
A Comprehensive Outlook on Dilated Cardiomyopathy (DCM): State-Of-The-Art Developments with Special Emphasis on OMICS-Based Approaches


Abstract
1. Introduction
2. Causes of DCM
2.1. Causes of DCM in Children and Newborns
2.1.1. Myocarditis
2.1.2. Selenium Deficiency
2.1.3. Malformation in Pulmonary Arteriovenous
2.1.4. Endocardial Fibroelastosis
2.1.5. Noncompacted Myocardium
2.1.6. Calcium Deficiency
2.1.7. Idiopathic DCM (IDCM)
2.1.8. Barth Syndrome
2.1.9. Familial/Genetic Pediatric DCM
2.2. Causes of DCM in Adults/Adolescents
2.2.1. Familial/Genetic
2.2.2. Alcohol
2.2.3. Myocarditis
2.2.4. Tachycardiomyopathy
2.2.5. Mitochondrial Diseases
2.2.6. Cardiomyopathy Associated with Right Ventricular Arrhythmia
2.2.7. Eosinophilic Myocarditis
2.2.8. Toxins
2.2.9. Peripartum Cardiomyopathy
2.2.10. Endocrinopathy
2.2.11. Nutritional Deficiency
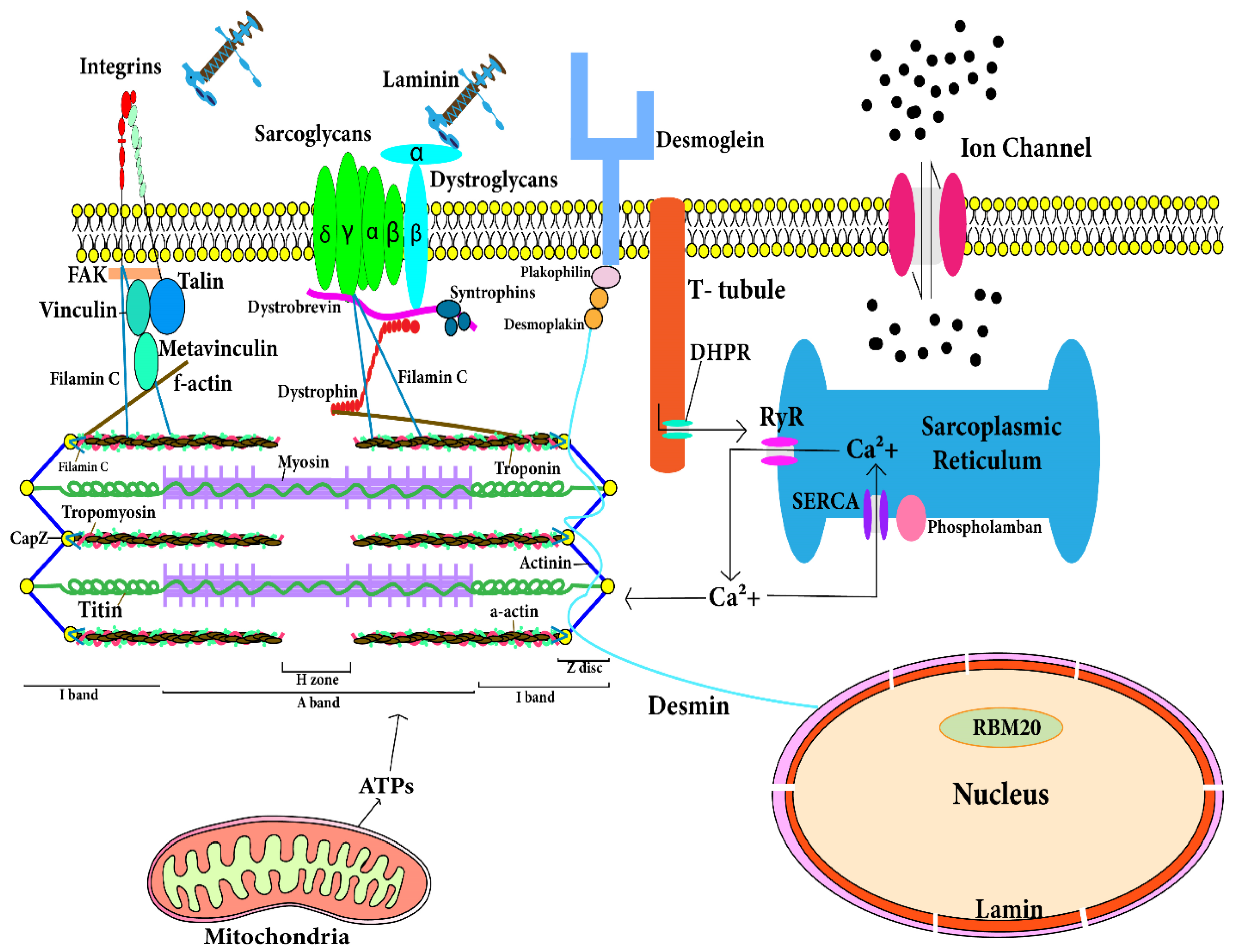
3. Genes Involved in Molecular Mechanisms of DCM
3.1. Troponin T Gene (TNNT2)
3.2. Cardiac Actin Alpha Gene (ACTC1)
3.3. Myosin Heavy Chain—β Gene (MYH7)
3.4. Cardiac Myosin Binding Protein C Gene (MYPBC3)
3.5. Tropomyosin α Gene (TPM1)
3.6. Titin Gene (TTN)
3.7. Desmin Gene (DES)
3.8. δ-Sarcoglycan Gene (SGCD)
3.9. Vinculin and Metavinculin Gene (VCL)
3.10. Lamin A and Lamin C Gene (LMNA)
3.11. Dystrophin Gene (DMD)
3.12. Tafazzin Gene (G4.5)
3.13. Phospholamban Gene (PLN)
3.14. Mitochondrial DNA
3.15. Filamin C (FLNC)
4. Mechanistic Insights for DCM Development
4.1. Signaling Pathways Involved in DCM
4.1.1. Cell Death Pathways in Cardiomyocytes
Extrinsic Pathway
Intrinsic Pathway
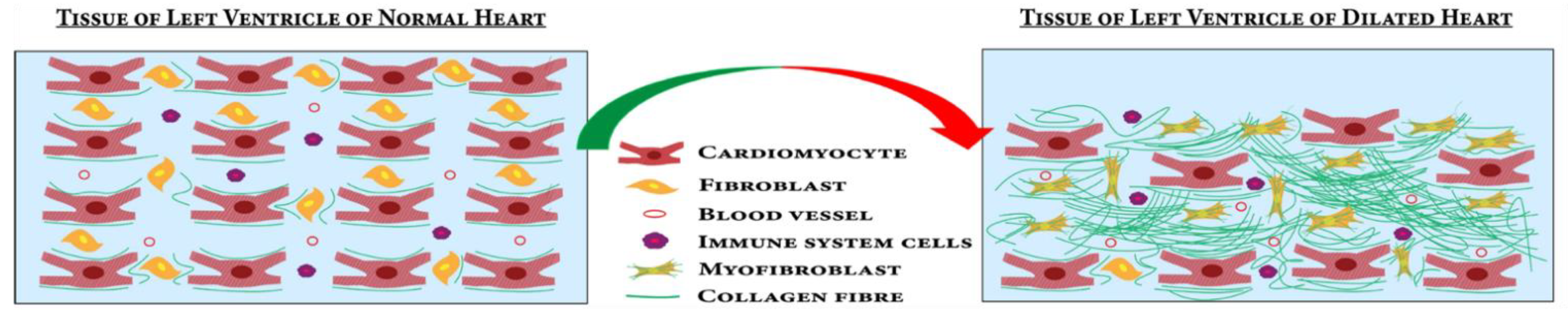
4.1.2. Fibrosis Pathways Associated with DCM
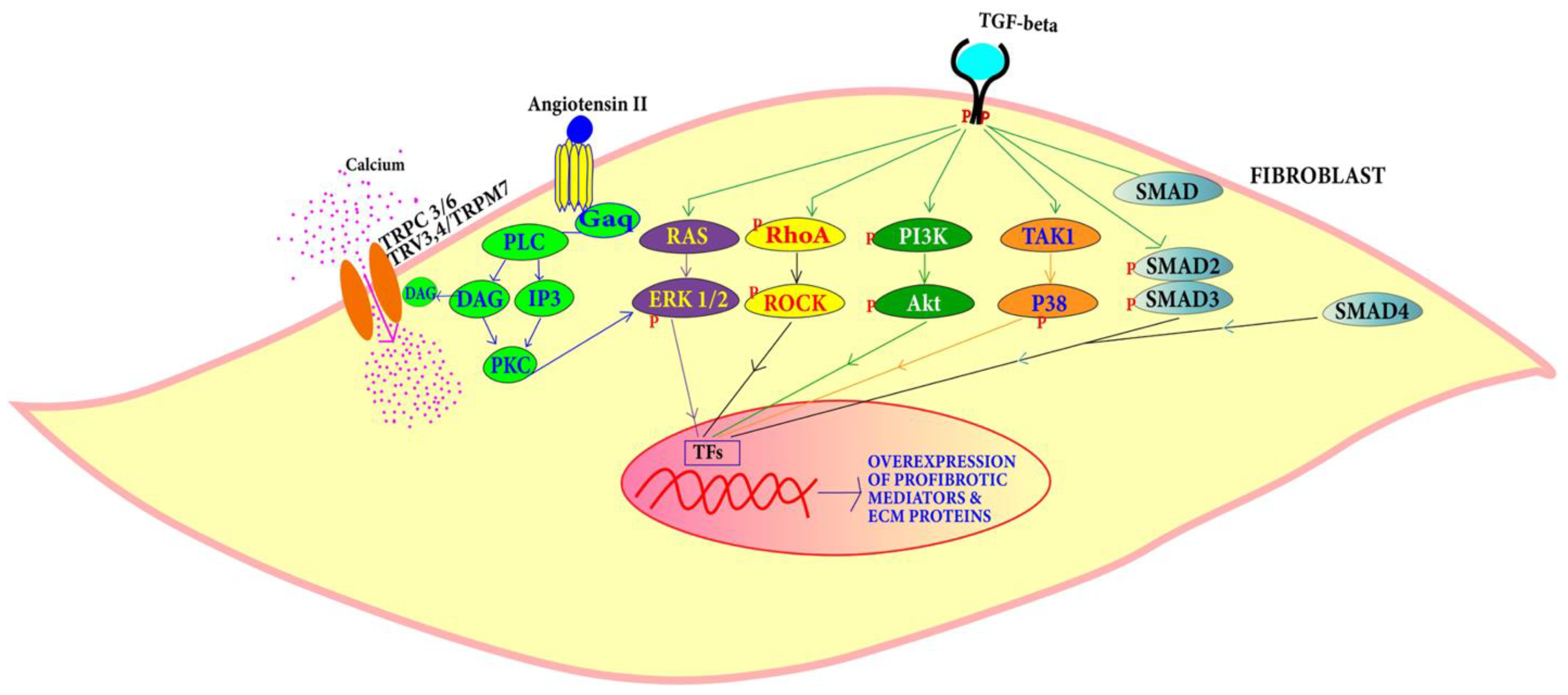
4.1.3. Angiotensin Pathway
5. Diagnosis of DCM
5.1. Clinical Investigation
5.2. Electrocardiography (ECG)
5.3. Echocardiography
5.4. Magnetic Resonance Imaging (MRI)
5.5. Serological Test for Virus
5.6. Endomyocardial Biopsy
5.7. Clinical Genetics of DCM
5.8. Pathological Tests for DCM Diagnosis
5.9. Protein Biomarkers of DCM
5.9.1. Brain Natriuretic Peptide (BNP)
5.9.2. ST2
5.9.3. Troponins T, I
5.9.4. Procollagen Type III
5.9.5. Matrix Metalloproteinase (MMP)
5.9.6. Galectin-3
6. Treatment Strategies of DCM
6.1. Angiotensin-Converting Enzyme (ACE) Inhibitors
6.2. Diuretics
6.3. Angiotensin II (Ang-II) Receptor Antagonists
6.4. Beta Blockers
6.5. Spironolactone
6.6. SGLT2 Inhibitors (SGLT2i)
6.7. Potential Novel Treatments of DCM
6.7.1. Cytokine Antagonists
6.7.2. Anticoagulants
6.7.3. Natriuretic Peptides
6.7.4. Stem Cell Therapy
6.7.5. Clinical Trials
6.8. Assisting Devices and Mechanical Support
6.8.1. Partial Left Ventriculectomy (PLV)
6.8.2. Left Ventricular Assist Devices (LVADs)
6.8.3. Multisite Ventricular Pacing
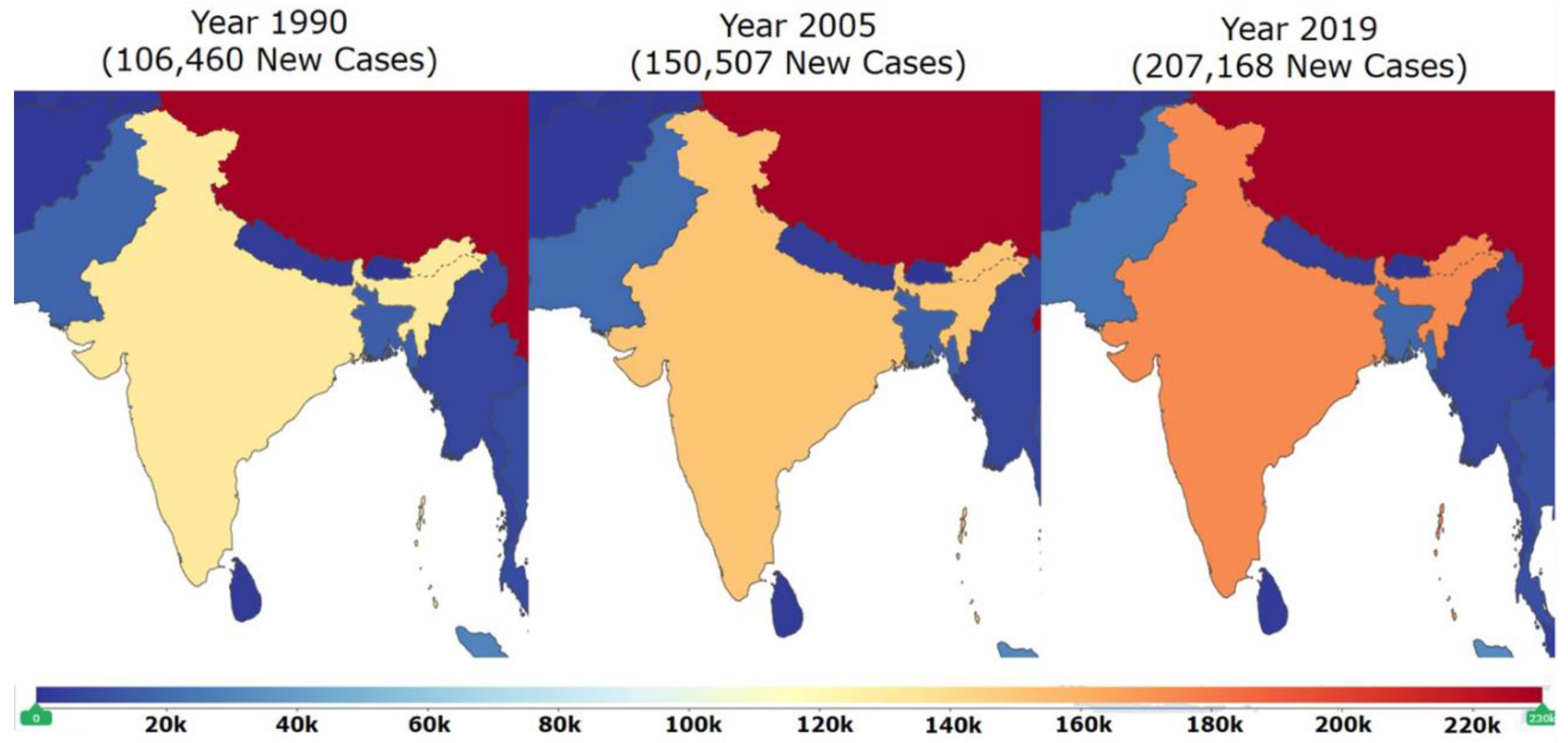
7. Epidemiological Studies in India on DCM
8. Limitations of Candidate-Gene Based Studies: Newer System-Level OMICS Approaches to the Rescue
9. Systems Biology Approach for Better Diagnosis of DCM
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kuhl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the european society of cardiology working group on myocardial and pericardial diseases. Eur. Hear J. 2007, 29, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B.; American Heart, A.; et al. Contemporary definitions and classification of the cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006, 113, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Ku, L.; Feiger, J.; Taylor, M.; Mestroni, L. Familial Dilated Cardiomyopathy. Circulation 2003, 108, e118–e121. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.C.; Calderon-Dominguez, M.; Mangas, A.; Campuzano, O.; Sarquella-Brugada, G.; Ramos, M.; Quezada-Feijoo, M.; Pinilla, J.M.G.; Robles-Mezcua, A.; Pacheco-Cruz, G.d.A.; et al. Circulating circRNA as biomarkers for dilated cardiomyopathy etiology. Klin. Wochenschr. 2021, 99, 1711–1725. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A.; Lowe, A.M.; Colan, S.D.; Sleeper, L.A.; Orav, E.J.; Clunie, S.; Messere, J.; Cox, G.F.; Lurie, P.R.; Hsu, D.; et al. Incidence, causes, and outcomes of dilated cardio-myopathy in children. Jama 2006, 296, 1867–1876. [Google Scholar] [CrossRef]
- Hazebroek, M.; Dennert, R.; Heymans, S. Idiopathic dilated cardiomyopathy: Possible triggers and treatment strategies. Neth. Hear J. 2012, 20, 332–335. [Google Scholar] [CrossRef]
- Weintraub, R.G.; Semsarian, C.; Macdonald, P. Dilated cardiomyopathy. Lancet 2017, 390, 400–414. [Google Scholar] [CrossRef]
- Morales, A.; Cowan, J.; Dagua, J.; Hershberger, R.E. Family History: An Essential Tool for Cardiovascular Genetic Medicine. Congest. Heart Fail. 2008, 14, 37–45. [Google Scholar] [CrossRef]
- Dilated Cardiomyopathy (DCM)|Pediatric Cardiomyopathy [Internet]. Available online: https://www.cincinnatichildrens.org/service/c/cardiomyopathy/types/dilated-cardiomyopathy (accessed on 21 April 2021).
- Hershberger, R.E.; Morales, A.; Siegfried, J.D. Clinical and genetic issues in dilated cardiomyopathy: A review for genetics professionals. Genet. Med. 2010, 12, 655–667. [Google Scholar] [CrossRef]
- Codd, M.B.; Sugrue, D.D.; Gersh, B.J.; Melton, L.J., 3rd. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975–1984. Circulation 1989, 80, 564–572. [Google Scholar] [CrossRef]
- Rakar, S.; Sinagra, G.; Di Lenarda, A.; Poletti, A.; Bussani, R.; Silvestri, F.; Camerini, F.; Heart Muscle Disease Study Group. Epidemiology of dilated cardiomyopathy: A prospective post-mortem study of 5252 necropsies. Eur. Heart J. 1997, 18, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef] [PubMed]
- Reichart, D.; Magnussen, C.; Zeller, T.; Blankenberg, S. Dilated cardiomyopathy: From epidemiologic to genetic phenotypes. J. Intern. Med. 2019, 286, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Institute for Health Metrics and Evaluation. Global Burden of Diseases (GBD) Compare. Available online: https://vizhub.healthdata.org/gbd-compare/ (accessed on 18 April 2021).
- Heymans, S.; Eriksson, U.; Lehtonen, J.; Cooper, L.T. The Quest for New Approaches in Myocarditis and Inflammatory Car-diomyopathy. J. Am. Coll. Cardiol. 2016, 68, 2348–2364. [Google Scholar] [CrossRef]
- Caforio, A.L.P.; Pankuweit, S.; Arbustini, E.; Basso, C.; Gimeno-Blanes, J.; Felix, S.B.; Fu, M.; Heliö, T.; Heymans, S.; Jahns, R.; et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 2636–2648. [Google Scholar] [CrossRef]
- Maisch, B.; Richter, A.; Sandmöller, A.; Portig, I.; Pankuweit, S. Inflammatory Dilated Cardiomyopathy (DCMI). Herz 2005, 30, 535–544. [Google Scholar] [CrossRef]
- Kindermann, I.; Kindermann, M.; Kandolf, R.; Klingel, K.; Bültmann, B.; Müller, T.; Lindinger, A.; Boühm, M. Predictors of Outcome in Patients with Suspected Myocarditis. Circulation 2008, 118, 639–648. [Google Scholar] [CrossRef]
- Mahrholdt, H.; Goedecke, C.; Wagner, A.; Meinhardt, G.; Athanasiadis, A.; Vogelsberg, H.; Fritz, P.; Klingel, K.; Kandolf, R.; Sechtem, U. Cardiovascular magnetic resonance assessment of human myocarditis: A comparison to histology and molecular pathology. Circulation 2004, 109, 1250–1258. [Google Scholar] [CrossRef]
- Kearney, M.T.; Cotton, J.; Richardson, P.J.; Shah, A. Viral myocarditis and dilated cardiomyopathy: Mechanisms, manifestations, and management. Postgrad. Med. J. 2001, 77, 4–10. [Google Scholar] [CrossRef]
- Oropeza-Moe, M.; Wisløff, H.; Bernhoft, A. Selenium deficiency associated porcine and human cardiomyopathies. J. Trace Elem. Med. Biol. 2015, 31, 148–156. [Google Scholar] [CrossRef]
- Tirumanisetty, P.; Abdullah, A.; Matos, M. Pulmonary arteriovenous malformation: A myriad of presentations. Chest 2018, 154, 1032A. [Google Scholar] [CrossRef]
- Seki, A.; Patel, S.; Ashraf, S.; Perens, G.; Fishbein, M.C. Primary endocardial fibroelastosis: An underappreciated cause of car-diomyopathy in children. Cardiovasc. Pathol. 2013, 22, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Yin, L. Non-compact cardiomyopathy or ventricular non-compact syndrome? J. Cardiovasc. Ultrasound 2014, 22, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, D.; Raychaudhuri, M. Infants with dilated cardiomyopathy and hypocalcemia. Indian J. Endocrinol. Metab. 2013, 17, 221–223. [Google Scholar] [CrossRef]
- Jefferies, J.L. Barth syndrome. Am. J. Med. Genet. Part C (Semin. Med. Genet.) 2013, 163, 198–205. [Google Scholar] [CrossRef]
- Aprikyan, A.A.; Khuchua, Z. Advances in the understanding of Barth syndrome. Br. J. Haematol. 2013, 161, 330–338. [Google Scholar] [CrossRef]
- Rampersaud, E.; Siegfried, J.D.; Norton, N.; Li, D.; Martin, E.; Hershberger, R.E. Rare variant mutations identified in pediatric patients with dilated cardiomyopathy. Prog. Pediatr. Cardiol. 2011, 31, 39–47. [Google Scholar] [CrossRef]
- Khan, R.S.; Pahl, E.; Dellefave-Castillo, L.; Rychlik, K.; Ing, A.; Yap, K.L.; Brew, C.; Johnston, J.R.; McNally, E.M.; Webster, G. Genotype and Cardiac Outcomes in Pediatric Dilated Cardiomyopathy. J. Am. Hear. Assoc. 2022, 11, e022854. [Google Scholar] [CrossRef]
- Luisa, M.; Francesca, B.; Anita, S.; Gianfranco, S.; Taylor, M.R. Genetic causes of Dilated Cardiomyopathy. Prog. Pediatr. Cardiol. 2014, 37, 13–18. [Google Scholar]
- Arbustini, E.; Narula, N.; Dec, G.W.; Reddy, K.S.; Greenberg, B.; Kushwaha, S.; Marwick, T.; Pinney, S.; Bellazzi, R.; Favalli, V.; et al. The MOGE(S) classification for a phenotype-genotype nomenclature of cardiomyopathy: Endorsed by the World Heart Federation. J. Am. Coll. Cardiol. 2013, 62, 2046–2072. [Google Scholar] [CrossRef]
- Schultheiss, H.P.; Fairweather, D.; Caforio, A.L.; Escher, F.; Hershberger, R.E.; Lipshultz, S.E.; Liu, P.P.; Matsumori, A.; Mazzanti, A.; McMurray, J.; et al. Dilated cardiomyopathy. Nat. Rev. Dis. Primers 2019, 5, 32. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Cowan, J.; Jordan, E.; Kinnamon, D.D. The Complex and Diverse Genetic Architecture of Dilated Cardiomyopathy. Circ. Res. 2021, 128, 1514–1532. [Google Scholar] [CrossRef] [PubMed]
- Maisch, B. Alcoholic cardiomyopathy: The result of dosage and individual predisposition. Herz 2016, 41, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, H.A. Tachycardia-induced Cardiomyopathy (Tachycardiomyopathy). Libyan J. Med. 2007, 2, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Bates, M.G.; Bourke, J.P.; Giordano, C.; D’Amati, G.; Turnbull, D.M.; Taylor, R.W. Cardiac involvement in mitochondrial DNA disease: Clinical spectrum, diagnosis, and management. Eur. Heart J. 2012, 33, 3023–3033. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Corrado, D.; Marcus, F.I.; Nava, A.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2009, 373, 1289–1300. [Google Scholar] [CrossRef]
- Bluett, R.; McDonnell, D.; O’Dowling, C.; Vaughan, C. Eosinophilic myocarditis as a first presentation of eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome). Case Rep. 2017, 2017, bcr-2017. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Murai, S.; Ohte, N. Dilated Cardiomyopathy with Eosinophilic Granulomatosis with Polyangiitis in Which Active Myocardial Inflammation Was Only Detected by Endomyocardial Biopsy. Intern. Med. 2018, 57, 2675–2679. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-X.; Yu, B.-L.; Peng, D.-Q.; Zhou, S.-H. Eosinophilic myocarditis due to Churg–Strauss syndrome mimicking reversible dilated cardiomyopathy. Hear Lung 2014, 43, 45–47. [Google Scholar] [CrossRef]
- Lipshultz, S.E.; Law, Y.M.; Asante-Korang, A.; Austin, E.D.; Dipchand, A.I.; Everitt, M.D.; Hsu, D.T.; Lin, K.Y.; Price, J.F.; Wilkinson, J.D.; et al. Cardiomyopathy in Children: Clas-sification and Diagnosis: A Scientific Statement from the American Heart Association. Circulation 2019, 140, e9–e68. [Google Scholar] [CrossRef]
- Pearson, G.D.; Veille, J.C.; Rahimtoola, S.; Hsia, J.; Oakley, C.M.; Hosenpud, J.D.; Ansari, A.; Baughman, K.L. Peripartum cardiomyopathy: National Heart, Lung, and Blood Institute and Office of Rare Diseases (National Institutes of Health) workshop recommendations and review. Jama 2000, 283, 1183–1188. [Google Scholar] [CrossRef] [PubMed]
- Klein, I.; Danzi, S. Thyroid disease and the heart. Circulation 2007, 116, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Mobine, H.R.; Baker, A.B.; Wang, L.; Wakimoto, H.; Jacobsen, K.C.; Seidman, C.E.; Seidman, J.G.; Edelman, E.R. Pheochromocytoma-induced cardiomyopathy is modulated by the synergistic effects of cell-secreted factors. Circ. Heart Fail. 2009, 2, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Bradley, T.J.; Slorach, C.; Mahmud, F.H.; Dunger, D.B.; Deanfield, J.; Deda, L.; Elia, Y.; Har, R.L.H.; Hui, W.; Moineddin, R.; et al. Early changes in cardiovascular structure and function in adolescents with type 1 diabetes. Cardiovasc. Diabetol. 2016, 15, 31. [Google Scholar] [CrossRef] [PubMed]
- Albakri, A. Nutritional deficiency cardiomyopathy: A review and pooled analysis of pathophysiology, diagnosis and clinical management. Res. Rev. Insights 2019, 3, 1–19. [Google Scholar] [CrossRef]
- Kamisago, M.; Sharma, S.D.; De Palma, S.R.; Solomon., S.; Sharma, P.; McDonough, B.; Smoot, L.; Mullen, M.P.; Woolf, P.K.; Wigle, E.D.; et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. ACC Curr. J. Rev. 2001, 10, 55. [Google Scholar] [CrossRef]
- Olson, T.M.; Michels, V.V.; Thibodeau, S.N.; Tai, Y.-S.; Keating, M.T. Actin Mutations in Dilated Cardiomyopathy, a Heritable Form of Heart Failure. Science 1998, 280, 750–752. [Google Scholar] [CrossRef]
- Daehmlow, S.; Erdmann, J.; Knueppel, T.; Gille, C.; Froemmel, C.; Hummel, M.; Hetzer, R.; Regitz-Zagrosek, V. Novel mutations in sarcomeric protein genes in dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 2002, 298, 116–120. [Google Scholar] [CrossRef]
- Olson, T.M.; Kishimoto, N.Y.; Whitby, F.G.; Michels, V.V. Mutations that alter the surface charge of alpha-tropomyosin are asso-ciated with dilated cardiomyopathy. J. Mol. Cell. Cardiol. 2001, 33, 723–732. [Google Scholar] [CrossRef]
- Gerull, B.; Gramlich, M.; Atherton, J.; McNabb, M.; Trombitás, K.; Sasse-Klaassen, S.; Seidman, J.G.; Seidman, C.; Granzier, H.; Labeit, S.; et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat. Genet. 2002, 30, 201–204. [Google Scholar] [CrossRef]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Verdonschot, J.A.; Hazebroek, M.R.; Krapels, I.P.; Henkens, M.T.; Raafs, A.; Wang, P.; Merken, J.J.; Claes, G.R.; Vanhoutte, E.K.; Wijngaard, A.V.D.; et al. Implications of Genetic Testing in Dilated Cardiomyopathy. Circ. Genom. Precis. Med. 2020, 13, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Tapscoft, T.; Gonzalez, O.; Burch, P.E.; Quiñones, M.A.; Zoghbi, W.A.; Hill, R.; Bachinski, L.L.; Mann, D.; Roberts, R. Desmin Mutation Responsible for Idiopathic Dilated Cardiomyopathy. Circulation 1999, 100, 461–464. [Google Scholar] [CrossRef]
- Tsubata, S.; Bowles, K.R.; Vatta, M.; Zintz, C.; Titus, J.; Muhonen, L.; Bowles, N.E.; Towbin, J.A. Mutations in the human delta-sarcoglycan gene in familial and sporadic dilated cardiomyopathy. J. Clin. Investig. 2000, 106, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Heling, A.; Zimmermann, R.; Kostin, S.; Maeno, Y.; Hein, S.; Devaux, B.; Bauer, E.; Kloüvekorn, W.P.; Schlepper, M.; Schaper, W.; et al. Increased expression of cytoskeletal, linkage, and extracellular proteins in failing human myocardium. Circ. Res. 2000, 86, 846–853. [Google Scholar] [CrossRef]
- Olson, T.M.; Illenberger, S.; Kishimoto, N.Y.; Huttelmaier, S.; Keating, M.T.; Jockusch, B.M. Metavinculin mutations alter actin interaction in dilated cardiomyopathy. Circulation 2002, 105, 431–437. [Google Scholar] [CrossRef]
- Fatkin, D.; Macrae, C.; Sasaki, T.; Wolff, M.R.; Porcu, M.; Frenneaux, M.; Atherton, J.; Vidaillet, H.J., Jr.; Spudich, S.; De Girolami, U.; et al. Missense Mutations in the Rod Domain of the Lamin A/C Gene as Causes of Dilated Cardiomyopathy and Conduction System Disease. N. Engl. J. Med. 1999, 341, 1715–1724. [Google Scholar] [CrossRef]
- DMD Gene: MedlinePlus Genetics. Available online: https://medlineplus.gov/genetics/gene/dmd/#references (accessed on 21 April 2021).
- Towbin, J.A.; Hejtmancik, J.F.; Brink, P.; Gelb, B.; Zhu, X.M.; Chamberlain, J.S.; McCabe, E.R.; Swift, M. X-linked dilated cardiomyopathy. Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation 1993, 87, 1854–1865. [Google Scholar] [CrossRef]
- Franz, W.-M.; Müller, M.; Muller, O.J.; Herrmann, R.; Rothmann, T.; Cremer, M.; Cohn, R.D.; Voit, T.; Katus, A.H. Association of nonsense mutation of dystrophin gene with disruption of sarcoglycan complex in X-linked dilated cardiomyopathy. Lancet 2000, 355, 1781–1785. [Google Scholar] [CrossRef]
- Bleyl, S.B.; Mumford, B.R.; Thompson, V.; Carey, J.C.; Pysher, T.J.; Chin, T.K.; Ward, K. Neonatal, lethal noncompaction of the left ventricular myocardium is allelic with Barth syndrome. Am. J. Hum. Genet. 1997, 61, 868–872. [Google Scholar] [CrossRef]
- Haghighi, K.; Kolokathis, F.; Gramolini, A.O.; Waggoner, J.R.; Pater, L.; Lynch, R.A.; Fan, G.C.; Tsiapras, D.; Parekh, R.R.; Dorn, G.W.; et al. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc. Natl. Acad. Sci. USA 2006, 103, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, J.P.; Kamisago, M.; Asahi, M.; Li, G.H.; Ahmad, F.; Mende, U.; Kranias, E.G.; MacLennan, D.H.; Seidman, J.G.; Seidman, C.E. Dilated Cardiomyopathy and Heart Failure Caused by a Mutation in Phospholamban. Science 2003, 299, 1410–1413. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Mott, J.L.; Farrar, P.; Ryerse, J.S.; Chang, S.-W.; Stevens, M.; Denniger, G.; Zassenhaus, H.P. Mitochondrial DNA mutations activate the mitochondrial apoptotic pathway and cause dilated cardiomyopathy. Cardiovasc. Res. 2003, 57, 147–157. [Google Scholar] [CrossRef]
- Arbustini, E.; Diegoli, M.; Fasani, R.; Grasso, M.; Morbini, P.; Banchieri, N.; Bellini, O.; Bello, B.D.; Pilotto, A.; Magrini, G.; et al. Mitochondrial DNA Mutations and Mitochondrial Abnormalities in Dilated Cardiomyopathy. Am. J. Pathol. 1998, 153, 1501–1510. [Google Scholar] [CrossRef]
- Begay, R.; Graw, S.L.; Sinagra, G.; Asimaki, A.; Rowland, T.J.; Slavov, D.B.; Gowan, K.; Jones, K.L.; Brun, F.; Merlo, M.; et al. Filamin C Truncation Mutations Are Associated with Arrhythmogenic Dilated Cardiomyopathy and Changes in the Cell–Cell Adhesion Structures. JACC Clin. Electrophysiol. 2018, 4, 504–514. [Google Scholar] [CrossRef]
- Agarwal, R.; Paulo, J.A.; Toepfer, C.N.; Ewoldt, J.K.; Sundaram, S.; Chopra, A.; Zhang, Q.; Gorham, J.; DePalma, S.R.; Chen, C.S.; et al. Filamin C Cardiomyopathy Variants Cause Protein and Lysosome Accumulation. Circ. Res. 2021, 129, 751–766. [Google Scholar] [CrossRef]
- Crocini, C.; Gotthardt, M. Cardiac sarcomere mechanics in health and disease. Biophys. Rev. 2021, 13, 637–652. [Google Scholar] [CrossRef]
- McNally, E.M.; Mestroni, L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef]
- Hong, B.K.; Kwon, H.M.; Byun, K.H.; Kim, D.; Choi, E.Y.; Kang, T.S.; Kang, S.; Chun, K.J.; Jang, Y.; Kim, H.S.; et al. Apoptosis in dilated cardiomyopathy. Korean J. Intern. Med. 2000, 15, 56–64. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4531747/ (accessed on 21 April 2021).
- Agapitos, E.; Kavantzas, N.; Nanas, J.; Margari, Z.; Bakouris, M.; Kassis, K.; Panolaridis, A.; Davaris, P. The myocardial fibrosis in patients with dilated cardiomyopathy: The application of image analysis in the myocardial biopsies. Gen. Diagn. Pathol. 1995, 141, 305–311. [Google Scholar]
- Shinde, A.V.; Frangogiannis, N.G. Fibroblasts in myocardial infarction: A role in inflammation and repair. J. Mol. Cell. Cardiol. 2014, 70, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.Y.; Miyamoto, S.D.; Schumacher, K.R. Dilated Cardiomyopathy. In Heart Failure in the Child and Young Adult: From Bench to Bedside; Elsevier: Amsterdam, The Netherlands, 2018; pp. 203–213. [Google Scholar]
- DCM-Symptoms and Causes-Mayo Clinic [Internet]. Available online: https://www.mayoclinic.org/diseases-conditions/dilated-cardiomyopathy/symptoms-causes/syc-20353149 (accessed on 21 April 2021).
- Twu, C.; Liu, N.Q.; Popik, W.; Bukrinsky, M.; Sayre, J.; Roberts, J.; Rania, S.; Bramhandam, V.; Roos, K.P.; MacLellan, W.R.; et al. Cardiomyocytes undergo apoptosis in human immunode-ficiency virus cardiomyopathy through mitochondrion and death receptor-controlled pathways. Proc. Natl. Acad. Sci. USA 2002, 99, 14386–14391. Available online: www.pnas.org (accessed on 21 April 2021).
- McGowan, J.V.; Chung, R.; Maulik, A.; Piotrowska, I.; Walker, J.M.; Yellon, D.M. Anthracycline Chemotherapy and Cardiotoxicity. Cardiovasc. Drugs Ther. 2017, 31, 63–75. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5346598/ (accessed on 21 April 2021).
- Wencker, D.; Chandra, M.; Nguyen, K.; Miao, W.; Garantziotis, S.; Factor, S.M.; Shirani, J.; Armstrong, R.C.; Kitsis, R.N. A mechanistic role for cardiac myocyte apoptosis in heart failure. J. Clin. Investig. 2003, 111, 1497–1504. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Liu, Y.; Cheng, Z. Signaling Pathways in Cardiac Myocyte Apoptosis. BioMed Res. Int. 2016, 2016, 9583268. [Google Scholar] [CrossRef]
- Ramaccini, D.; Montoya-Uribe, V.; Aan, F.J.; Modesti, L.; Potes, Y.; Wieckowski, M.R.; Krga, I.; Glibetić, M.; Pinton, P.; Giorgi, C.; et al. Mitochondrial Function and Dysfunction in Dilated Cardiomyopathy. Front. Cell Dev. Biol. 2021, 8, 1800. [Google Scholar] [CrossRef]
- Mitra, A.; Basak, T.; Datta, K.; Naskar, S.; Sengupta, S.; Sarkar, S. Role of α-crystallin B as a regulatory switch in modulating cardiomyocyte apoptosis by mitochondria or endoplasmic reticulum during cardiac hypertrophy and myocardial infarction. Cell Death Dis. 2013, 4, e582. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming growth factor (TGF)-β signaling in cardiac remodeling. J. Mol. Cell. Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef]
- Li, R.K.; Li, G.; Mickle, D.A.; Weisel, R.D.; Merante, F.; Luss, H.; Rao, V.; Christakis, G.T.; Williams, W.G. Overexpression of transforming growth factor-beta1 and insulinlike growth factor-I in patients with idiopathic hypertrophic cardiomyopathy. Circulation 1997, 96, 874–881. [Google Scholar] [CrossRef]
- Schneiders, D.; Heger, J.; Best, P.; Taimor, G.; Piper, H.M. SMAD proteins are involved in apoptosis induction in ventricular cardiomyocytes. Cardiovasc. Res. 2005, 67, 87–96. [Google Scholar] [CrossRef]
- Huntgeburth, M.; Tiemann, K.; Shahverdyan, R.; Schlüter, K.-D.; Schreckenberg, R.; Gross, M.-L.; Mödersheim, S.; Caglayan, E.; Müller-Ehmsen, J.; Ghanem, A.; et al. Transforming Growth Factor β1 Oppositely Regulates the Hypertrophic and Contractile Response to β-Adrenergic Stimulation in the Heart. PLoS ONE 2011, 6, e26628. [Google Scholar] [CrossRef]
- Pellieux, C.; Foletti, A.; Peduto, G.; Aubert, J.F.; Nussberger, J.; Beermann, F.; Brunner, H.R.; Pedrazzini, T. Dilated cardiomyopathy and impaired cardiac hypertrophic response to angiotensin II in mice lacking FGF-2. J. Clin. Investig. 2001, 108, 1843–1851. [Google Scholar] [CrossRef]
- Merl-Pham, J.; Basak, T.; Knüppel, L.; Ramanujam, D.; Athanason, M.; Behr, J.; Engelhardt, S.; Eickelberg, O.; Hauck, S.M.; Vanacore, R.; et al. Quantitative proteomic profiling of extracellular matrix and site-specific collagen post-translational modifications in an in vitro model of lung fibrosis. Matrix Biol. Plus 2019, 1, 100005. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhou, G.; Hu, M.C.; Yao, Z.; Tan, T.H. Activation of the hematopoietic progenitor kinase-1 (HPK1)-dependent, stress-activated c-Jun N-terminal kinase (JNK) pathway by transforming growth factor beta (TGF-β)-activated kinase (TAK1), a kinase mediator of TGF beta signal transduction. J. Biol. Chem. 1997, 272, 22771–22775. [Google Scholar] [CrossRef] [PubMed]
- Courcelles, M.; Frémin, C.; Voisin, L.; Lemieux, S.; Meloche, S.; Thibault, P. Phosphoproteome dynamics reveal novel ERK1/2 MAP kinase substrates with broad spectrum of functions. Mol. Syst. Biol. 2013, 9, 669. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Liao, J.K. Rho Kinases and Cardiac Remodeling. Circ. J. Off. J. Jpn. Circ. Soc. 2016, 80, 1491–1498. [Google Scholar] [CrossRef] [PubMed]
- Wieder, N.; Greka, A. Calcium, TRPC channels, and regulation of the actin cytoskeleton in podocytes: Towards a future of targeted therapies. Pediatric Nephrol. 2016, 31, 1047–1054. [Google Scholar] [CrossRef]
- Dilated Cardiomyopathy (DCM)-Cardiomyopathy UK [Internet]. Available online: https://www.cardiomyopathy.org/dilated-cardiomyopathy/intro (accessed on 20 April 2021).
- Duran, J.; Martinez, A.; Adler, E. Cardiovascular Manifestations of Mitochondrial Disease. Biology 2019, 8, 34. [Google Scholar] [CrossRef]
- Dilated Cardiomyopathy (DCM) • LITFL • ECG Library Diagnosis [Internet]. Available online: https://litfl.com/dilated-cardiomyopathy-dcm-ecg-library/ (accessed on 20 April 2021).
- Finocchiaro, G.; Merlo, M.; Sheikh, N.; De Angelis, G.; Papadakis, M.; Olivotto, I.; Rapezzi, C.; Carr-White, G.; Sharma, S.; Mestroni, L.; et al. The electrocardiogram in the diagnosis and management of patients with dilated cardiomyopathy. Eur. J. Hear Fail. 2020, 22, 1097–1107. [Google Scholar] [CrossRef]
- Mathew, T.; Williams, L.; Navaratnam, G.; Rana, B.; Wheeler, R.; Collins, K.; Harkness, A.; Jones, R.; Knight, D.; O’Gallagher, K.; et al. Diagnosis and assessment of dilated cardio-myopathy: A guideline protocol from the British Society of Echocardiography. Echo Res. Pract. 2017, 4, G1–G13. [Google Scholar] [CrossRef]
- Green, J.J.; Berger, J.S.; Kramer, C.M.; Salerno, M. Prognostic Value of Late Gadolinium Enhancement in Clinical Outcomes for Hypertrophic Cardiomyopathy. JACC Cardiovasc. Imaging 2012, 5, 370–377. [Google Scholar] [CrossRef]
- Japp, A.G.; Gulati, A.; Cook, S.A.; Cowie, M.; Prasad, S.K. The Diagnosis and Evaluation of Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2996–3010. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.A.; Lapeyre, A.C.; Cooper, L.T. Current Role of Endomyocardial Biopsy in the Management of Dilated Cardiomyopathy and Myocarditis. Mayo Clin. Proc. 2001, 76, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Jordan, E.; Peterson, L.; Ai, T.; Asatryan, B.; Bronicki, L.; Brown, E.; Celeghin, R.; Edwards, M.; Fan, J.; Ingles, J.; et al. Evidence-Based Assessment of Genes in Dilated Cardiomyopathy. Circulation 2021, 144, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Rapezzi, C.; Arbustini, E.; Caforio, A.L.P.; Charron, P.; Blanes, J.G.; Heliö, T.; Linhart, A.; Mogensen, J.; Pinto, Y.; Ristic, A.; et al. Diagnostic work-up in cardiomyopathies: Bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Hear J. 2012, 34, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- Suresh, C.P.; Saha, A.; Kaur, M.; Kumar, R.; Dubey, N.K.; Basak, T.; Tanwar, V.S.; Bhardwaj, G.; Sengupta, S.; Batra, V.V.; et al. Differentially expressed urinary biomarkers in children with idiopathic nephrotic syndrome. Clin. Exp. Nephrol. 2015, 20, 273–283. [Google Scholar] [CrossRef]
- Felker, G.M.; Hasselblad, V.; Hernandez, A.F.; O’Connor, C.M. Biomarker-guided therapy in chronic heart failure: A meta-analysis of randomized controlled trials. Am. Hear J. 2009, 158, 422–430. [Google Scholar] [CrossRef]
- Januzzi, J.L.; Peacock, W.F.; Maisel, A.S.; Chae, C.U.; Jesse, R.L.; Baggish, A.L.; O’Donoghue, M.; Sakhuja, R.; Chen, A.A.; van Kimmenade, R.R.; et al. Measurement of the Interleukin Family Member ST2 in Patients with Acute Dyspnea: Results from the PRIDE (Pro-Brain Natriuretic Peptide Investigation of Dyspnea in the Emergency Department) Study. J. Am. Coll. Cardiol. 2007, 50, 607–613. [Google Scholar] [CrossRef]
- DeFilippi, C.R.; de Lemos, J.A.; Christenson, R.H.; Gottdiener, J.S.; Kop, W.J.; Zhan, M.; Seliger, S.L. Association of serial measures of cardiac troponin T using a sensitive assay with incident heart failure and cardiovascular mortality in older adults. Jama 2010, 304, 2494–2502. [Google Scholar] [CrossRef]
- Cicoira, M.; Rossi, A.; Bonapace, S.; Zanolla, L.; Golia, G.; Franceschini, L.; Caruso, B.; Marino, P.N.; Zardini, P. Independent and additional prognostic value of aminoterminal propeptide of type III procollagen circulating levels in patients with chronic heart failure. J. Card. Fail. 2004, 10, 403–411. [Google Scholar] [CrossRef]
- Yan, A.T.; Yan, R.T.; Spinale, F.G.; Afzal, R.; Gunasinghe, H.R.; Arnold, M.; Demers, C.; Mckelvie, R.S.; Liu, P.P. Plasma Matrix Metalloproteinase-9 Level Is Correlated with Left Ventricular Volumes and Ejection Fraction in Patients with Heart Failure. J. Card. Fail. 2006, 12, 514–519. [Google Scholar] [CrossRef]
- Kapur, N.K.; Heffernan, K.S.; Yunis, A.A.; Parpos, P.; Kiernan, M.S.; Sahasrabudhe, N.A.; Kimmelstiel, C.D.; Kass, D.A.; Karas, R.H.; Mendelsohn, M.E. Usefulness of Soluble Endoglin as a Noninvasive Measure of Left Ventricular Filling Pressure in Heart Failure. Am. J. Cardiol. 2010, 106, 1770–1776. [Google Scholar] [CrossRef] [PubMed]
- Herman, L.L.; Padala, S.A.; Annamaraju, P.; Bashir, K. Angiotensin Converting Enzyme Inhibitors (ACEI); StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Packer, M.; Poole-Wilson, P.A.; Armstrong, P.W.; Cleland, J.G.; Horowitz, J.D.; Massie, B.M.; Rydén, L.; Thygesen, K.; Uretsky, B.F. Comparative effects of low and high doses of the angiotensin-converting enzyme inhibitor, lisinopril, on morbidity and mortality in chronic heart failure. ATLAS Study Group. Circulation 1999, 100, 2312–2318. [Google Scholar] [CrossRef]
- Diuretics [Internet]. Available online: https://www.cvpharmacology.com/diuretic/diuretics (accessed on 19 April 2021).
- Ballew, J.R.; Fink, G.D. Characterization of the antihypertensive effect of a thiazide diuretic in angiotensin II-induced hy-pertension. J. Hypertens. 2001, 19, 1601–1606. [Google Scholar] [CrossRef] [PubMed]
- Barreras, A.; Gurk-Turner, C. Angiotensin Ii Receptor Blockers. Bayl. Univ. Med Cent. Proc. 2003, 16, 123–126. [Google Scholar] [CrossRef]
- Beta-Blockers for Heart Disease: Benefits, Risks, and More [Internet]. Available online: https://www.healthline.com/health/heart-disease/beta-blockers (accessed on 19 April 2021).
- Böhm, M.; Maack, C. Treatment of heart failure with beta-blockers. Mechanisms and results. Basic Res. Cardiol. 2000, 95, I15–I24. [Google Scholar] [CrossRef]
- James, P.A.; Oparil, S.; Carter, B.L.; Cushman, W.C.; Dennison-Himmelfarb, C.; Handler, J.; Lackland, D.T.; LeFevre, M.L.; MacKenzie, T.D.; Ogedegbe, O.; et al. 2014 evidence-based guideline for the management of high blood pressure in adults: Report from the panel members appointed to the Eighth Joint National Committee (JNC 8). Jama 2014, 311, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Sica, D.A. Pharmacokinetics and Pharmacodynamics of Mineralocorticoid Blocking Agents and their Effects on Potassium Homeostasis. Hear Fail. Rev. 2005, 10, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.; Shubrook, J.H. Sodium glucose co-transporter 2 inhibitors in the treatment of type 2 diabetes mellitus. Am. Coll. Osteopath. Fam. Physicians 2015, 7, 10–30. [Google Scholar]
- Zou, H.; Zhou, B.; Xu, G. SGLT2 inhibitors: A novel choice for the combination therapy in diabetic kidney disease. Cardiovasc. Diabetol. 2017, 16, 65. [Google Scholar] [CrossRef]
- Blum, A.; Miller, H. Pathophysiological role of cytokines in congestive heart failure. Annu. Rev. Med. 2001, 52, 15–27. [Google Scholar] [CrossRef]
- Hassan, I.; Dorjay, K.; Anwar, P. Pentoxifylline and its applications in dermatology. Indian Dermatol. Online J. 2014, 5, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.J.; Roux, S. Bosentan: A dual endothelin receptor antagonist. Expert Opin. Investig. Drugs 2002, 11, 991–1002. [Google Scholar] [PubMed]
- Koniaris, L.S.; Goldhaber, S.Z. Anticoagulation in dilated cardiomyopathy. J. Am. Coll. Cardi-Ology 1998, 31, 745–748. [Google Scholar] [CrossRef]
- Riegger, A.J.G. Atrial Natriuretic Peptide in Heart Failure. In Endocrinology of the Heart; Springer: Berlin, Heidelberg, 1989; pp. 85–89. [Google Scholar]
- Seth, S.; Bhargava, B.; Narang, R.; Ray, R.; Mohanty, S.; Gulati, G.; Kumar, L.; Airan, B.; Venugopal, P. The ABCD (Autologous Bone Marrow Cells in Dilated Cardiomyopathy) Trial. J. Am. Coll. Cardiol. 2010, 55, 1643–1644. [Google Scholar] [CrossRef] [PubMed]
- U.S. National Library of Medicine. Clinical Trials.gov. Available online: https://www.clinicaltrials.gov/ (accessed on 17 September 2020).
- Yoshii, S.; Hosaka, S.; Suzuki, S.; Osawa, H.; Akashi, O.; Tada, Y.; Sugiyama, H.; Hoshiai, M.; Tan, T.; Kadono, T.; et al. Indications for partial left ventriculectomy in pediatric dilated cardiomyopathy. Circ. J. 2002, 66, 337–340. [Google Scholar] [CrossRef][Green Version]
- Franco-Cereceda, A.; McCarthy, P.M.; Blackstone, E.H.; Hoercher, K.J.; White, J.A.; Young, J.B.; Starling, R.C. Partial left ventriculectomy for dilated cardiomyopathy: Is this an alternative to transplantation? J. Thorac. Cardiovasc. Surg. 2001, 121, 879–893. [Google Scholar] [CrossRef] [PubMed]
- Mancini, D.; Colombo, P.C. Left ventricular assist devices: A rapidly evolving alternative to transplant. J. Am. Coll. Cardiol. 2015, 65, 2542–2555. [Google Scholar] [CrossRef]
- Sipahi, I.; Fang, J.C. QRS duration criteria to select patients for cardiac resynchronization therapy: Crt should be reserved for a QRS duration ≥150 ms: Pro. Circ. Arrhythmia Electrophysiol. 2013, 6, 436–441. [Google Scholar] [CrossRef]
- Dhandapany, P.S.; Sadayappan, S.; Xue, Y.; Powell, G.T.; Rani, D.S.; Nallari, P.; Rai, T.S.; Khullar, M.; Soares, P.; Bahl, A.; et al. A common MYBPC3 (cardiac myosin binding protein C) variant associated with cardiomyopathies in South Asia. Nat. Genet. 2009, 41, 187–191. [Google Scholar] [CrossRef]
- Das, S.; Biswas, A.; Kapoor, M.; Bhargava, B.; Seth, S.; Rao, V.R. Epidemiology of Cardiomyopathy-A Clinical and Genetic Study of Dilated Cardiomyopathy: The EPOCH-D study. J. Pract. Cardiovasc. Sci. 2015, 1, 30–34. [Google Scholar]
- Kothari, S.S.; Dhopeshwarkar, R.A.; Saxena, A.; Juneja, R. Dilated cardiomyopathy in Indian children. Indian Hear J. 2003, 55, 147–151. [Google Scholar]
- Dilated Cardiomyopathy in Indian Children-PubMed [Internet]. Available online: https://pubmed.ncbi.nlm.nih.gov/12921329/ (accessed on 19 April 2021).
- Rai, T.S.; Ahmad, S.; Ahluwalia, T.S.; Ahuja, M.; Bahl, A.; Saikia, U.N.; Singh, B.; Talwar, K.K.; Khullar, M. Genetic and clinical profile of Indian patients of idiopathic restrictive cardiomyopathy with and without hypertrophy. Mol. Cell. Biochem. 2009, 331, 187–192. [Google Scholar] [CrossRef]
- Nallari, P.; Tanjore, R.; RangaRaju, A.; Vadapalli, S.; Remersu, S.; Narsimhan, C. Genetic variations of β-MYH7 in hypertrophic cardiomyopathy and dilated cardiomyopathy. Indian J. Hum. Genet. 2010, 16, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Perundurai, D.; Dhandapany, P.S.; Ahluwalia, T.S.; Bhardwaj, M.; Bahl, A.; Talwar, K.K.; Nair, K.; Rathinavel, A.; Khullar, M. ACE I/D polymorphism in Indian patients with hypertrophic cardiomyopathy and dilated cardiomyopathy. Mol. Cell. Biochem. 2007, 311, 67–72. [Google Scholar] [CrossRef]
- Jadhav, K.B.; Karpe, K.K.; Maramattom, B.V. An Indian family with an Emery-Dreifuss myopathy and familial dilated cardiomyopathy due to a novel LMNA mutation. Ann. Indian Acad. Neurol. 2012, 15, 344–346. [Google Scholar] [CrossRef] [PubMed]
- Matsa, L.S.; Rangaraju, A.; Vengaldas, V.; Latifi, M.; Jahromi, H.M.; Ananthapur, V.; Nallari, P. Haplotypes of NOS3 Gene Polymorphisms in Dilated Cardiomyopathy. PLoS ONE 2013, 8, e70523. [Google Scholar] [CrossRef] [PubMed]
- Matsa, L.S.; Sagurthi, S.R.; Ananthapur, V.; Nalla, S.; Nallari, P. Endothelin 1 gene as a modifier in dilated cardiomyopathy. Gene 2014, 548, 256–262. [Google Scholar] [CrossRef]
- Rani, D.S.; Dhandapany, P.S.; Nallari, P.; Narasimhan, C.; Thangaraj, K. A Novel Arginine to Tryptophan (R144W) Mutation in Troponin T (cTnT) Gene in an Indian Multigenerational Family with Dilated Cardiomyopathy (FDCM). PLoS ONE 2014, 9, e101451. [Google Scholar] [CrossRef]
- Deshmukh, A.; Deshmukh, A.; Deshmukh, G.; Garg, P.K. A pilot study of dilated cardiomyopathy (DCM) in western Uttar Pradesh, India: A four year review. Med.-Leg. Update 2011, 11, 1. [Google Scholar]
- Rana, H.; Rathod, C.; Chavda, P.; Patel, S.; Deshpande, S. Clinical profile of Dilated Cardiomyopathy patients presenting to a tertiary care hospital Clinical profile of Dilated Cardiomyopathy patients presenting to a tertiary care hospital from central Gujarat. Int J Res Med. 2015, 4, 143–146. [Google Scholar]
- Saha, K.K.; Kumar, A.; Sneha, K.; Kumar, P.; Mishra, A.; Tiwari, M.; Roshan, R. A Clinical Study of Dilated Cardiomyopathy with Correlation to Electrocardiography and Echocardiography: A Cross Sectional Study. Int. J. Sci. Study 2018, 5, 91–95. Available online: www.ijss-sn.com.
- Ushasree, B.; Shivani, V.; Venkateshwari, A.; Jain, R.K.; Narsimhan, C.; Nallari, P. Epidemiology and genetics of dilated cardio-myopathy in the Indian context. Indian J. Med. Sci. 2009, 63, 288–296. [Google Scholar] [PubMed]
- Shairgojri, A.A.; Mohiuddin, K.; Bashir, A.; Jan, M. Childhood cardiomyopathies: A study in tertiary care hospital of Kashmir. Int. J. Contemp. Pediatr. 2021, 8, 289–294. [Google Scholar] [CrossRef]
- Histopathological Changes Seen in The Heart Samples of Dcm Patients from North India a Cadaveric Study, IJSR-International Journal of Scientific Research(IJSR), IJSR|World Wide Journals [Internet]. Available online: https://www.worldwidejournals.com/international-journal-of-scientific-research-(IJSR)/article/histopathological-changes-seen-in-the-heart-samples-of-dcm-patients-from-north-india-a-cadaveric-study/MTA4NjE=/ (accessed on 22 April 2021).
- Bhawani, G.; Balije, S.; Kumar, A.; Murthy, K.S.N.; Kumari, N. Effect of hypertension at presentation on prognosis in patients with dilated cardiomyopathy presenting with normal renal angiogram. Indian J. Med Res. 2016, 144, 281–287. [Google Scholar] [CrossRef]
- Clinical, E.C.G. EKG, Profile of Patients with DCM [Internet]. Available online: http://iscindia.co.in/Uploads/635343547865542527.PDF (accessed on 22 April 2021).
- Das, S.; Biswas, A.; Kapoor, M.; Seth, S.; Bhargava, B.; Rao, V. A novel donor site mutation in LMNA gene leading to severe form of Dilated Cardiomyopathy in a proband of a family from Bihar, India. Mol. Cytogenet. 2014, 7, P35. [Google Scholar] [CrossRef]
- Sonowal, N.; Vanamali, D.R. Clinical profile of patients with dilated cardiomyopathy in a tertiary care center in north east India. J. Evol. Med. Dent. Sci. 2014, 3, 8378–8386. [Google Scholar] [CrossRef]
- Paul, R.; Nandi, S.; Sinha, P.K. Epidemiological study of dilated cardiomyopathy from eastern India with special reference to left atrial size. Int. J. Med. Res. Heal Sci. 2014, 3, 639. [Google Scholar] [CrossRef]
- Reich, D.; Thangaraj, K.; Patterson, N.; Price, A.L.; Singh, L. Reconstructing Indian population history. Nature 2009, 461, 489–494. [Google Scholar] [CrossRef]
- Moorjani, P.; Thangaraj, K.; Patterson, N.; Lipson, M.; Loh, P.-R.; Govindaraj, P.; Berger, B.; Reich, D.; Singh, L. Genetic Evidence for Recent Population Mixture in India. Am. J. Hum. Genet. 2013, 93, 422–438. [Google Scholar] [CrossRef]
- Basak, T.; Varshney, S.; Akhtar, S.; Sengupta, S. Understanding different facets of cardiovascular diseases based on model sys-tems to human studies: A proteomic and metabolomic perspective. J. Proteom. 2015, 127, 50–60, Elsevier. [Google Scholar] [CrossRef]
- Basak, T.; Varshney, S.; Hamid, Z.; Ghosh, S.; Seth, S.; Sengupta, S. Identification of metabolic markers in coronary artery disease using an untargeted LC-MS based metabolomic approach. J. Proteom. 2015, 127, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Wang, Z.; Hu, C.; Zhang, C.; Kovatcheva-Datchary, P.; Yu, D.; Liu, S.; Ren, F.; Wang, X.; Li, Y.; et al. Integrated Metabolomics and Lipidomics Analyses Reveal Metabolic Reprogramming in Human Glioma with IDH1 Mutation. J. Proteome Res. 2018, 18, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Louridas, G.E.; Lourida, K.G. Systems Biology and Biomechanical Model of Heart Failure. Curr. Cardiol. Rev. 2012, 8, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, I.; Rosa, I.; Bravo, S.B.; Guitián, E.; Pérez-Serra, A.; Campuzano, O.; Brugada, R.; Mangas, A.; García, Á.; Toro, R. Proteomic identification of putative biomarkers for early detection of sudden cardiac death in a family with a LMNA gene mutation causing dilated cardiomyopathy. J. Proteom. 2016, 148, 75–84. [Google Scholar] [CrossRef]
- Egerstedt, A.; Berntsson, J.; Smith, M.L.; Gidlöf, O.; Nilsson, R.; Benson, M.; Wells, Q.S.; Celik, S.; Lejonberg, C.; Farrell, L.; et al. Profiling of the plasma proteome across different stages of human heart failure. Nat. Commun. 2019, 10, 5830. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symptoms of DCM in Adults | Symptoms of DCM in Children/Newborns |
|---|---|
| Dyspnea and fatigue | Poor appetite/Problems in feeding |
| Dizziness | Poor growth |
| Syncope | Excessive sweating during feeding |
| Edema | Sweating while doing any activity |
| Palpitations | Palpitations |
| Controlled weight loss/gain | Fast breathing/difficult breathing |
| Excessive Sweating | Edema |
| Abdominal discomfort | |
| Nausea | |
| Anorexia | |
| Cachexia |
| Cytoskeletal and Sarcomeric Genes | |
|---|---|
| Titin (TTN) | Cardiac actin alpha |
| Desmin (DES) | Troponin T2, I3, C1 (TNNT2, TNNI3, TNNC1) |
| Lamin A/C (LMNA) | β-myosin heavy chain (MHY7) |
| α-actinin-2 (ACTN2) | Tropomyosin-1 (TPM1) |
| Actin binding LIM domain protein (ABLIM1) | Phospholamban (PLN) |
| Nebulette (NEBL) | Myosin binding protein C (MYBPC3) |
| Myopalladin (MYPN) | Sodium channel protein type 5 subunit alpha (SCN5A) |
| Filamin C (FLNC) | BCL2-associated athanogene 3 (BAG3) |
| δ sarcoglycan (SGCD) | |
| Vinculin (VCL) | |
| Z band alternatively spliced PDZ domain protein (ZASP) | |
| Dystrophin gene (DMD) |
| View | Measurement | Description |
|---|---|---|
| Plax (parasternal long axis) M-mode | LVIDd (left ventricular internal dimension in diastole). LVPWd (left ventricular posterior (inferolateral) wall thickness in diastole). IVSd (interventricular septal thickness diastole). | Left ventricle cavity size >112% (2 S.D.) based on surface area and age. Left ventricle cavity size >117% is specific criterion in screening. |
| Fraction shortening | FS < 25% | |
| Mitral valve E-septal separation | Normal range—0–5.3 mm. Reduced systolic function is indicated by value above 7 mm. | |
| PLAX (parasternal long axis) color flow Doppler | Mitral regurgitation |
|
| A4C (apical four chamber) PW | Diastolic Dysfunction | Diastolic dysfunction is present in patients with EF < 45%. |
| A4C 2D | Left ventricle ejection fraction | EF < 45% indicates DCM |
| Sphericity index | Normal > 1.5, decrease in sphericity index value to near 1 indicates DCM [97]. |
| Pathological Tests | Other Tests in Specific Indications |
|---|---|
| Erythrocyte sedimentation rate (ESR) | Coronary angiography |
| Viral serology | Blood content tests—carnitine, pyruvate, lactate, autoantibodies, selenium, acylcarnitine profile and drug screening. |
| Creatine kinase (CK) | Red cell transketolase (beri beri) |
| Liver function tests | Urine |
| Renal function | Enteroviruses test |
| Serum ferritin/iron/transferring | Infective screening (HIV/hepatitis C) |
| Thyroid function tests | Organic acid/amino acids |
| Drug | Title of Project | Description | Disease or Condition | Location |
|---|---|---|---|---|
| ARRY-371797, (p38 inhibitor) | A rollover study of ARRY-371797 in patients with LMNA-related dilated cardiomyopathy | Assessment of effectiveness of drug ARRY-371797 is being investigated in this clinical trial. | LMNA-related dilated cardiomyopathy | University of Colorado Aurora, Colorado, United States Johns Hopkins University Baltimore, Maryland, United States |
| ARRY-371797 | A study of ARRY-371797 in patients with symptomatic dilated cardiomyopathy due to a lamin A/C gene mutation | This study is a placebo controlled, dose-dependent efficacy assessment of ARRY-371797 drug on LMNA gene mutation dilated cardiomyopathy patients. | Lamin A/C gene mutation dilated cardiomyopathy | Pfizer Investigational Site Birmingham, Alabama, United States. CB Flock Research Corporation Mobile, Alabama, United States, and 64 more. |
| Ivabradine | Pulse reduction on beta-blocker and Ivabradine therapy | Ivabradine improves ejection fraction by reducing heart rate independently from beta-blockade. | Dilated cardiomyopathy ventricular remodeling electrical remodeling | University of Colorado Anschutz Medical Campus Aurora, Colorado, United States The Ohio State University Wexner Medical Center Columbus, Ohio, United States |
| Ifetroban | Oral Ifetroban in subjects with Duchenne muscular dystrophy (DMD) | X-linked Duchenne muscular dystrophy (DMD) is a fatal genetic disorder. This lacks effective treatment therapy. Ifetroban is assessed in this study for the treatment of DMD. | Duchenne muscular dystrophy cardiomyopathy dilated cardiomyopathy | Mattel Children’s Hospital Los Angeles, California, United States and 3 more. |
| Age Group | Male or Female (%) | Number of Cardiomyopathy Patients | Study Description | Sample Collection | Focus Population | Reference |
|---|---|---|---|---|---|---|
| 41.7 ± 16.5 | 38.70% males | 80 DCM | 40% familial, 48% sporadic. LMNA (c. 639 + G > C) associated novel splice site and MYH7 (c. 2769 > T) rare varient found. | EPOCH-D Study, AIIMS, New Delhi | General [151] | Das et al., 2015 |
| 15–67 | 35.3% males | 61 DCM | 61% DCM patients with MYH7 gene (exon 8–24) mutation with a novel p.Gly377Ser mutation found. | Nehru Hospital, Chandigarh | Asian Indians [136] | Rai et al., 2009 |
| 45.41 ± 14.35 | 54% males | 51 DCM | Influence of D allele in DCM development associated with ACE I/D | PGIMER, Chandigarh, Rajaji Government hospital, Madurai, and, Sri Chitra Tirunal Institute of Medical Sciences and Technology, Trivandrum | General [138] | Rai et al., 2008 |
| Juvenile–Adult | Undefined | 97 DCM | Common exon link of MYH7 gene IN DCM and HCM. Homozygous condition develops DCM. | CARE Hospitals, Mahavir hospitals and Niloufer hospitals, Hyderabad | General [137] | Tanjore et al., 2010 |
| 53 year old | 1 male | DCM Case Study | Familial DCM along with Emery-Dreifuss myopathy associated with mutation in LMNA gene. | Lourdes Heart Institute and Neuro Center, Kochi, Kerala | Kerala [139] | Jadhav et al., 2012 [22] |
| >25 years old | Undefined | 115 DCM | Influence of endothelial NOS3 gene in DCM manifestation by oxidative stress production. | CARE hospitals, Krishna Institute of Medical Sciences (KIMS) and Niloufer hospital for Children, Hyderabad, India | General [140] | Matsa et al., 2013 |
| 33.2± 16.1 | 70% male 30% females | 115 DCM | Screening of EDN1 gene and two rare genetic variations found. Insertion variations (+138 A) corresponding to heterozygotic condition leads to 4-fold increased risk of DCM. | CARE hospitals, Krishna Institute of Medical Sciences (KIMS) and Niloufer Hospital for Children, Hyderabad, India | General [141] | Matsa et al., 2014 |
| NA | NA | 147 DCM | Role of novel mutation R144W in Toponin T binding domain found by overall exon analysis in DCM development. | NA | South Indian [142] | Rani et al., 2014 |
| 2.9 ± 3.07 | 50 males | 80 DCM | Pediatric DCM was explored in this observational study. | General [135] | Kothari et al., 2003 | |
| Above 60 | 66.6% Males 33.4% females | 100 DCM | 4-year study of DCM charecterization based on cause. DCM in peripartum females accounted for 9%, DCM in smokers was 65%, 30% alcoholic DCM. | M.M.C. Muzaffarnagar, U.P, & SIMS, Hapur, UP | Western Uttar Pradesh [143] | Deshmukh et al., 2011 |
| Above 60 | 56.6% males 43.25% females | 30 DCM | Cross-sectional study of DCM over 1 year period. Ischemic DCM 33.3%+ diabetic cardiomyopathy (23.3%) peripartum cardiomyopathy (16.6%) 6.6 Alcoholic and miscellaneous DCM | Rajendra Institute of Medical Sciences, Ranchi. | India General [145] | Saha et al., 2018 |
| Above 13 | 43% male, 56.66% females | 180 DCM | 3-year DCM etiology study. Diabetes 13.33% alcohol 23.33% postpartum 15.00% idiopathic 30.00% inflammation 03.33% nutritional 6.66% multifactorial 06.66% | GMERS General Hospital, Gotri, Vadodara. | Indian General [144] | Rana et al., 2015 |
| 6 months–Adult | 62.61% males 37.38% females | 107 DCM | Parental consanguinity 22.42% alcoholic/smoking DCM | KIMS and Niloufer Hospital for children, Hyderabad | Indian General [146] | Ushasree et al., 2009 |
| 1 month–18 | 52.65% males 47.6% female | 19 DCM | Hospital based observational profile of DCM in children. Study period of two and half years, charecterization made on symptoms, gender preponderance parental consanguinity and myocarditis. | Post Graduate Department of Pediatrics Government Medical College, Srinagar | North (Kashmir) [147] | Shairgojri et al., 2021 |
| 18–80 | 61.29% males 38.71% females | 31 DCM | Observational study characterizing DCM on the basis of its causes. Alcoholic DCM 23.33% + viral DCM 03.33% peripartum cardiomyopathy 22.58% idiopathic DCM 41.93% familial DCM 9.6% | Jorhat Medical College and Hospital, Jorhat, Assam | North East India [152] | Sonowal et al., 2014 |
| 15–75 | 18.3% females | 60 DCM | An analytical and observational study showed 35 patients DCM with hypertension and 25 DCM patients without hypertension | Cardiology, GSL Medical College and Hospital, Rajahmundry India | Andhra Pradesh [149] | Balije et al., 2016 |
| 50 ± 15 | 65.45% males 34% females | 55 DCM | 1-year clinical and incidence profiling of idiopathic DCM | JN Medical College, Aligarh | General [150] | Ahmad et al., 2005 |
| All | 61.4% males 38.6% females | 70 DCM | Pilot study to determine different demographic parameters in DCM patients. 27% alcoholic 46% smokers in the cohort | Tertiary Medical College of Eastern India | Eastern India [153] | Paul et al., 2014 |
| 10–70 years | NA | 10 DCM | Measuring the gross morphological changes in heart wall of DCM patients | Institute of Medical Sciences, BHU, Varanasi (U.P) | North Indian [148] | Prasenjit Bose et al., 2017 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarohi, V.; Srivastava, S.; Basak, T. A Comprehensive Outlook on Dilated Cardiomyopathy (DCM): State-Of-The-Art Developments with Special Emphasis on OMICS-Based Approaches. J. Cardiovasc. Dev. Dis. 2022, 9, 174. https://doi.org/10.3390/jcdd9060174
Sarohi V, Srivastava S, Basak T. A Comprehensive Outlook on Dilated Cardiomyopathy (DCM): State-Of-The-Art Developments with Special Emphasis on OMICS-Based Approaches. Journal of Cardiovascular Development and Disease. 2022; 9(6):174. https://doi.org/10.3390/jcdd9060174
Chicago/Turabian StyleSarohi, Vivek, Shriya Srivastava, and Trayambak Basak. 2022. "A Comprehensive Outlook on Dilated Cardiomyopathy (DCM): State-Of-The-Art Developments with Special Emphasis on OMICS-Based Approaches" Journal of Cardiovascular Development and Disease 9, no. 6: 174. https://doi.org/10.3390/jcdd9060174
APA StyleSarohi, V., Srivastava, S., & Basak, T. (2022). A Comprehensive Outlook on Dilated Cardiomyopathy (DCM): State-Of-The-Art Developments with Special Emphasis on OMICS-Based Approaches. Journal of Cardiovascular Development and Disease, 9(6), 174. https://doi.org/10.3390/jcdd9060174