. Soc. Trans. 2007, 35, 1529–1532. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tam, P.; Gee, K.; Piechocinski, M.; Macreadie, I. Candida glabrata, Friend and Foe. J. Fungi 2015, 1, 277-292. https://doi.org/10.3390/jof1020277
Tam P, Gee K, Piechocinski M, Macreadie I. Candida glabrata, Friend and Foe. Journal of Fungi. 2015; 1(2):277-292. https://doi.org/10.3390/jof1020277
Chicago/Turabian StyleTam, Phyllix, Kirsten Gee, Miryam Piechocinski, and Ian Macreadie. 2015. "Candida glabrata, Friend and Foe" Journal of Fungi 1, no. 2: 277-292. https://doi.org/10.3390/jof1020277
APA StyleTam, P., Gee, K., Piechocinski, M., & Macreadie, I. (2015). Candida glabrata, Friend and Foe. Journal of Fungi, 1(2), 277-292. https://doi.org/10.3390/jof1020277