
The Importance of Telomere Shortening for Atherosclerosis and Mortality


Abstract
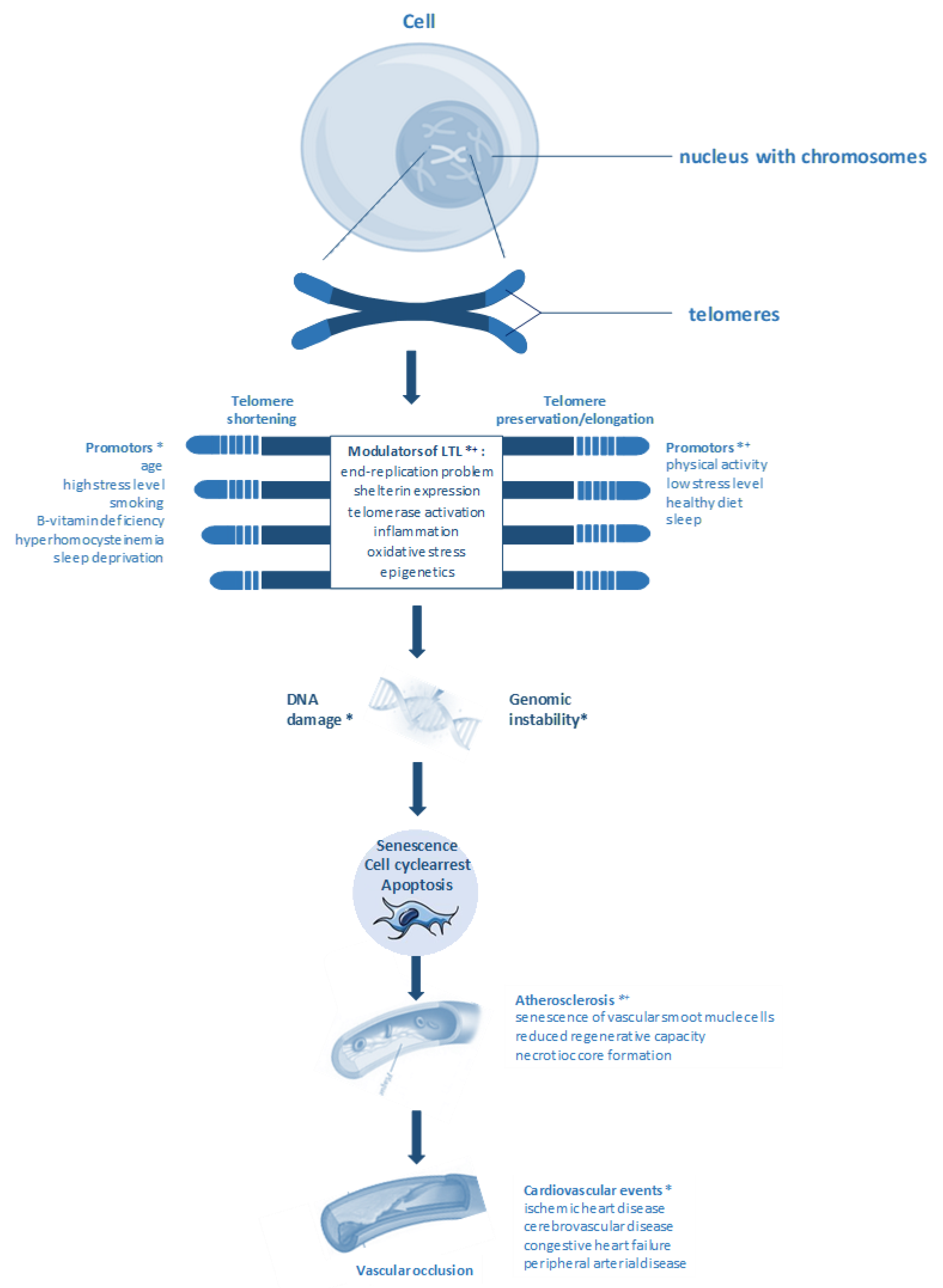
1. Introduction
2. Telomere Length and Age
3. Telomere Length, Cardiovascular Disease (CVD), and Mortality
4. Telomere Length and Atherosclerosis
5. Telomere Shortening and CVD in Pre-Clinical Models
6. B-vitamins, Homocysteine, Telomere Length, and CVD
7. Markers of Inflammation, Oxidative Stress, HCY, LTL, and Mortality
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vijg, J.; Suh, Y. Genome instability and aging. Annu. Rev. Physiol. 2013, 75, 645–668. [Google Scholar] [CrossRef]
- Lombard, D.B.; Chua, K.F.; Mostoslavsky, R.; Franco, S.; Gostissa, M.; Alt, F.W. DNA repair, genome stability, and aging. Cell 2005, 120, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Blasco, M.A. Telomere length, stem cells and aging. Nat. Chem. Biol. 2007, 3, 640–649. [Google Scholar] [CrossRef]
- Blasco, M.A. Telomeres and human disease: Ageing, cancer and beyond. Nat. Rev. Genet. 2005, 6, 611–622. [Google Scholar] [CrossRef]
- Muller, H. The remaking of chromosomes. Collect. Net 1938, 13, 181–198. [Google Scholar]
- McClintock, B. The stability of broken ends of chromosomes in Zea mays. Genetics 1941, 26, 234. [Google Scholar] [PubMed]
- de Lange, T. Shelterin-mediated telomere protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef] [PubMed]
- de Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, U.; Griese, E.-U.; Schwab, M.; Fritz, P.; Thon, K.-P.; Klotz, U. Telomere length in different tissues of elderly patients. Mech. Ageing Dev. 2000, 119, 89–99. [Google Scholar] [CrossRef]
- Yeh, J.-K.; Wang, C.-Y. Telomeres and telomerase in cardiovascular diseases. Genes 2016, 7, 58. [Google Scholar] [CrossRef]
- Fitzpatrick, A.L.; Kronmal, R.A.; Kimura, M.; Gardner, J.P.; Psaty, B.M.; Jenny, N.S.; Tracy, R.P.; Hardikar, S.; Aviv, A. Leukocyte telomere length and mortality in the cardiovascular health study. J. Gerontol. Ser. Biomed. Sci. Med. Sci. 2011, 66, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Dlouha, D.; Maluskova, J.; Lesna, I.K.; Lanska, V.; Hubacek, J. Comparison of the relative telomere length measured in leukocytes and eleven different human tissues. Physiol. Res. 2014, 63 (Suppl. 3), S343–350. [Google Scholar]
- Wilson, W.R.; Herbert, K.E.; Mistry, Y.; Stevens, S.E.; Patel, H.R.; Hastings, R.A.; Thompson, M.M.; Williams, B. Blood leucocyte telomere DNA content predicts vascular telomere DNA content in humans with and without vascular disease. Eur. Heart J. 2008, 29, 2689–2694. [Google Scholar] [CrossRef] [PubMed]
- Willeit, P.; Willeit, J.; Brandstätter, A.; Ehrlenbach, S.; Mayr, A.; Gasperi, A.; Weger, S.; Oberhollenzer, F.; Reindl, M.; Kronenberg, F. Cellular aging reflected by leukocyte telomere length predicts advanced atherosclerosis and cardiovascular disease risk. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1649–1656. [Google Scholar] [CrossRef] [PubMed]
- Mons, U.; Müezzinler, A.; Schöttker, B.; Dieffenbach, A.K.; Butterbach, K.; Schick, M.; Peasey, A.; De Vivo, I.; Trichopoulou, A.; Boffetta, P.; et al. Leukocyte telomere length and all-cause, cardiovascular disease, and cancer mortality: Results from individual-participant-data meta-analysis of 2 large prospective cohort studies. Am. J. Epidemiol. 2017, 185, 1317–1326. [Google Scholar] [CrossRef]
- Needham, B.L.; Rehkopf, D.; Adler, N.; Gregorich, S.; Lin, J.; Blackburn, E.H.; Epel, E.S. Leukocyte telomere length and mortality in the National Health and Nutrition Examination Survey, 1999–2002. Epidemiology 2015, 26, 528. [Google Scholar] [CrossRef]
- Rode, L.; Nordestgaard, B.G.; Bojesen, S.E. Peripheral blood leukocyte telomere length and mortality among 64 637 individuals from the general population. J. Nat. Cancer Inst. 2015, 107, djv074. [Google Scholar] [CrossRef]
- Cawthon, R.M.; Smith, K.R.; O’Brien, E.; Sivatchenko, A.; Kerber, R.A. Association between telomere length in blood and mortality in people aged 60 years or older. Lancet 2003, 361, 393–395. [Google Scholar] [CrossRef]
- Herrmann, M.; Pusceddu, I.; März, W.; Herrmann, W. Telomere biology and age-related diseases. Clin. Chem. Lab. Med. 2018, 56, 1210–1222. [Google Scholar] [CrossRef]
- Müezzinler, A.; Zaineddin, A.K.; Brenner, H. A systematic review of leukocyte telomere length and age in adults. Ageing Res. Rev. 2013, 12, 509–519. [Google Scholar] [CrossRef]
- Pusceddu, I.; Kleber, M.; Delgado, G.; Herrmann, W.; März, W.; Herrmann, M. Telomere length and mortality in the ludwigshafen risk and cardiovascular health study. PLoS ONE 2018, 13, e0198373. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Kimura, M.; Kim, S.; Cao, X.; Srinivasan, S.R.; Berenson, G.; Kark, J.; Aviv, A. Longitudinal versus cross-sectional evaluations of leukocyte telomere length dynamics: Age-dependent telomere shortening is the rule. J. Gerontol. Ser. Biomed. Sci. Med. Sci. 2011, 66, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Ehrlenbach, S.; Willeit, P.; Kiechl, S.; Willeit, J.; Reindl, M.; Schanda, K.; Kronenberg, F.; Brandstätter, A. Influences on the reduction of relative telomere length over 10 years in the population-based bruneck study: Introduction of a well-controlled high-throughput assay. Int. J. Epidemiol. 2009, 38, 1725–1734. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, T.; Codd, V.; Brouilette, S.; Thijs, L.; González, A.; Jin, Y.; Richart, T.; van der Harst, P.; Díez, J.; Staessen, J.A.; et al. Association between left ventricular mass and telomere length in a population study. Am. J. Epidemiol. 2010, 172, 440–450. [Google Scholar] [CrossRef]
- Rius-Ottenheim, N.; Houben, J.M.; Kromhout, D.; Kafatos, A.; van der Mast, R.C.; Zitman, F.G.; Geleijnse, J.M.; Hageman, G.J.; Giltay, E.J. Telomere length and mental well-being in elderly men from the Netherlands and Greece. Behav. Genet. 2012, 42, 278–286. [Google Scholar] [CrossRef]
- Benetos, A.; Verhulst, S.; Labat, C.; Lai, T.P.; Girerd, N.; Toupance, S.; Zannad, F.; Rossignol, P.; Aviv, A. Telomere length tracking in children and their parents: Implications for adult onset diseases. FASEB J. 2019, 33, 14248–14253. [Google Scholar] [CrossRef]
- Aviv, A.; Chen, W.; Gardner, J.P.; Kimura, M.; Brimacombe, M.; Cao, X.; Srinivasan, S.R.; Berenson, G.S. Leukocyte telomere dynamics: Longitudinal findings among young adults in the bogalusa heart study. Am. J. Epidemiol. 2009, 169, 323–329. [Google Scholar] [CrossRef]
- Martinez-Delgado, B.; Yanowsky, K.; Inglada-Perez, L.; Domingo, S.; Urioste, M.; Osorio, A.; Benitez, J. Genetic anticipation is associated with telomere shortening in hereditary breast cancer. PLoS Genet. 2011, 7, e1002182. [Google Scholar] [CrossRef]
- Benitez-Buelga, C.; Sanchez-Barroso, L.; Gallardo, M.; Apellániz-Ruiz, M.; Inglada-Perez, L.; Yanowski, K.; Carrillo, J.; Garcia-Estevez, L.; Calvo, I.; Perona, R. Impact of chemotherapy on telomere length in sporadic and familial breast cancer patients. Breast Cancer Res. Treat. 2015, 149, 385–394. [Google Scholar] [CrossRef]
- Weischer, M.; Bojesen, S.E.; Nordestgaard, B.G. Telomere shortening unrelated to smoking, body weight, physical activity, and alcohol intake: 4576 general population individuals with repeat measurements 10 years apart. PLoS Genet. 2014, 10, e1004191. [Google Scholar] [CrossRef]
- Farzaneh-Far, R.; Lin, J.; Epel, E.; Lapham, K.; Blackburn, E.; Whooley, M.A. Telomere length trajectory and its determinants in persons with coronary artery disease: Longitudinal findings from the heart and soul study. PLoS ONE 2010, 5, e8612. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, C.; Petersen, H.C.; Graakjaer, J.; Andersen-Ranberg, K.; Vaupel, J.W.; Bohr, V.A.; Kølvraa, S.; Christensen, K. No association between telomere length and survival among the elderly and oldest old. Epidemiology 2006, 17, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Svensson, J.; Karlsson, M.K.; Ljunggren, Ö.; Tivesten, Å.; Mellström, D.; Movérare-Skrtic, S. Leukocyte telomere length is not associated with mortality in older men. Exp. Gerontol. 2014, 57, 6–12. [Google Scholar] [CrossRef]
- Brouilette, S.W.; Moore, J.S.; McMahon, A.D.; Thompson, J.R.; Ford, I.; Shepherd, J.; Packard, C.J.; Samani, N.J. Telomere length, risk of coronary heart disease, and statin treatment in the west of scotland primary prevention study: A nested case-control study. Lancet 2007, 369, 107–114. [Google Scholar] [CrossRef]
- Van Der Harst, P.; van der Steege, G.; de Boer, R.A.; Voors, A.A.; Hall, A.S.; Mulder, M.J.; van Gilst, W.H.; van Veldhuisen, D.J.; Group, M.-H.S. Telomere length of circulating leukocytes is decreased in patients with chronic heart failure. J. Am. Coll. Cardiol. 2007, 49, 1459–1464. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhu, Z.; Zhou, L.; Yang, M. The joint effects of frailty and telomere length for predicting mortality in older adults: The national health and nutrition examination survey 1999–2002. Aging Clin. Exp. Res. 2019, 1–9. [Google Scholar] [CrossRef]
- Margaritis, M.; Sanna, F.; Lazaros, G.; Akoumianakis, I.; Patel, S.; Antonopoulos, A.S.; Duke, C.; Herdman, L.; Psarros, C.; Oikonomou, E.K. Predictive value of telomere length on outcome following acute myocardial infarction: Evidence for contrasting effects of vascular vs. blood oxidative stress. Eur. Heart J. 2017, 38, 3094–3104. [Google Scholar] [CrossRef]
- Wang, Q.; Zhan, Y.; Pedersen, N.L.; Fang, F.; Hägg, S. Telomere length and all-cause mortality: A meta-analysis. Ageing Res. Rev. 2018, 48, 11–20. [Google Scholar] [CrossRef]
- Martínez, P.; Blasco, M.A. Heart-breaking telomeres. Circ. Res. 2018, 123, 787–802. [Google Scholar] [CrossRef]
- Calado, R.T.; Yewdell, W.T.; Wilkerson, K.L.; Regal, J.A.; Kajigaya, S.; Stratakis, C.A.; Young, N.S. Sex hormones, acting on the TERT gene, increase telomerase activity in human primary hematopoietic cells. Blood 2009, 114, 2236–2243. [Google Scholar] [CrossRef]
- Bär, C.; Huber, N.; Beier, F.; Blasco, M.A. Therapeutic effect of androgen therapy in a mouse model of aplastic anemia produced by short telomeres. Haematologica 2015, 100, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Townsley, D.M.; Dumitriu, B.; Liu, D.; Biancotto, A.; Weinstein, B.; Chen, C.; Hardy, N.; Mihalek, A.D.; Lingala, S.; Kim, Y.J. Danazol treatment for telomere diseases. N. Engl. J. Med. 2016, 374, 1922–1931. [Google Scholar] [CrossRef] [PubMed]
- Bär, C.; Blasco, M.A. Telomeres and telomerase as therapeutic targets to prevent and treat age-related diseases. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Bär, C.; Povedano, J.M.; Serrano, R.; Benitez-Buelga, C.; Popkes, M.; Formentini, I.; Bobadilla, M.; Bosch, F.; Blasco, M.A. Telomerase gene therapy rescues telomere length, bone marrow aplasia, and survival in mice with aplastic anemia. Blood J. Am. Soc. Hematol. 2016, 127, 1770–1779. [Google Scholar] [CrossRef]
- Povedano, J.M.; Martinez, P.; Serrano, R.; Tejera, Á.; Gómez-López, G.; Bobadilla, M.; Flores, J.M.; Bosch, F.; Blasco, M.A. Therapeutic effects of telomerase in mice with pulmonary fibrosis induced by damage to the lungs and short telomeres. Elife 2018, 7, e31299. [Google Scholar] [CrossRef]
- Martínez, P.; Blasco, M.A. Telomere-driven diseases and telomere-targeting therapies. J. Cell Biol. 2017, 216, 875–887. [Google Scholar] [CrossRef]
- González-Suárez, E.; Samper, E.; Ramírez, A.; Flores, J.M.; Martín-Caballero, J.; Jorcano, J.L.; Blasco, M.A. Increased epidermal tumors and increased skin wound healing in transgenic mice overexpressing the catalytic subunit of telomerase, mTERT, in basal keratinocytes. EMBO J. 2001, 20, 2619–2630. [Google Scholar] [CrossRef]
- Muñoz-Lorente, M.A.; Martínez, P.; Tejera, Á.; Whittemore, K.; Moisés-Silva, A.C.; Bosch, F.; Blasco, M.A. AAV9-mediated telomerase activation does not accelerate tumorigenesis in the context of oncogenic K-Ras-induced lung cancer. PLoS Genet. 2018, 14, e1007562. [Google Scholar] [CrossRef]
- Bernardes de Jesus, B.; Schneeberger, K.; Vera, E.; Tejera, A.; Harley, C.B.; Blasco, M.A. The telomerase activator TA-65 elongates short telomeres and increases health span of adult/old mice without increasing cancer incidence. Aging Cell 2011, 10, 604–621. [Google Scholar] [CrossRef]
- Chen, S.; Lin, J.; Matsuguchi, T.; Blackburn, E.; Yeh, F.; Best, L.G.; Devereux, R.B.; Lee, E.T.; Howard, B.V.; Roman, M.J.; et al. Short leukocyte telomere length predicts incidence and progression of carotid atherosclerosis in American Indians: The strong heart family study. Aging 2014, 6, 414–427. [Google Scholar] [CrossRef]
- O’Donnell, C.J.; Demissie, S.; Kimura, M.; Levy, D.; Gardner, J.P.; White, C.; D’Agostino, R.B.; Wolf, P.A.; Polak, J.; Cupples, L.A.; et al. Leukocyte telomere length and carotid artery intimal medial thickness. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1165–1171. [Google Scholar] [CrossRef] [PubMed]
- De Meyer, T.; Rietzschel, E.R.; De Buyzere, M.L.; Langlois, M.R.; De Bacquer, D.; Segers, P.; Van Damme, P.; De Backer, G.G.; Van Oostveldt, P.; Van Criekinge, W.; et al. Systemic telomere length and preclinical atherosclerosis: The Asklepios study. Eur. Heart J. 2009, 30, 3074–3081. [Google Scholar] [CrossRef] [PubMed]
- Calvert, P.A.; Liew, T.V.; Gorenne, I.; Clarke, M.; Costopoulos, C.; Obaid, D.R.; O’Sullivan, M.; Shapiro, L.M.; McNab, D.C.; Densem, C.G.; et al. Leukocyte telomere length is associated with high-risk plaques on virtual histology intravascular ultrasound and increased proinflammatory activity. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Huzen, J.; Peeters, W.; Boer, R.A.d.; Moll, F.L.; Wong, L.S.M.; Codd, V.; Kleijn, D.P.V.d.; Smet, B.J.G.L.d.; Veldhuisen, D.J.v.; Samani, N.J.; et al. Circulating leukocyte and carotid atherosclerotic plaque telomere length. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1219–1225. [Google Scholar] [CrossRef] [PubMed]
- Panayiotou, A.G.; Nicolaides, A.N.; Griffin, M.; Tyllis, T.; Georgiou, N.; Bond, D.; Martin, R.M.; Hoppensteadt, D.; Fareed, J.; Humphries, S.E. Leukocyte telomere length is associated with measures of subclinical atherosclerosis. Atherosclerosis 2010, 211, 176–181. [Google Scholar] [CrossRef]
- Benetos, A.; Gardner, J.P.; Zureik, M.; Labat, C.; Xiaobin, L.; Adamopoulos, C.; Temmar, M.; Bean, K.E.; Thomas, F.; Aviv, A. Short telomeres are associated with increased carotid atherosclerosis in hypertensive subjects. Hypertension 2004, 43, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Mainous, A.G., 3rd; Codd, V.; Diaz, V.A.; Schoepf, U.J.; Everett, C.J.; Player, M.S.; Samani, N.J. Leukocyte telomere length and coronary artery calcification. Atherosclerosis 2010, 210, 262–267. [Google Scholar] [CrossRef]
- Fernández-Alvira, J.M.; Fuster, V.; Dorado, B.; Soberón, N.; Flores, I.; Gallardo, M.; Pocock, S.; Blasco, M.A.; Andrés, V. Short telomere load, telomere length, and subclinical atherosclerosis: The PESA study. J. Am. Coll. Cardiol. 2016, 67, 2467–2476. [Google Scholar] [CrossRef]
- Farzaneh-Far, R.; Cawthon, R.M.; Na, B.; Browner, W.S.; Schiller, N.B.; Whooley, M.A. Prognostic value of leukocyte telomere length in patients with stable coronary artery disease: Data from the heart and soul study. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1379–1384. [Google Scholar] [CrossRef]
- Astrup, A.S.; Tarnow, L.; Jorsal, A.; Lajer, M.; Nzietchueng, R.; Benetos, A.; Rossing, P.; Parving, H.H. Telomere length predicts all-cause mortality in patients with type 1 diabetes. Diabetologia 2010, 53, 45–48. [Google Scholar] [CrossRef]
- Humphrey, L.L.; Fu, R.; Rogers, K.; Freeman, M.; Helfand, M. Homocysteine level and coronary heart disease incidence: A systematic review and meta-analysis. Mayo Clin. Proc. 2008, 83, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Richards, J.B.; Valdes, A.M.; Gardner, J.P.; Kato, B.S.; Siva, A.; Kimura, M.; Lu, X.; Brown, M.J.; Aviv, A.; Spector, T.D. Homocysteine levels and leukocyte telomere length. Atherosclerosis 2008, 200, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Prescott, J.; Giovannucci, E.; Hankinson, S.E.; Rosner, B.; De Vivo, I. One-carbon metabolism factors and leukocyte telomere length. Am. J. Clin. Nutr. 2013, 97, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Masi, S.; D’Aiuto, F.; Cooper, J.; Salpea, K.; Stephens, J.W.; Hurel, S.J.; Deanfield, J.E.; Humphries, S.E. Telomere length, antioxidant status and incidence of ischaemic heart disease in type 2 diabetes. Int. J. Cardiol. 2016, 216, 159–164. [Google Scholar] [CrossRef]
- Steer, S.E.; Williams, F.M.; Kato, B.; Gardner, J.P.; Norman, P.J.; Hall, M.A.; Kimura, M.; Vaughan, R.; Aviv, A.; Spector, T.D. Reduced telomere length in rheumatoid arthritis is independent of disease activity and duration. Ann. Rheum. Dis 2007, 66, 476–480. [Google Scholar] [CrossRef]
- Pusceddu, I.; Herrmann, W.; Kleber, M.E.; Scharnagl, H.; Hoffmann, M.M.; Winklhofer-Roob, B.M.; März, W.; Herrmann, M. Subclinical inflammation, telomere shortening, homocysteine, vitamin B6, and mortality: The ludwigshafen risk and cardiovascular health study. Eur. J. Nutr. 2020, 59, 1399–1411. [Google Scholar] [CrossRef]
- O’Donovan, A.; Pantell, M.S.; Puterman, E.; Dhabhar, F.S.; Blackburn, E.H.; Yaffe, K.; Cawthon, R.M.; Opresko, P.L.; Hsueh, W.C.; Satterfield, S.; et al. Cumulative inflammatory load is associated with short leukocyte telomere length in the health, aging and body composition study. PLoS ONE 2011, 6, e19687. [Google Scholar] [CrossRef]
- Werner, C.M.; Furster, T.; Widmann, T.; Poss, J.; Roggia, C.; Hanhoun, M.; Scharhag, J.; Buchner, N.; Meyer, T.; Kindermann, W.; et al. Physical exercise prevents cellular senescence in circulating leukocytes and in the vessel wall. Circulation 2009, 120, 2438–2447. [Google Scholar] [CrossRef]
- Werner, C.; Hanhoun, M.; Widmann, T.; Kazakov, A.; Semenov, A.; Pöss, J.; Bauersachs, J.; Thum, T.; Pfreundschuh, M.; Müller, P.; et al. Effects of physical exercise on myocardial telomere-regulating proteins, survival pathways, and apoptosis. J. Am. Coll. Cardiol. 2008, 52, 470. [Google Scholar] [CrossRef]
- Ludlow, A.T.; Witkowski, S.; Marshall, M.R.; Wang, J.; Lima, L.C.; Guth, L.M.; Spangenburg, E.E.; Roth, S.M. Chronic exercise modifies age-related telomere dynamics in a tissue-specific fashion. J. Gerontol. Biol. Sci. Med. Sci. 2012, 67, 911–926. [Google Scholar] [CrossRef]
- Ludlow, A.T.; Lima, L.C.J.; Wang, J.; Hanson, E.D.; Guth, L.M.; Spangenburg, E.E.; Roth, S.M. Exercise alters mRNA expression of telomere-repeat binding factor 1 in skeletal muscle via p38 MAPK. J. Appl. Physiol. 2012, 113, 1737–1746. [Google Scholar] [CrossRef] [PubMed]
- Tomás-Loba, A.; Flores, I.; Fernández-Marcos, P.J.; Cayuela, M.L.; Maraver, A.; Tejera, A.; Borrás, C.; Matheu, A.; Klatt, P.; Flores, J.M.; et al. Telomerase reverse transcriptase delays aging in cancer-resistant mice. Cell 2008, 135, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Bernardes de Jesus, B.; Blasco, M.A. Aging by telomere loss can be reversed. Cell Stem Cell 2011, 8, 3–4. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jaskelioff, M.; Muller, F.L.; Paik, J.-H.; Thomas, E.; Jiang, S.; Adams, A.C.; Sahin, E.; Kost-Alimova, M.; Protopopov, A.; Cadiñanos, J.; et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature 2011, 469, 102–106. [Google Scholar] [CrossRef]
- Ludlow, A.T.; Gratidao, L.; Ludlow, L.W.; Spangenburg, E.E.; Roth, S.M. Acute exercise activates p38 MAPK and increases the expression of telomere-protective genes in cardiac muscle. Exp. Physiol. 2017, 102, 397–410. [Google Scholar] [CrossRef]
- Tie, G.; Messina, K.E.; Yan, J.; Messina, J.A.; Messina, L.M. Hypercholesterolemia induces oxidant stress that accelerates the ageing of hematopoietic stem cells. J. Am. Heart Assoc. 2014, 3, e000241. [Google Scholar] [CrossRef]
- Wang, J.; Uryga, A.K.; Reinhold, J.; Figg, N.; Baker, L.; Finigan, A.; Gray, K.; Kumar, S.; Clarke, M.; Bennett, M. Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation 2015, 132, 1909–1919. [Google Scholar] [CrossRef]
- Fan, R.; Zhang, A.; Zhong, F. Association between homocysteine levels and all-cause mortality: A dose-response meta-analysis of prospective studies. Sci. Rep. 2017, 7, 4769. [Google Scholar] [CrossRef]
- Wu, Y.; Huang, Y.; Hu, Y.; Zhong, J.; He, Z.; Li, W.; Yang, Y.; Xu, D.; Wu, S. Hyperhomocysteinemia is an independent risk factor in young patients with coronary artery disease in southern China. Herz 2013, 38, 779–784. [Google Scholar] [CrossRef]
- Alam, N.; Khan, H.I.; Chowdhury, A.W.; Haque, M.S.; Ali, M.S.; Sabah, K.M.; Amin, M.G. Elevated serum homocysteine level has a positive correlation with serum cardiac troponin I in patients with acute myocardial infarction. Bangladesh Med. Res. Counc. Bull. 2012, 38, 9–13. [Google Scholar] [CrossRef]
- Wei, M.; Wang, L.; Liu, Y.-S.; Zheng, M.-Q.; Ma, F.-F.; Qi, Y.-C.; Liu, G. Homocysteine as a potential predictive factor for high major adverse cardiovascular events risk in female patients with premature acute coronary syndrome. Medicine 2019, 98, e18019. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Li, L.; Shang, X.-M.; Tan, Z.; Geng, X.-B.; Zhao, B.-Q.; Tian, M.-R. Analysis of factors related to short-term prognosis in patients undergoing percutaneous coronary intervention for acute myocardial infarction. Exp. Ther. Med. 2013, 5, 1206–1210. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Qian, G.; Xue, H.; Guo, J.; Chen, L.; Yang, X.; Shen, M.; Dong, W.; Chen, Y. Hyperhomocysteinemia is an independent predictor of long-term clinical outcomes in Chinese octogenarians with acute coronary syndrome. Clin. Interv. Aging 2015, 10, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, P.; Alam, S.F. Role of homocysteine in the development of cardiovascular disease. Nutr. J. 2015, 14, 6. [Google Scholar] [CrossRef] [PubMed]
- Marković Boras, M.; Čaušević, A.; Brizić, I.; Mikulić, I.; Vasilj, M.; Jelić Knezović, N. A relation of serum homocysteine, uric acid and C-reactive protein level in patients with acute myocardial infarction. Med. Glas. 2018, 15, 101–108. [Google Scholar] [CrossRef]
- Chambers, J.C.; McGregor, A.; Jean-Marie, J.; Obeid, O.A.; Kooner, J.S. Demonstration of rapid onset vascular endothelial dysfunction after hyperhomocysteinemia: An effect reversible with vitamin C therapy. Circulation 1999, 99, 1156–1160. [Google Scholar] [CrossRef]
- Eberhardt, R.T.; Forgione, M.A.; Cap, A.; Leopold, J.A.; Rudd, M.A.; Trolliet, M.; Heydrick, S.; Stark, R.; Klings, E.S.; Moldovan, N.I.; et al. Endothelial dysfunction in a murine model of mild hyperhomocyst(e)inemia. J. Clin. Invest. 2000, 106, 483–491. [Google Scholar] [CrossRef]
- Weiss, N.; Heydrick, S.; Zhang, Y.Y.; Bierl, C.; Cap, A.; Loscalzo, J. Cellular redox state and endothelial dysfunction in mildly hyperhomocysteinemic cystathionine beta-synthase-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 34–41. [Google Scholar] [CrossRef]
- Topal, G.; Brunet, A.; Millanvoye, E.; Boucher, J.L.; Rendu, F.; Devynck, M.A.; David-Dufilho, M. Homocysteine induces oxidative stress by uncoupling of no synthase activity through reduction of tetrahydrobiopterin. Free Radic. Biol. Med. 2004, 36, 1532–1541. [Google Scholar] [CrossRef]
- Papatheodorou, L.; Weiss, N. Vascular oxidant stress and inflammation in hyperhomocysteinemia. Antioxid. Redox Signal. 2007, 9, 1941–1958. [Google Scholar] [CrossRef]
- Kim, C.S.; Kim, Y.R.; Naqvi, A.; Kumar, S.; Hoffman, T.A.; Jung, S.B.; Kumar, A.; Jeon, B.H.; McNamara, D.M.; Irani, K. Homocysteine promotes human endothelial cell dysfunction via site-specific epigenetic regulation of p66shc. Cardiovasc. Res. 2011, 92, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cui, L.; Joseph, J.; Jiang, B.; Pimental, D.; Handy, D.E.; Liao, R.; Loscalzo, J. Homocysteine induces cardiomyocyte dysfunction and apoptosis through p38 MAPK-mediated increase in oxidant stress. J. Mol. Cell Cardiol. 2012, 52, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.N.; Liang, Y.R.; Lin, W.B.; Yao, Z.R.; Chen, D.B.; Chen, P.S.; Ouyang, J. Homocysteine induced oxidative stress in human umbilical vein endothelial cells via regulating methylation of SORBS1. Eur. Rev. Med. Pharm. Sci. 2018, 22, 6948–6958. [Google Scholar] [CrossRef]
- Rane, G.; Koh, W.P.; Kanchi, M.M.; Wang, R.; Yuan, J.M.; Wang, X. Association between leukocyte telomere length and plasma homocysteine in a singapore chinese population. Rejuvenation Res. 2015, 18, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Shin, C.; Baik, I. Leukocyte telomere length is associated with serum vitamin B12 and homocysteine levels in older adults with the presence of systemic inflammation. Clin. Nutr. Res. 2016, 5, 7–14. [Google Scholar] [CrossRef]
- Milne, E.; O’Callaghan, N.; Ramankutty, P.; de Klerk, N.H.; Greenop, K.R.; Armstrong, B.K.; Miller, M.; Fenech, M. Plasma micronutrient levels and telomere length in children. Nutrition 2015, 31, 331–336. [Google Scholar] [CrossRef]
- Nomura, S.J.; Robien, K.; Zota, A.R. Serum folate, vitamin B-12, vitamin A, γ-tocopherol, α-tocopherol, and carotenoids do not modify associations between cadmium exposure and leukocyte telomere length in the general US adult population. J. Nutr. 2017, 147, 538–548. [Google Scholar] [CrossRef]
- Stanger, O.; Herrmann, W.; Pietrzik, K.; Fowler, B.; Geisel, J.; Dierkes, J.; Weger, M. DACH-LIGA homocystein (german, austrian and swiss homocysteine society): Consensus paper on the rational clinical use of homocysteine, folic acid and B-vitamins in cardiovascular and thrombotic diseases: Guidelines and recommendations. Clin. Chem. Lab. Med. 2003, 41, 1392–1403. [Google Scholar] [CrossRef]
- Stabler, S.P. Clinical practice. Vitamin B12 deficiency. N. Engl. J. Med. 2013, 368, 149–160. [Google Scholar] [CrossRef]
- Herrmann, W.; Obeid, R. Utility and limitations of biochemical markers of vitamin B12 deficiency. Eur. J. Clin. Invest. 2013, 43, 231–237. [Google Scholar] [CrossRef]
- Herrmann, W.; Obeid, R. Vitamins in the Prevention of Human Diseases, 1st ed.; De Gruyter: Berlin, Germany, 2011. [Google Scholar]
- Herrmann, W.; Obeid, R. Hyperhomocysteinemia and response of methionine cycle intermediates to vitamin treatment in renal patients. Clin. Chem. Lab. Med. 2005, 43, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Friso, S.; Udali, S.; De Santis, D.; Choi, S.W. One-carbon metabolism and epigenetics. Mol. Asp. Med. 2017, 54, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Mendioroz, M.; Puebla-Guedea, M.; Montero-Marín, J.; Urdánoz-Casado, A.; Blanco-Luquin, I.; Roldán, M.; Labarga, A.; García-Campayo, J. Telomere length correlates with subtelomeric DNA methylation in long-term mindfulness practitioners. Sci. Rep. 2020, 10, 4564. [Google Scholar] [CrossRef] [PubMed]
- Novakovic, B.; Napier, C.E.; Vryer, R.; Dimitriadis, E.; Manuelpillai, U.; Sharkey, A.; Craig, J.M.; Reddel, R.R.; Saffery, R. DNA methylation mediated up-regulation of TERRA non-coding RNA is coincident with elongated telomeres in the human placenta. Mol. Hum. Reprod. 2016, 22, 791–799. [Google Scholar] [CrossRef]
- Hu, H.; Li, B.; Duan, S. The alteration of subtelomeric DNA methylation in aging-related diseases. Front. Genet. 2018, 9, 697. [Google Scholar] [CrossRef]
- Sibrian-Vazquez, M.; Escobedo, J.O.; Lim, S.; Samoei, G.K.; Strongin, R.M. Homocystamides promote free-radical and oxidative damage to proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 551–554. [Google Scholar] [CrossRef]
- Pusceddu, I.; Herrmann, W.; Kleber, M.E.; Scharnagl, H.; März, W.; Herrmann, M. Telomere length, vitamin B12 and mortality in persons undergoing coronary angiography: The ludwigshafen risk and cardiovascular health study. Aging 2019, 11, 7083–7097. [Google Scholar] [CrossRef]
- Chiriacò, M.; Georgiopoulos, G.; Duranti, E.; Antonioli, L.; Puxeddu, I.; Nannipieri, M.; Rosada, J.; Blandizzi, C.; Taddei, S.; Virdis, A.; et al. Inflammation and vascular ageing: From telomeres to novel emerging mechanisms. High. Blood Press. Cardiovasc. Prev. 2019, 26, 321–329. [Google Scholar] [CrossRef]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Flores, I.; Canela, A.; Vera, E.; Tejera, A.; Cotsarelis, G.; Blasco, M.A. The longest telomeres: A general signature of adult stem cell compartments. Genes Dev. 2008, 22, 654–667. [Google Scholar] [CrossRef]
- Yui, J.; Chiu, C.P.; Lansdorp, P.M. Telomerase activity in candidate stem cells from fetal liver and adult bone marrow. Blood 1998, 91, 3255–3262. [Google Scholar] [CrossRef] [PubMed]
- von Zglinicki, T. Role of oxidative stress in telomere length regulation and replicative senescence. Ann. N. Y. Acad. Sci. 2000, 908, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Bekaert, S.; De Meyer, T.; Rietzschel, E.R.; De Buyzere, M.L.; De Bacquer, D.; Langlois, M.; Segers, P.; Cooman, L.; Van Damme, P.; Cassiman, P.; et al. Telomere length and cardiovascular risk factors in a middle-aged population free of overt cardiovascular disease. Aging Cell 2007, 6, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Fouquerel, E.; Lormand, J.; Bose, A.; Lee, H.T.; Kim, G.S.; Li, J.; Sobol, R.W.; Freudenthal, B.D.; Myong, S.; Opresko, P.L. Oxidative guanine base damage regulates human telomerase activity. Nat. Struct. Mol. Biol. 2016, 23, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Lindhardsen, J.; Ahlehoff, O.; Gislason, G.H.; Madsen, O.R.; Olesen, J.B.; Torp-Pedersen, C.; Hansen, P.R. The risk of myocardial infarction in rheumatoid arthritis and diabetes mellitus: A Danish nationwide cohort study. Ann. Rheum. Dis. 2011, 70, 929–934. [Google Scholar] [CrossRef]
- van Halm, V.P.; Peters, M.J.; Voskuyl, A.E.; Boers, M.; Lems, W.F.; Visser, M.; Stehouwer, C.D.; Spijkerman, A.M.; Dekker, J.M.; Nijpels, G.; et al. Rheumatoid arthritis versus diabetes as a risk factor for cardiovascular disease: A cross-sectional study, the CARRE investigation. Ann. Rheum. Dis. 2009, 68, 1395–1400. [Google Scholar] [CrossRef]
- Gamal, R.M.; Hammam, N.; Zakary, M.M.; Abdelaziz, M.M.; Razek, M.R.A.; Mohamed, M.S.E.; Emad, Y.; Elnaggar, M.G.; Furst, D.E. Telomere dysfunction-related serological markers and oxidative stress markers in rheumatoid arthritis patients: Correlation with diseases activity. Clin. Rheumatol. 2018, 37, 3239–3246. [Google Scholar] [CrossRef]
- Sampson, M.J.; Winterbone, M.S.; Hughes, J.C.; Dozio, N.; Hughes, D.A. Monocyte telomere shortening and oxidative DNA damage in type 2 diabetes. Diabetes Care 2006, 29, 283–289. [Google Scholar] [CrossRef]
- Masi, S.; Gkranias, N.; Li, K.; Salpea, K.D.; Parkar, M.; Orlandi, M.; Suvan, J.E.; Eng, H.L.; Taddei, S.; Patel, K.; et al. Association between short leukocyte telomere length, endotoxemia, and severe periodontitis in people with diabetes: A cross-sectional survey. Diabetes Care 2014, 37, 1140–1147. [Google Scholar] [CrossRef]
- García-Redondo, A.B.; Aguado, A.; Briones, A.M.; Salaices, M. NADPH oxidases and vascular remodeling in cardiovascular diseases. Pharm. Res. 2016, 114, 110–120. [Google Scholar] [CrossRef]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH oxidases in vascular pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef]
- Sorescu, D.; Weiss, D.; Lassègue, B.; Clempus, R.E.; Szöcs, K.; Sorescu, G.P.; Valppu, L.; Quinn, M.T.; Lambeth, J.D.; Vega, J.D.; et al. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation 2002, 105, 1429–1435. [Google Scholar] [CrossRef]
- Kalinina, N.; Agrotis, A.; Tararak, E.; Antropova, Y.; Kanellakis, P.; Ilyinskaya, O.; Quinn, M.T.; Smirnov, V.; Bobik, A. Cytochrome b558-dependent NAD(P)H oxidase-phox units in smooth muscle and macrophages of atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 2037–2043. [Google Scholar] [CrossRef] [PubMed]
- Madrigal-Matute, J.; Fernandez-Laso, V.; Sastre, C.; Llamas-Granda, P.; Egido, J.; Martin-Ventura, J.L.; Zalba, G.; Blanco-Colio, L.M. TWEAK/Fn14 interaction promotes oxidative stress through NADPH oxidase activation in macrophages. Cardiovasc Res. 2015, 108, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Pejenaute, Á.; Cortés, A.; Marqués, J.; Montero, L.; Beloqui, Ó.; Fortuño, A.; Martí, A.; Orbe, J.; Zalba, G. NADPH oxidase overactivity underlies telomere shortening in human atherosclerosis. Int. J. Mol. Sci. 2020, 21, 1434. [Google Scholar] [CrossRef]
- Billard, P.; Poncet, D.A. Replication stress at telomeric and mitochondrial DNA: Common origins and consequences on ageing. Int. J. Mol. Sci. 2019, 20, 4959. [Google Scholar] [CrossRef]
- Sies, H. Role of metabolic H2O2 generation: Redox signaling and oxidative stress. J. Biol. Chem. 2014, 289, 8735–8741. [Google Scholar] [CrossRef]
- Sanderson, S.L.; Simon, A.K. In aged primary T cells, mitochondrial stress contributes to telomere attrition measured by a novel imaging flow cytometry assay. Aging Cell 2017, 16, 1234–1243. [Google Scholar] [CrossRef]
- Liu, L.; Trimarchi, J.R.; Smith, P.J.; Keefe, D.L. Mitochondrial dysfunction leads to telomere attrition and genomic instability. Aging Cell 2002, 1, 40–46. [Google Scholar] [CrossRef]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef]
- Kolling, J.; Scherer, E.B.; da Cunha, A.A.; da Cunha, M.J.; Wyse, A.T. Homocysteine induces oxidative-nitrative stress in heart of rats: Prevention by folic acid. Cardiovasc. Toxicol. 2011, 11, 67–73. [Google Scholar] [CrossRef]
- Matté, C.; Mackedanz, V.; Stefanello, F.M.; Scherer, E.B.; Andreazza, A.C.; Zanotto, C.; Moro, A.M.; Garcia, S.C.; Gonçalves, C.A.; Erdtmann, B.; et al. Chronic hyperhomocysteinemia alters antioxidant defenses and increases DNA damage in brain and blood of rats: Protective effect of folic acid. Neurochem. Int. 2009, 54, 7–13. [Google Scholar] [CrossRef]
- Li, J.J.; Li, Q.; Du, H.P.; Wang, Y.L.; You, S.J.; Wang, F.; Xu, X.S.; Cheng, J.; Cao, Y.J.; Liu, C.F.; et al. Homocysteine triggers inflammatory responses in macrophages through inhibiting CSE-H2S signaling via DNA hypermethylation of CSE promoter. Int. J. Mol. Sci. 2015, 16, 12560–12577. [Google Scholar] [CrossRef]
- Steele, M.L.; Fuller, S.; Maczurek, A.E.; Kersaitis, C.; Ooi, L.; Münch, G. Chronic inflammation alters production and release of glutathione and related thiols in human U373 astroglial cells. Cell Mol. Neurobiol. 2013, 33, 19–30. [Google Scholar] [CrossRef]
- Ulvik, A.; Midttun, Ø.; Pedersen, E.R.; Eussen, S.J.; Nygård, O.; Ueland, P.M. Evidence for increased catabolism of vitamin B-6 during systemic inflammation. Am. J. Clin. Nutr. 2014, 100, 250–255. [Google Scholar] [CrossRef]
- Wong, J.Y.; De Vivo, I.; Lin, X.; Fang, S.C.; Christiani, D.C. The relationship between inflammatory biomarkers and telomere length in an occupational prospective cohort study. PLoS ONE 2014, 9, e87348. [Google Scholar] [CrossRef]
- Tan, R.; Lan, L. Guarding chromosomes from oxidative DNA damage to the very end. Acta Biochim. Biophys. Sin. 2016, 48, 617–622. [Google Scholar] [CrossRef]
- Opresko, P.L.; Fan, J.; Danzy, S.; Wilson, D.M., 3rd; Bohr, V.A. Oxidative damage in telomeric DNA disrupts recognition by TRF1 and TRF2. Nucleic Acids Res. 2005, 33, 1230–1239. [Google Scholar] [CrossRef]
- Lin, N.; Qin, S.; Luo, S.; Cui, S.; Huang, G.; Zhang, X. Homocysteine induces cytotoxicity and proliferation inhibition in neural stem cells via DNA methylation in vitro. FEBS J. 2014, 281, 2088–2096. [Google Scholar] [CrossRef]
- Zhang, D.; Sun, X.; Liu, J.; Xie, X.; Cui, W.; Zhu, Y. Homocysteine accelerates senescence of endothelial cells via DNA hypomethylation of human telomerase reverse transcriptase. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 71–78. [Google Scholar] [CrossRef]
- Farcas, R.; Schneider, E.; Frauenknecht, K.; Kondova, I.; Bontrop, R.; Bohl, J.; Navarro, B.; Metzler, M.; Zischler, H.; Zechner, U.; et al. Differences in DNA methylation patterns and expression of the CCRK gene in human and nonhuman primate cortices. Mol. Biol Evol. 2009, 26, 1379–1389. [Google Scholar] [CrossRef]
- Svenson, U.; Nordfjäll, K.; Baird, D.; Roger, L.; Osterman, P.; Hellenius, M.L.; Roos, G. Blood cell telomere length is a dynamic feature. PLoS ONE 2011, 6, e21485. [Google Scholar] [CrossRef]
- Gutierrez-Rodrigues, F.; Santana-Lemos, B.A.; Scheucher, P.S.; Alves-Paiva, R.M.; Calado, R.T. Direct comparison of flow-FISH and qPCR as diagnostic tests for telomere length measurement in humans. PLoS ONE 2014, 9, e113747. [Google Scholar] [CrossRef]
- Behrens, Y.L.; Thomay, K.; Hagedorn, M.; Ebersold, J.; Henrich, L.; Nustede, R.; Schlegelberger, B.; Göhring, G. Comparison of different methods for telomere length measurement in whole blood and blood cell subsets: Recommendations for telomere length measurement in hematological diseases. Genes Chromosomes Cancer 2017, 56, 700–708. [Google Scholar] [CrossRef]
- Aviv, A.; Hunt, S.C.; Lin, J.; Cao, X.; Kimura, M.; Blackburn, E. Impartial comparative analysis of measurement of leukocyte telomere length/DNA content by Southern blots and qPCR. Nucleic Acids Res. 2011, 39, e134. [Google Scholar] [CrossRef]
- Lai, T.P.; Wright, W.E.; Shay, J.W. Comparison of telomere length measurement methods. Philos. Trans. R. Soc. 2018, 373. [Google Scholar] [CrossRef]
- Montpetit, A.J.; Alhareeri, A.A.; Montpetit, M.; Starkweather, A.R.; Elmore, L.W.; Filler, K.; Mohanraj, L.; Burton, C.W.; Menzies, V.S.; Lyon, D.E.; et al. Telomere length: A review of methods for measurement. Nurs. Res. 2014, 63, 289–299. [Google Scholar] [CrossRef]


{kind=link}
| Topic | Reference | Type of Study | Participants | Follow-up | Primary Outcome |
| Telomere length & age | [23] | Longitudinal population-based | 510 males and females mean age at baseline 60 y | 10 y | LTL reduction of 45.5 bp/y |
| [24] | Longitudinal | 334 randomly selected flemish males and females, mean age at baseline 51.9 y | average 7.4 y | Significant LTL reduction, results provided as T/S ratio | |
| [22] | Longitudinal | 271 males and females, Caucasian and African Americans, mean age at baseline 31.9 y | average 12.4 y | LTL reduction of 31 bp/y | |
| [25] | Longitudinal | 75 Dutch men, mean age at baseline 77.6 y | 7 y | LTL reduction of 45.5 bp/y | |
| [26] | Longitudinal | 67 children, mean age at baseline 11.4 y, 99 of their parents, mean age at baseline 43.4 y | 14 y | LTL reduction in children 40.7 bp/y LTL reduction in adults 20.3 bp/y | |
| [20] | meta-analysis of 124 cross-sectional studies | 124 cross-sectional studies, age range 0–104 y, participants (range): 23–12,409 | n/a | weighted mean loss rate: 21.9 bp/y weighted median loss rate: 30.3 bp/y | |
| Telomere length & all-cause mortality | [21] | prospectice cohort study | 3316 patients hospitalized for elective coronary angiography, mean age 62.7 y | median 9.9 y | LTL quartiles 2–4 vs. 1 (shortest telomeres): HR(95% CI) 0.82 (0.71–0.92) |
| [15] | prospectice cohort study | 8633 females from the Nuses Health study, mean age at baseline 59 y, 3566 males and females from the ESTHER study, mean age at baseline 61.9 y | 18.4 y | shortest vs. longest LTL quintile: HR (95% CI) 1.23 (1.04–1.46) | |
| [17] | prospectice cohort study | 64,637 participants from the Copenhagen City Heart Study, Copenhagen General Population Study | median 7 y | shortest vs. the longest decile of LTL: HR (95% CI) 1.40 (1.25–1.57) | |
| [38] | meta-analysis of 25 prospective cohort studies | 12,083 participants, 2517 deaths | n/a | per 1 SD LTL decrement: HR (95% CI) 1.09 (1.06-1.13); shortest vs longest LTL quartile: HR (95% CI) 1.26 (1.15–1.38) | |
| Telomere length & cardiovascular mortality | [14] | populatin-based prospectice cohort study | 800 males and females, mean age at baseline 62.7 y | 10 y | shortest vs. the longest tertile of LTL: HR 3.04 (95% CI: 1.13–8.19) |
| [21] | prospectice cohort study | 3316 patients hospitalized for elective coronary angiography, mean age 62.7 y | median 9.9 y | LTL quartiles 2–4 vs. 1 (shortest telomeres): HR(95% CI) 0.84; (0.72–0.97) | |
| [15] | prospectice cohort study | 8633 females from the Nuses Health study, mean age at baseline 59 y, 3566 males and females from the ESTHER study, mean age at baseline 61.9 y | 18.4 y | shortest vs. longest LTL quintile: HR (95% CI) 1.10 (0.88–1.37) | |
| [17] | prospectice cohort study | 64,637 participants from the Copenhagen City Heart Study, Copenhagen General Population Study | median 7 y | per 200 bp reduction of LTL: HR(95% CI) 1.02 (1.01–1.03) | |
| Telomere length & atherosclerosis | [50] | prospectice cohort study | 2819 participants, were free of overt CVD, mean age at baseline 38.5 y | average 5.5 y | shortes vs. longest LTL tertile: HR(95% CI) for incident plaque 1.49 (1.09–2.03) HR(95% CI) plaque progression 1.61 (1.26–2.07) |
| [14] | populatin-based prospectice cohort study | 800 males and females, mean age at baseline 62.7 y | 10 y | shortest vs. the longest tertile of LTL: HR 3.18 (95% CI: 1.66–6.08) composite CVD end points (stroke, myocardial infarction, vascular death) | |
| [58] | cross-sectional | 1459 participants without CVD at recruitment, age at baseline 40–54 y | n/a | Average LTL and short telomere load are no significant predictors of total and femoral plaques | |
| [57] | cross-sectional | 325 subjects free of diabetes, coronary artery disease, stroke and cancer, age 40–64 years | n/a | Shortest vs. longest tertile of LTL: OR (95% CI) 2.36 (1.23–4.52) for having coronary artery calcification (after adjustment for age, race, gender, metabolic syndrome) | |
| [52] | cross-sectional | 2509 participants withoutestablished CVD, aged approximately 35–55 | n/a | LTL is neither an independent determinant of intima-media-thickness nor plaque presence | |
| Telomere length, HCY and B-vitamins | [61] | meta-analysis of 26 studies | n/a | n/a | estimated RR (95% CI) for coronary heart disease events associated with each 5-μmol/L increase in homocysteine 1.18 (1.10–1.26) |
| [62] | cross-sectional population-based cohort study | 1319 healthy subjects, mean age 49 y | n/a | adjusted LTL difference in the highest and lowest tertile of HCY was 111 base pairs (corresponding to 6.0 years of telomeric aging) | |
| [63] | cross-sectional cohort study | 1715 females | n/a | no sognificant association between LTL, HCY and B-vitamins | |
| Telomere length, oxidative stress and inflammation | [64] | cross-sectional and prospective cohort-study | 489 type 2 diabetics, mean age 67 y | 10 y | at baseline correltation between LTL and total antioxidant staus (r = 0.106, p = 0.024), lower TAOS and shorter LTL at baseline predicted increased risk of incident ischemic heart disease |
| [65] | cross-sectional | 176 patients with rheumatoid arthritis and 1151 controls | n/a | in rheumatoid arthritis patients significantly lower LTL, LTL unrelated to disease duration, CRP or rheumatoid factor | |
| [66] | cross-sectional | 2968 patients hospitalized for elective coronary angiography, mean age 63.5 y | n/a | Subjects with the longest telomeres had lower concentrations of HCY, IL-6, and hs-CRP | |
| [67] | cross-sectional | 1962 adults; age range: 70–79 y | n/a | OR (95% CI) for LTL in the shortest tertile: 1.3 (1.1–1.7) for subjects with IL-6 in top tertile 1.5 (1.2–1.9) for subjects with TNF-a in top tertile 1.8 (1.3–2.4) for subjects IL-6 + TNF-a in top tertile |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrmann, W.; Herrmann, M. The Importance of Telomere Shortening for Atherosclerosis and Mortality. J. Cardiovasc. Dev. Dis. 2020, 7, 29. https://doi.org/10.3390/jcdd7030029
Herrmann W, Herrmann M. The Importance of Telomere Shortening for Atherosclerosis and Mortality. Journal of Cardiovascular Development and Disease. 2020; 7(3):29. https://doi.org/10.3390/jcdd7030029
Chicago/Turabian StyleHerrmann, Wolfgang, and Markus Herrmann. 2020. "The Importance of Telomere Shortening for Atherosclerosis and Mortality" Journal of Cardiovascular Development and Disease 7, no. 3: 29. https://doi.org/10.3390/jcdd7030029
APA StyleHerrmann, W., & Herrmann, M. (2020). The Importance of Telomere Shortening for Atherosclerosis and Mortality. Journal of Cardiovascular Development and Disease, 7(3), 29. https://doi.org/10.3390/jcdd7030029