The Non-Fibrillar Side of Fibrosis: Contribution of the Basement Membrane, Proteoglycans, and Glycoproteins to Myocardial Fibrosis


Abstract
1. Introduction
2. Fibrillar ECM in the Myocardium
3. Basement Membrane Proteins in Myocardial Fibrosis
3.1. Fibronectin (FN) Is a Dimeric Glycoprotein in the ECM
3.2. Laminin Is One of the Main Proteins in the Basement Membrane
3.3. Collagen IV Forms a Network with Laminin in the Basement Membrane
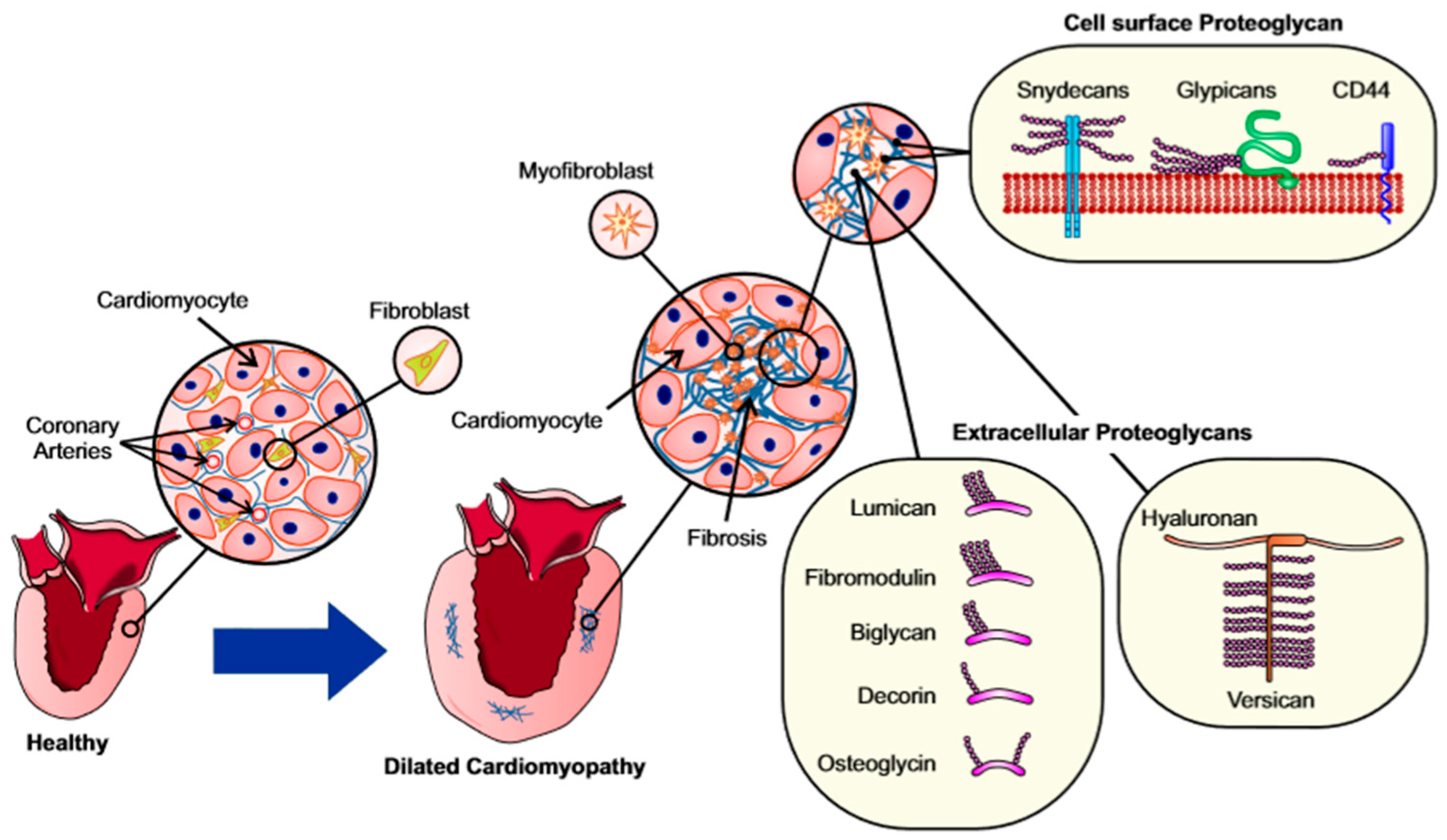
4. Proteoglycans and Glycoproteins in Fibrosis
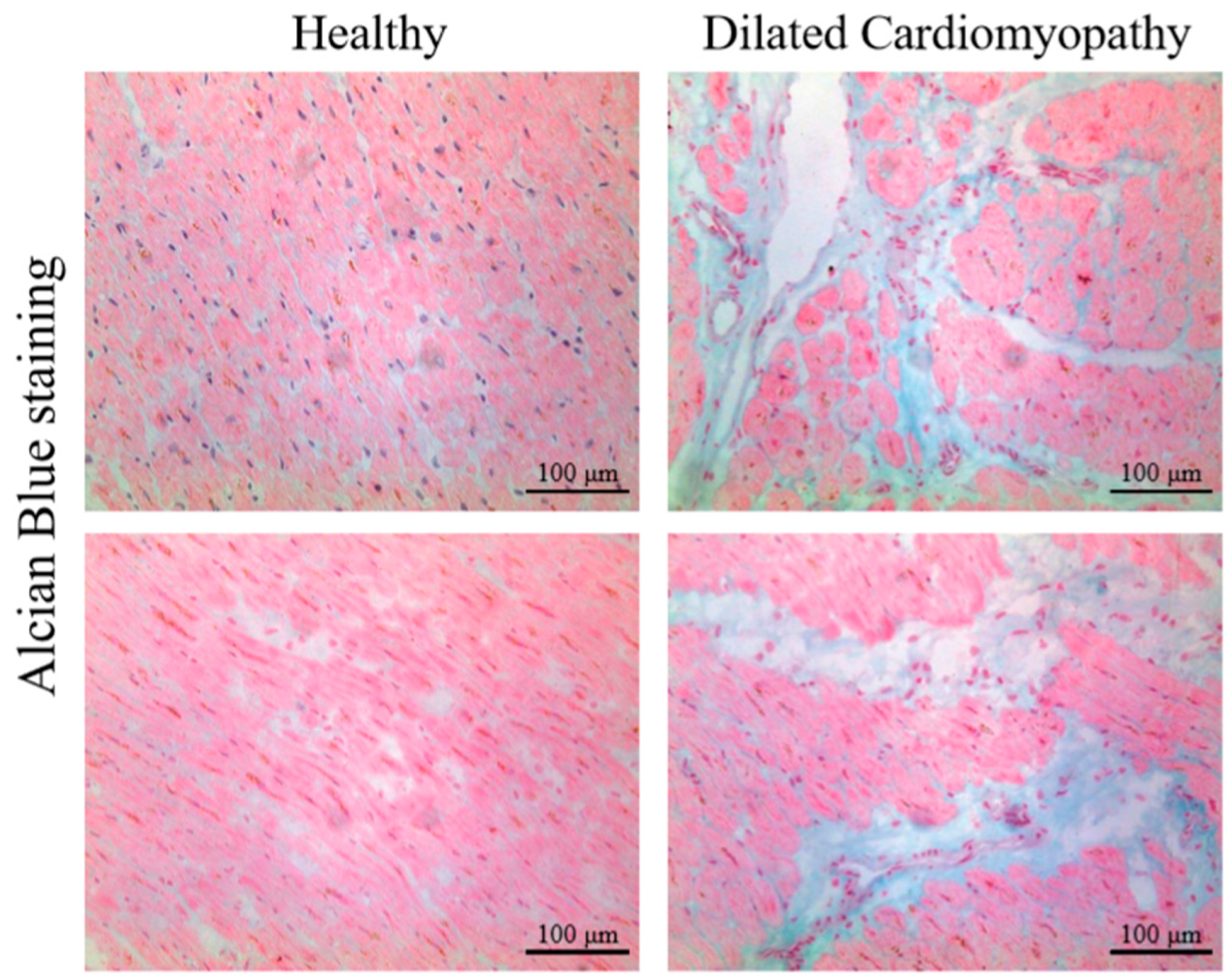
4.1. Proteoglycans and Glycosaminoglycans
4.2. Cell Surface Proteoglycans
4.3. Extracellular Proteoglycans (Hyalectins)
4.4. Basement Membrane Proteoglycans
4.5. Small Leucine Rich Proteoglycans
4.6. Fibromodulin and Osteoglycin
5. Glycoproteins
6. Matricryptins Can Regulate Fibrosis

{kind=link}
{kind=link}
| Matricrptins | Expression in Fibrotic Heart Disease | Role in Fibrotic Heart Disease |
|---|---|---|
| p1158/59 | Cleavage product of collagen 1α1 by MMP2, found in both human and mouse plasma post-MI [170]. | A pro-angiogenic factor that promotes early ECM deposition post-MI, reducing adverse LV remodeling [170]. |
| Arresten | Cleavage product of collagen IVα1 by MT1-MMP and MT2-MMP [166]. Expression is decreased 1-day post-MI in rats but increased in an I/R pig model [174,175]. | An anti-angiogenic and pro-apoptotic molecule [176,177]. |
| Canstatin | Cleavage product of collagen IVα2 by MT1-MMP and MT2-MMP [166]. Expression is decreased 1-day post-MI [174]. | Stimulates the migration of rat cardiac fibroblasts through the increased expression of MMP2 [171]. |
| Tumstatin | Cleavage product of collagen IVα3 by MMP9 [166]. Expression is increased after cardiac pressure overload [173]. | Stimulates the proliferation and migration of cardiac fibroblasts [173]. |
| Tetrastatin | Cleavage product of collagen IV α4 [166]. | Unknown. |
| Pentastatin | Cleavage product of collagen IV α5 [166]. | Unknown. |
| Hexastatin | Cleavage product of collagen IV α6 [166]. | Unknown. |
| Endostatin | Cleavage product of collagen XVIII [166]. Expression increased in rats post-MI [167]. | Anti-angiogenic but inhibition promotes adverse remodeling and fibrosis in rats post-MI [167]. Promotes proliferation, migration and wound healing in cardiac fibroblasts [169]. |
| Endorepellin | Cleavage product of perlecan, a proteoglycan [178]. | Anti-angiogenic and pro-fibrogenic [178]. |
7. General Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pitt, B.; Zannad, F. The Detection of Myocardial Fibrosis. Circulation Cardiovasc. Imaging 2012, 5, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Aspects Med. 2019, 65, 70–99. [Google Scholar] [CrossRef] [PubMed]
- De Boer, R.A.; De Keulenaer, G.; Bauersachs, J.; Brutsaert, D.; Cleland, J.G.; Diez, J.; Du, X.J.; Ford, P.; Heinzel, F.R.; Lipson, K.E.; et al. Towards better definition, quantification and treatment of fibrosis in heart failure. A scientific roadmap by the Committee of Translational Research of the Heart Failure Association (HFA) of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Creemers, E.E.; Kassiri, Z. Matrix as an interstitial transport system. Circ. Res. 2014, 114, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Querejeta, R.; López, B.; González, A.; Sánchez, E.; Larman, M.; Martínez Ubago José, L.; Díez, J. Increased Collagen Type I Synthesis in Patients With Heart Failure of Hypertensive Origin. Circulation 2004, 110, 1263–1268. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Takawale, A.; Lee, J.; Kassiri, Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair 2012, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Cleutjens, J.P.; Verluyten, M.J.; Smiths, J.F.; Daemen, M.J. Collagen remodeling after myocardial infarction in the rat heart. Am. J. Pathol. 1995, 147, 325–338. [Google Scholar]
- Weber, K.T. Cardiac interstitium in health and disease: The fibrillar collagen network. J. Am. Coll. Cardiol. 1989, 13, 1637–1652. [Google Scholar] [CrossRef]
- Mukherjee, D.; Sen, S. Alteration of collagen phenotypes in ischemic cardiomyopathy. J. Clin. Invest. 1991, 88, 1141–1146. [Google Scholar] [CrossRef]
- Skjøt-Arkil, H.; Clausen, R.E.; Rasmussen, L.M.; Wang, W.; Wang, Y.; Zheng, Q.; Mickley, H.; Saaby, L.; Diederichsen, A.C.P.; Lambrechtsen, J.; et al. Acute Myocardial Infarction and Pulmonary Diseases Result in Two Different Degradation Profiles of Elastin as Quantified by Two Novel ELISAs. PloS ONE 2013, 8, e60936. [Google Scholar] [CrossRef]
- Yu, Y.; Yin, G.; Bao, S.; Guo, Z. Kinetic alterations of collagen and elastic fibres and their association with cardiac function in acute myocardial infarction. Mol. Med. Rep. 2018, 17, 3519–3526. [Google Scholar] [CrossRef] [PubMed]
- Ruzicka, M.; Keeley, F.W.; Leenen, F.H. The renin-angiotensin system and volume overload-induced changes in cardiac collagen and elastin. Circulation 1994, 90, 1989–1996. [Google Scholar] [CrossRef] [PubMed]
- Cocciolone, A.J.; Hawes, J.Z.; Staiculescu, M.C.; Johnson, E.O.; Murshed, M.; Wagenseil, J.E. Elastin, arterial mechanics, and cardiovascular disease. Am. J. Physiol. Heart Circul. Physiol. 2018, 315, H189–H205. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Yau Terrence, M.; Weisel Richard, D.; Kiani Chris, G.; Li, R.-K. Elastin Stabilizes an Infarct and Preserves Ventricular Function. Circulation 2005, 112, I–81–I–88. [Google Scholar]
- 15 Mamuya, W.S.; Brecher, P. Fibronectin expression in the normal and hypertrophic rat heart. J. Clin. Invest. 1992, 89, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, M.M.; Janssen, A.M.; Daemen, M.J.; Rappaport, L.; Samuel, J.L.; Contard, F.; Smits, J.F.; Cleutjens, J.P. Increased expression of fibronectin isoforms after myocardial infarction in rats. J. Mol. Cell Cardiol. 1997, 29, 2533–2543. [Google Scholar] [CrossRef]
- Kohan, M.; Muro, A.F.; White, E.S.; Berkman, N. EDA-containing cellular fibronectin induces fibroblast differentiation through binding to alpha4beta7 integrin receptor and MAPK/Erk 1/2-dependent signaling. FASEB J. 2010, 24, 4503–4512. [Google Scholar] [CrossRef]
- Sottile, J.; Shi, F.; Rublyevska, I.; Chiang, H.Y.; Lust, J.; Chandler, J. Fibronectin-dependent collagen I deposition modulates the cell response to fibronectin. Am. J. Physiol. Cell Physiol. 2007, 293, C1934–C1946. [Google Scholar] [CrossRef]
- Schellings Mark, W.M.; Vanhoutte, D.; van Almen Geert, C.; Swinnen, M.; Leenders Joost, J.G.; Kubben, N.; van Leeuwen Rick, E.W.; Hofstra, L.; Heymans, S.; Pinto Yigal, M. Syndecan-1 Amplifies Angiotensin II–Induced Cardiac Fibrosis. Hypertension 2010, 55, 249–256. [Google Scholar] [CrossRef]
- Finsen, A.V.; Woldbaek, P.R.; Li, J.; Wu, J.; Lyberg, T.; Tonnessen, T.; Christensen, G. Increased syndecan expression following myocardial infarction indicates a role in cardiac remodeling. Physiol. Genomics 2004, 16, 301–308. [Google Scholar] [CrossRef]
- Vanhoutte, D.; Schellings Mark, W.M.; Götte, M.; Swinnen, M.; Herias, V.; Wild Martin, K.; Vestweber, D.; Chorianopoulos, E.; Cortés, V.; Rigotti, A.; et al. Increased Expression of Syndecan-1 Protects Against Cardiac Dilatation and Dysfunction After Myocardial Infarction. Circulation 2007, 115, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Herum, K.M.; Lunde, I.G.; McCulloch, A.D.; Christensen, G. The Soft- and Hard-Heartedness of Cardiac Fibroblasts: Mechanotransduction Signaling Pathways in Fibrosis of the Heart. J. Clin. Med. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Herum, K.M.; Lunde, I.G.; Skrbic, B.; Florholmen, G.; Behmen, D.; Sjaastad, I.; Carlson, C.R.; Gomez, M.F.; Christensen, G. Syndecan-4 signaling via NFAT regulates extracellular matrix production and cardiac myofibroblast differentiation in response to mechanical stress. J. Mol. Cell Cardiol. 2013, 54, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Finsen, A.V.; Lunde, I.G.; Sjaastad, I.; Ostli, E.K.; Lyngra, M.; Jarstadmarken, H.O.; Hasic, A.; Nygard, S.; Wilcox-Adelman, S.A.; Goetinck, P.F.; et al. Syndecan-4 is essential for development of concentric myocardial hypertrophy via stretch-induced activation of the calcineurin-NFAT pathway. PLoS ONE 2011, 6, e28302. [Google Scholar] [CrossRef] [PubMed]
- Herum, K.M.; Lunde, I.G.; Skrbic, B.; Louch, W.E.; Hasic, A.; Boye, S.; Unger, A.; Brorson, S.H.; Sjaastad, I.; Tonnessen, T.; et al. Syndecan-4 is a key determinant of collagen cross-linking and passive myocardial stiffness in the pressure-overloaded heart. Cardiovasc. Res. 2015. [Google Scholar] [CrossRef]
- Melleby, A.O.; Strand, M.E.; Romaine, A.; Herum, K.M.; Skrbic, B.; Dahl, C.P.; Sjaastad, I.; Fiane, A.E.; Filmus, J.; Christensen, G.; et al. The Heparan Sulfate Proteoglycan Glypican-6 Is Upregulated in the Failing Heart, and Regulates Cardiomyocyte Growth through ERK1/2 Signaling. PLoS ONE 2016, 11, e0165079. [Google Scholar] [CrossRef]
- Hatano, S.; Kimata, K.; Hiraiwa, N.; Kusakabe, M.; Isogai, Z.; Adachi, E.; Shinomura, T.; Watanabe, H. Versican/PG-M is essential for ventricular septal formation subsequent to cardiac atrioventricular cushion development. Glycobiology 2012, 22, 1268–1277. [Google Scholar] [CrossRef]
- Nandadasa, S.; Foulcer, S.; Apte, S.S. The multiple, complex roles of versican and its proteolytic turnover by ADAMTS proteases during embryogenesis. Matrix Biol. 2014, 35, 34–41. [Google Scholar] [CrossRef]
- Vistnes, M.; Aronsen, J.M.; Lunde, I.G.; Sjaastad, I.; Carlson, C.R.; Christensen, G. Pentosan polysulfate decreases myocardial expression of the extracellular matrix enzyme ADAMTS4 and improves cardiac function in vivo in rats subjected to pressure overload by aortic banding. PLoS ONE 2014, 9, e89621. [Google Scholar] [CrossRef]
- Wight, T.N.; Kang, I.; Merrilees, M.J. Versican and the control of inflammation. Matrix Biol. 2014, 35, 152–161. [Google Scholar] [CrossRef]
- Nakahama, M.; Murakami, T.; Kusachi, S.; Naito, I.; Takeda, K.; Ohnishi, H.; Komatsubara, I.; Oka, T.; Ninomiya, Y.; Tsuji, T. Expression of perlecan proteoglycan in the infarct zone of mouse myocardial infarction. J. Mol. Cell Cardiol. 2000, 32, 1087–1100. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, A.; Cooper, C.R.; Gomes, R.R., Jr. Soluble perlecan domain I enhances vascular endothelial growth factor-165 activity and receptor phosphorylation in human bone marrow endothelial cells. BMC Biochem. 2010, 11, 43. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ballesteros, S.; Varona, L.; Valera, M.; Gutierrez, J.P.; Cervantes, I. Cross-validation analysis for genetic evaluation models for ranking in endurance horses. Animal 2018, 12, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, A.N.; Schlessinger, J.; Hubbard, S.R.; Mohammadi, M. Structural basis for FGF receptor dimerization and activation. Cell 1999, 98, 641–650. [Google Scholar] [CrossRef]
- Schlessinger, J.; Plotnikov, A.N.; Ibrahimi, O.A.; Eliseenkova, A.V.; Yeh, B.K.; Yayon, A.; Linhardt, R.J.; Mohammadi, M. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell 2000, 6, 743–750. [Google Scholar] [CrossRef]
- Zoeller, J.J.; Whitelock, J.M.; Iozzo, R.V. Perlecan regulates developmental angiogenesis by modulating the VEGF-VEGFR2 axis. Matrix Biol. 2009, 28, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Aviezer, D.; Hecht, D.; Safran, M.; Eisinger, M.; David, G.; Yayon, A. Perlecan, basal lamina proteoglycan, promotes basic fibroblast growth factor-receptor binding, mitogenesis, and angiogenesis. Cell 1994, 79, 1005–1013. [Google Scholar] [CrossRef]
- Engebretsen, K.V.; Lunde, I.G.; Strand, M.E.; Waehre, A.; Sjaastad, I.; Marstein, H.S.; Skrbic, B.; Dahl, C.P.; Askevold, E.T.; Christensen, G.; et al. Lumican is increased in experimental and clinical heart failure, and its production by cardiac fibroblasts is induced by mechanical and proinflammatory stimuli. FEBS J. 2013, 280, 2382–2398. [Google Scholar] [CrossRef] [PubMed]
- Beetz, N.; Rommel, C.; Schnick, T.; Neumann, E.; Lother, A.; Monroy-Ordonez, E.B.; Zeeb, M.; Preissl, S.; Gilsbach, R.; Melchior-Becker, A.; et al. Ablation of biglycan attenuates cardiac hypertrophy and fibrosis after left ventricular pressure overload. J. Mol. Cell. Cardiol. 2016, 101, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Westermann, D.; Mersmann, J.; Melchior, A.; Freudenberger, T.; Petrik, C.; Schaefer, L.; Lullmann-Rauch, R.; Lettau, O.; Jacoby, C.; Schrader, J.; et al. Biglycan is required for adaptive remodeling after myocardial infarction. Circulation 2008, 117, 1269–1276. [Google Scholar] [CrossRef]
- Gubbiotti, M.A.; Vallet, S.D.; Ricard-Blum, S.; Iozzo, R.V. Decorin interacting network: A comprehensive analysis of decorin-binding partners and their versatile functions. Matrix Biol. 2016, 55, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Wang, P.; Zhao, C.X.; Tang, J.; Xiao, X.; Wang, D.W. Decorin gene delivery inhibits cardiac fibrosis in spontaneously hypertensive rats by modulation of transforming growth factor-beta/Smad and p38 mitogen-activated protein kinase signaling pathways. Hum. Gene Ther. 2009, 20, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- Naito, Z. The Role of Small Leucine-rich Proteoglycan (SLRP) Family in Pathological Lesions and Cancer Cell Growth. J. Nippon Med. School 2005, 72, 137–145. [Google Scholar] [CrossRef]
- Harris, B.S.; Zhang, Y.; Card, L.; Rivera, L.B.; Brekken, R.A.; Bradshaw, A.D. SPARC regulates collagen interaction with cardiac fibroblast cell surfaces. Am. J. Physiol. Heart Circul. Physiol. 2011, 301, H841–H847. [Google Scholar] [CrossRef] [PubMed]
- Sangaletti, S.; Di Carlo, E.; Gariboldi, S.; Miotti, S.; Cappetti, B.; Parenza, M.; Rumio, C.; Brekken, R.A.; Chiodoni, C.; Colombo, M.P. Macrophage-derived SPARC bridges tumor cell-extracellular matrix interactions toward metastasis. Cancer Res. 2008, 68, 9050–9059. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, A.D.; Baicu, C.F.; Rentz, T.J.; Van Laer, A.O.; Boggs, J.; Lacy, J.M.; Zile, M.R. Pressure overload-induced alterations in fibrillar collagen content and myocardial diastolic function: Role of secreted protein acidic and rich in cysteine (SPARC) in post-synthetic procollagen processing. Circulation 2009, 119, 269–280. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Bujak, M.; Zymek, P.; Ren, G.; Entman, M.L.; Frangogiannis, N.G. Extracellular matrix remodeling in canine and mouse myocardial infarcts. Cell Tissue Res. 2006, 324, 475–488. [Google Scholar] [CrossRef]
- Komatsubara, I.; Murakami, T.; Kusachi, S.; Nakamura, K.; Hirohata, S.; Hayashi, J.; Takemoto, S.; Suezawa, C.; Ninomiya, Y.; Shiratori, Y. Spatially and temporally different expression of osteonectin and osteopontin in the infarct zone of experimentally induced myocardial infarction in rats. Cardiovasc. Pathol. 2003, 12, 186–194. [Google Scholar] [CrossRef]
- McDonald, L.T.; Zile, M.R.; Zhang, Y.; Van Laer, A.O.; Baicu, C.F.; Stroud, R.E.; Jones, J.A.; LaRue, A.C.; Bradshaw, A.D. Increased macrophage-derived SPARC precedes collagen deposition in myocardial fibrosis. Am. J. Physiol. Heart Circul. Physiol. 2018, 315, H92–H100. [Google Scholar] [CrossRef]
- Jana, S.; Zhang, H.; Lopaschuk, G.D.; Freed, D.H.; Sergi, C.; Kantor, P.F.; Oudit, G.Y.; Kassiri, Z. Disparate Remodeling of the Extracellular Matrix and Proteoglycans in Failing Pediatric Versus Adult Hearts. J. Am. Heart Assoc. 2018, 7, e010427. [Google Scholar] [CrossRef]
- Agah, A.; Kyriakides, T.R.; Lawler, J.; Bornstein, P. The lack of thrombospondin-1 (TSP1) dictates the course of wound healing in double-TSP1/TSP2-null mice. Am. J. Pathol. 2002, 161, 831–839. [Google Scholar] [CrossRef]
- Rosini, S.; Pugh, N.; Bonna, A.M.; Hulmes, D.J.S.; Farndale, R.W.; Adams, J.C. Thrombospondin-1 promotes matrix homeostasis by interacting with collagen and lysyl oxidase precursors and collagen cross-linking sites. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef]
- Bein, K.; Simons, M. Thrombospondin type 1 repeats interact with matrix metalloproteinase 2. Regulation of metalloproteinase activity. J. Biol. Chem. 2000, 275, 32167–32173. [Google Scholar] [CrossRef]
- Yang, Z.; Kyriakides, T.R.; Bornstein, P. Matricellular proteins as modulators of cell-matrix interactions: Adhesive defect in thrombospondin 2-null fibroblasts is a consequence of increased levels of matrix metalloproteinase-2. Mol. Biol. Cell 2000, 11, 3353–3364. [Google Scholar] [CrossRef]
- Emonard, H.; Bellon, G.; Troeberg, L.; Berton, A.; Robinet, A.; Henriet, P.; Marbaix, E.; Kirkegaard, K.; Patthy, L.; Eeckhout, Y.; et al. Low density lipoprotein receptor-related protein mediates endocytic clearance of pro-MMP-2.TIMP-2 complex through a thrombospondin-independent mechanism. J. Biol. Chem. 2004, 279, 54944–54951. [Google Scholar] [CrossRef] [PubMed]
- Schroen, B.; Heymans, S.; Sharma, U.; Blankesteijn, W.M.; Pokharel, S.; Cleutjens Jack, P.M.; Porter, J.G.; Evelo Chris, T.A.; Duisters, R.; van Leeuwen Rick, E.W.; et al. Thrombospondin-2 Is Essential for Myocardial Matrix Integrity. Circulation Res. 2004, 95, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Adolph, K.W. Relative Abundance of Thrombospondin 2 and Thrombospondin 3 mRNAs in Human Tissues. Biochem. Biophys. Res. Commun. 1999, 258, 792–796. [Google Scholar] [CrossRef] [PubMed]
- Lawler, J.; Duquette, M.; Whittaker, C.A.; Adams, J.C.; McHenry, K.; DeSimone, D.W. Identification and characterization of thrombospondin-4, a new member of the thrombospondin gene family. J. Cell Biol. 1993, 120, 1059. [Google Scholar] [CrossRef] [PubMed]
- Stenina-Adognravi, O. Thrombospondins: Old players, new games. Curr. Opin. Lipidol. 2013, 24, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Schips, T.G.; Vanhoutte, D.; Vo, A.; Correll, R.N.; Brody, M.J.; Khalil, H.; Karch, J.; Tjondrokoesoemo, A.; Sargent, M.A.; Maillet, M.; et al. Thrombospondin-3 augments injury-induced cardiomyopathy by intracellular integrin inhibition and sarcolemmal instability. Nat. Commun. 2019, 10, 76. [Google Scholar] [CrossRef] [PubMed]
- Frolova, E.G.; Sopko, N.; Blech, L.; Popovic, Z.B.; Li, J.; Vasanji, A.; Drumm, C.; Krukovets, I.; Jain, M.K.; Penn, M.S.; et al. Thrombospondin-4 regulates fibrosis and remodeling of the myocardium in response to pressure overload. FASEB J. 2012, 26, 2363–2373. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.M.; Maillet, M.; Vanhoutte, D.; Schloemer, A.; Sargent, M.A.; Blair, N.S.; Lynch, K.A.; Okada, T.; Aronow, B.J.; Osinska, H.; et al. A Thrombospondin-Dependent Pathway for a Protective ER Stress Response. Cell 2012, 149, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Narouz-Ott, L.; Maurer, P.; Nitsche, D.P.; Smyth, N.; Paulsson, M. Thrombospondin-4 binds specifically to both collagenous and non-collagenous extracellular matrix proteins via its C-terminal domains. J. Biol. Chem. 2000, 275, 37110–37117. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, M.; Nakamura, K.; Kii, I.; Kashima, T.; Amizuka, N.; Li, M.; Saito, M.; Fukuda, K.; Nishiyama, T.; Kitajima, S.; et al. Periostin is essential for cardiac healing after acute myocardial infarction. J. Exp. Med. 2008, 205, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Xu, J.; Kaiser, R.A.; Melendez, J.; Hambleton, M.; Sargent, M.A.; Lorts, A.; Brunskill, E.W.; Dorn, G.W., 2nd; Conway, S.J.; et al. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ. Res. 2007, 101, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Ashley, S.L.; Wilke, C.A.; Kim, K.K.; Moore, B.B. Periostin regulates fibrocyte function to promote myofibroblast differentiation and lung fibrosis. Mucosal. Immunol. 2017, 10, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Willems, I.E.; Arends, J.W.; Daemen, M.J. Tenascin and fibronectin expression in healing human myocardial scars. J. Pathol. 1996, 179, 321–325. [Google Scholar] [CrossRef]
- Tamaoki, M.; Imanaka-Yoshida, K.; Yokoyama, K.; Nishioka, T.; Inada, H.; Hiroe, M.; Sakakura, T.; Yoshida, T. Tenascin-C regulates recruitment of myofibroblasts during tissue repair after myocardial injury. Am. J. Pathol 2005, 167, 71–80. [Google Scholar] [CrossRef]
- Chablais, F.; Jazwinska, A. The regenerative capacity of the zebrafish heart is dependent on TGF-beta signaling. Development 2012, 139, 1921–1930. [Google Scholar] [CrossRef]
- Imanaka-Yoshida, K.; Hiroe, M.; Yoshida, T. Interaction between cell and extracellular matrix in heart disease: Multiple roles of tenascin-C in tissue remodeling. Histol. Histopathol. 2004, 19, 517–525. [Google Scholar]
- Frangogiannis, N.G.; Shimoni, S.; Chang, S.M.; Ren, G.; Dewald, O.; Gersch, C.; Shan, K.; Aggeli, C.; Reardon, M.; Letsou, G.V.; et al. Active interstitial remodeling: An important process in the hibernating human myocardium. J. Am. Coll. Cardiol. 2002, 39, 1468–1474. [Google Scholar] [CrossRef]
- Nishioka, T.; Onishi, K.; Shimojo, N.; Nagano, Y.; Matsusaka, H.; Ikeuchi, M.; Ide, T.; Tsutsui, H.; Hiroe, M.; Yoshida, T.; et al. Tenascin-C may aggravate left ventricular remodeling and function after myocardial infarction in mice. Am. J. Physiol Heart Circ. Physiol. 2010, 298, H1072–H1078. [Google Scholar] [CrossRef]
- Song, L.; Wang, L.; Li, F.; Yukht, A.; Qin, M.; Ruther, H.; Yang, M.; Chaux, A.; Shah, P.K.; Sharifi, B.G. Bone Marrow-Derived Tenascin-C Attenuates Cardiac Hypertrophy by Controlling Inflammation. J. Am. College Cardiol. 2017, 70, 1601. [Google Scholar] [CrossRef]
- Prockop, D.J.; Kivirikko, K.I. Collagens: Molecular biology, diseases, and potentials for therapy. Annu Rev. Biochem. 1995, 64, 403–434. [Google Scholar] [CrossRef]
- Shamhart, P.E.; Meszaros, J.G. Non-fibrillar collagens: Key mediators of post-infarction cardiac remodeling? J. Mol. Cell Cardiol. 2010, 48, 530–537. [Google Scholar] [CrossRef]
- Segura, A.M.; Frazier, O.H.; Buja, L.M. Fibrosis and heart failure. Heart Fail. Rev. 2012. [Google Scholar] [CrossRef]
- Takawale, A.; Sakamuri, S.S.; Kassiri, Z. Extracellular matrix communication and turnover in cardiac physiology and pathology. Compr Physiol. 2015, 5, 687–719. [Google Scholar]
- Kadler, K.E.; Baldock, C.; Bella, J.; Boot-Handford, R.P. Collagens at a glance. J. Cell Sci. 2007, 120, 1955–1958. [Google Scholar] [CrossRef]
- Zuurmond, A.M.; van der Slot-Verhoeven, A.J.; van Dura, E.A.; De Groot, J.; Bank, R.A. Minoxidil exerts different inhibitory effects on gene expression of lysyl hydroxylase 1, 2, and 3: Implications for collagen cross-linking and treatment of fibrosis. Matrix Biol. 2005, 24, 261–270. [Google Scholar] [CrossRef]
- Eyre, D.; Shao, P.; Weis, M.A.; Steinmann, B. The kyphoscoliotic type of Ehlers-Danlos syndrome (type VI): Differential effects on the hydroxylation of lysine in collagens I and II revealed by analysis of cross-linked telopeptides from urine. Mol. Genet. Metab. 2002, 76, 211–216. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Matricellular proteins in cardiac adaptation and disease. Physiol. Rev. 2012, 92, 635–688. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ. Res. 2019, 125, 117–146. [Google Scholar] [CrossRef]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef]
- Gonzalez, A.; Schelbert, E.B.; Diez, J.; Butler, J. Myocardial Interstitial Fibrosis in Heart Failure: Biological and Translational Perspectives. J. Am. Coll Cardiol. 2018, 71, 1696–1706. [Google Scholar] [CrossRef]
- Ross, R.S.; Borg, T.K. Integrins and the myocardium. Circ. Res. 2001, 88, 1112–1119. [Google Scholar] [CrossRef]
- Manso, A.M.; Elsherif, L.; Kang, S.M.; Ross, R.S. Integrins, membrane-type matrix metalloproteinases and ADAMs: Potential implications for cardiac remodeling. Cardiovasc Res. 2006, 69, 574–584. [Google Scholar] [CrossRef]
- Kuhn, K. Basement membrane (type IV) collagen. Matrix Biol. 1995, 14, 439–445. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Lee, D.Y.; White, E.S.; Cui, Z.; Larios, J.M.; Chacon, R.; Horowitz, J.C.; Day, R.M.; Thomas, P.E. Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J. Biol. Chem. 2003, 278, 12384–12389. [Google Scholar] [CrossRef]
- Sapudom, J.; Rubner, S.; Martin, S.; Thoenes, S.; Anderegg, U.; Pompe, T. The interplay of fibronectin functionalization and TGF-beta1 presence on fibroblast proliferation, differentiation and migration in 3D matrices. Biomater. Sci. 2015, 3, 1291–1301. [Google Scholar] [CrossRef]
- Van Dijk, A.; Niessen, H.W.M.; Ursem, W.; Twisk, J.W.R.; Visser, F.C.; van Milligen, F.J. Accumulation of fibronectin in the heart after myocardial infarction: A putative stimulator of adhesion and proliferation of adipose-derived stem cells. Cell Tissue Res. 2008, 332, 289–298. [Google Scholar] [CrossRef]
- Valiente-Alandi, I.; Potter Sarah, J.; Salvador Ane, M.; Schafer Allison, E.; Schips, T.; Carrillo-Salinas, F.; Gibson Aaron, M.; Nieman Michelle, L.; Perkins, C.; Sargent Michelle, A.; et al. Inhibiting Fibronectin Attenuates Fibrosis and Improves Cardiac Function in a Model of Heart Failure. Circulation 2018, 138, 1236–1252. [Google Scholar] [CrossRef]
- Konstandin, M.H.; Volkers, M.; Collins, B.; Quijada, P.; Quintana, M.; De La Torre, A.; Ormachea, L.; Din, S.; Gude, N.; Toko, H.; et al. Fibronectin contributes to pathological cardiac hypertrophy but not physiological growth. Basic Res. Cardiol. 2013, 108, 375. [Google Scholar] [CrossRef]
- Fogelgren, B.; Polgar, N.; Szauter, K.M.; Ujfaludi, Z.; Laczko, R.; Fong, K.S.; Csiszar, K. Cellular fibronectin binds to lysyl oxidase with high affinity and is critical for its proteolytic activation. J. Biol. Chem. 2005, 280, 24690–24697. [Google Scholar] [CrossRef]
- Huang, G.; Zhang, Y.; Kim, B.; Ge, G.; Annis, D.S.; Mosher, D.F.; Greenspan, D.S. Fibronectin binds and enhances the activity of bone morphogenetic protein 1. J. Biol Chem. 2009, 284, 25879–25888. [Google Scholar] [CrossRef]
- Aumailley, M.; Bruckner-Tuderman, L.; Carter, W.G.; Deutzmann, R.; Edgar, D.; Ekblom, P.; Engel, J.; Engvall, E.; Hohenester, E.; Jones, J.C.; et al. A simplified laminin nomenclature. Matrix Biol. 2005, 24, 326–332. [Google Scholar] [CrossRef]
- Yurchenco, P.D.; Cheng, Y.S.; Colognato, H. Laminin forms an independent network in basement membranes. J. Cell Biol. 1992, 117, 1119–1133. [Google Scholar] [CrossRef]
- McKee, K.K.; Harrison, D.; Capizzi, S.; Yurchenco, P.D. Role of laminin terminal globular domains in basement membrane assembly. J. Biol. Chem. 2007, 282, 21437–21447. [Google Scholar] [CrossRef]
- Yurchenco, P.D.; Cheng, Y.S. Self-assembly and calcium-binding sites in laminin. A three-arm interaction model. J. Biol. Chem. 1993, 268, 17286–17299. [Google Scholar]
- Woodley, D.T.; Rao, C.N.; Hassell, J.R.; Liotta, L.A.; Martin, G.R.; Kleinman, H.K. Interactions of basement membrane components. Biochim Biophys Acta 1983, 761, 278–283. [Google Scholar] [CrossRef]
- Garbe, J.H.; Gohring, W.; Mann, K.; Timpl, R.; Sasaki, T. Complete sequence, recombinant analysis and binding to laminins and sulphated ligands of the N-terminal domains of laminin alpha3B and alpha5 chains. Biochem. J. 2002, 362, 213–221. [Google Scholar] [CrossRef]
- Aumailley, M.; Wiedemann, H.; Mann, K.; Timpl, R. Binding of nidogen and the laminin-nidogen complex to basement membrane collagen type IV. Eur. J. Biochem. 1989, 184, 241–248. [Google Scholar] [CrossRef]
- Hollfelder, D.; Frasch, M.; Reim, I. Distinct functions of the laminin beta LN domain and collagen IV during cardiac extracellular matrix formation and stabilization of alary muscle attachments revealed by EMS mutagenesis in Drosophila. BMC Dev. Biol. 2014, 14, 26. [Google Scholar] [CrossRef]
- Tisi, D.; Talts, J.F.; Timpl, R.; Hohenester, E. Structure of the C-terminal laminin G-like domain pair of the laminin α2 chain harbouring binding sites for alpha-dystroglycan and heparin. EMBO J. 2000, 19, 1432–1440. [Google Scholar] [CrossRef]
- Talts, J.F.; Andac, Z.; Göhring, W.; Brancaccio, A.; Timpl, R. Binding of the G domains of laminin alpha1 and alpha2 chains and perlecan to heparin, sulfatides, alpha-dystroglycan and several extracellular matrix proteins. EMBO J. 1999, 18, 863–870. [Google Scholar] [CrossRef]
- Nishiuchi, R.; Takagi, J.; Hayashi, M.; Ido, H.; Yagi, Y.; Sanzen, N.; Tsuji, T.; Yamada, M.; Sekiguchi, K. Ligand-binding specificities of laminin-binding integrins: A comprehensive survey of laminin-integrin interactions using recombinant alpha3beta1, alpha6beta1, alpha7beta1 and alpha6beta4 integrins. Matrix Biol. 2006, 25, 189–197. [Google Scholar] [CrossRef]
- Smyth, N.; Vatansever, H.S.; Murray, P.; Meyer, M.; Frie, C.; Paulsson, M.; Edgar, D. Absence of basement membranes after targeting the LAMC1 gene results in embryonic lethality due to failure of endoderm differentiation. J. Cell. Biol. 1999, 144, 151–160. [Google Scholar] [CrossRef]
- Yurchenco, P.D.; Amenta, P.S.; Patton, B.L. Basement membrane assembly, stability and activities observed through a developmental lens. Matrix Biol. 2004, 22, 521–538. [Google Scholar] [CrossRef]
- Wang, J.; Hoshijima, M.; Lam, J.; Zhou, Z.; Jokiel, A.; Dalton, N.D.; Hultenby, K.; Ruiz-Lozano, P.; Ross, J., Jr.; Tryggvason, K.; et al. Cardiomyopathy associated with microcirculation dysfunction in laminin alpha4 chain-deficient mice. J. Biol. Chem. 2006, 281, 213–220. [Google Scholar] [CrossRef]
- Stipp, C.S. Laminin-binding integrins and their tetraspanin partners as potential antimetastatic targets. Expert Rev. Mol. Med. 2010, 12, e3. [Google Scholar] [CrossRef]
- Campbell, I.D.; Humphries, M.J. Integrin structure, activation, and interactions. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef]
- Yurchenco, P.D.; Ruben, G.C. Basement membrane structure in situ: Evidence for lateral associations in the type IV collagen network. J. Cell Biol. 1987, 105, 2559–2568. [Google Scholar] [CrossRef]
- Timpl, R.; Wiedemann, H.; van Delden, V.; Furthmayr, H.; Kuhn, K. A network model for the organization of type IV collagen molecules in basement membranes. Eur. J. Biochem 1981, 120, 203–211. [Google Scholar] [CrossRef]
- Weber, S.; Engel, J.; Wiedemann, H.; Glanville, R.W.; Timpl, R. Subunit structure and assembly of the globular domain of basement-membrane collagen type IV. Eur, J. Biochem 1984, 139, 401–410. [Google Scholar] [CrossRef]
- Boutaud, A.; Borza, D.B.; Bondar, O.; Gunwar, S.; Netzer, K.O.; Singh, N.; Ninomiya, Y.; Sado, Y.; Noelken, M.E.; Hudson, B.G. Type IV collagen of the glomerular basement membrane. Evidence that the chain specificity of network assembly is encoded by the noncollagenous NC1 domains. J. Biol. Chem. 2000, 275, 30716–30724. [Google Scholar] [CrossRef]
- Yurchenco, P.D.; Furthmayr, H. Self-assembly of basement membrane collagen. Biochemistry 1984, 23, 1839–1850. [Google Scholar] [CrossRef]
- Poschl, E.; Schlotzer-Schrehardt, U.; Brachvogel, B.; Saito, K.; Ninomiya, Y.; Mayer, U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development 2004, 131, 1619–1628. [Google Scholar] [CrossRef]
- Murakami, M.; Kusachi, S.; Nakahama, M.; Naito, I.; Murakami, T.; Doi, M.; Kondo, J.; Higashi, T.; Ninomiya, Y.; Tsuji, T. Expression of the alpha 1 and alpha 2 chains of type IV collagen in the infarct zone of rat myocardial infarction. J. Mol. Cell Cardiol. 1998, 30, 1191–1202. [Google Scholar] [CrossRef]
- Ohsato, K.; Shimizu, M.; Sugihara, N.; Ino, H.; Yoshio, H.; Takagi, Y.; Takeda, R. Type IV collagen in hypertrophic cardiomyopathy. Jpn. Heart J. 1994, 35, 311–321. [Google Scholar] [CrossRef][Green Version]
- Albert, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. The Extracellular Matrix of Animals. Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Peysselon, F.; Ricard-Blum, S. Heparin-protein interactions: From affinity and kinetics to biological roles. Application to an interaction network regulating angiogenesis. Matrix Biol. 2014, 35, 73–81. [Google Scholar] [CrossRef]
- Karamanos, N.K.; Tzanakakis, G.N. Glycosaminoglycans: From "cellular glue" to novel therapeutical agents. Curr. Opin. Pharmacol. 2012, 12, 220–222. [Google Scholar] [CrossRef][Green Version]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Syndecan-1: A critical mediator in cardiac fibrosis. Hypertension 2010, 55, 233–235. [Google Scholar] [CrossRef]
- Matsui, Y.; Ikesue, M.; Danzaki, K.; Morimoto, J.; Sato, M.; Tanaka, S.; Kojima, T.; Tsutsui, H.; Uede, T. Syndecan-4 prevents cardiac rupture and dysfunction after myocardial infarction. Circ. Res. 2011, 108, 1328–1339. [Google Scholar] [CrossRef]
- Lei, J.; Xue, S.; Wu, W.; Zhou, S.; Zhang, Y.; Yuan, G.; Wang, J. Sdc1 Overexpression Inhibits the p38 MAPK Pathway and Lessens Fibrotic Ventricular Remodeling in MI Rats. Inflammation 2013, 36, 603–615. [Google Scholar] [CrossRef]
- Tromp, J.; van der Pol, A.; Klip, I.T.; de Boer, R.A.; Jaarsma, T.; van Gilst, W.H.; Voors, A.A.; van Veldhuisen, D.J.; van der Meer, P. Fibrosis marker syndecan-1 and outcome in patients with heart failure with reduced and preserved ejection fraction. Circ. Heart Fail. 2014, 7, 457–462. [Google Scholar] [CrossRef]
- Gopal, S.; Søgaard, P.; Multhaupt, H.A.B.; Pataki, C.; Okina, E.; Xian, X.; Pedersen, M.E.; Stevens, T.; Griesbeck, O.; Park, P.W.; et al. Transmembrane proteoglycans control stretch-activated channels to set cytosolic calcium levels. J. Cell Biol. 2015, 210, 1199. [Google Scholar] [CrossRef]
- Davis, J.; Burr, A.R.; Davis, G.F.; Birnbaumer, L.; Molkentin, J.D. A TRPC6-dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev. Cell 2012, 23, 705–715. [Google Scholar] [CrossRef]
- Filmus, J.; Capurro, M.; Rast, J. Glypicans. Genome Biol. 2008, 9, 224. [Google Scholar] [CrossRef]
- Filmus, J.; Selleck, S.B. Glypicans: Proteoglycans with a surprise. J. Clin. Invest. 2001, 108, 497–501. [Google Scholar] [CrossRef]
- Yasmin; Maskari, R.A.; McEniery, C.M.; Cleary, S.E.; Li, Y.; Siew, K.; Figg, N.L.; Khir, A.W.; Cockcroft, J.R.; Wilkinson, I.B.; et al. The matrix proteins aggrecan and fibulin-1 play a key role in determining aortic stiffness. Sci. Rep. 2018, 8, 8550. [Google Scholar] [CrossRef]
- Yamaguchi, Y. Lecticans: Organizers of the brain extracellular matrix. Cell Mol. Life Sci. 2000, 57, 276–289. [Google Scholar] [CrossRef]
- Inatani, M.; Tanihara, H.; Oohira, A.; Honjo, M.; Kido, N.; Honda, Y. Upregulated Expression of Neurocan, a Nervous Tissue Specific Proteoglycan, in Transient Retinal Ischemia. Invest. Ophthalmol. Visual Sci. 2000, 41, 2748–2754. [Google Scholar]
- Asher, R.A.; Morgenstern, D.A.; Fidler, P.S.; Adcock, K.H.; Oohira, A.; Braistead, J.E.; Levine, J.M.; Margolis, R.U.; Rogers, J.H.; Fawcett, J.W. Neurocan is upregulated in injured brain and in cytokine-treated astrocytes. J. Neurosci. 2000, 20, 2427–2438. [Google Scholar] [CrossRef]
- Dongaonkar, R.M.; Stewart, R.H.; Geissler, H.J.; Laine, G.A. Myocardial microvascular permeability, interstitial oedema, and compromised cardiac function. Cardiovasc. Res. 2010, 87, 331–339. [Google Scholar] [CrossRef]
- Brooks, W.W.; Shen, S.S.; Conrad, C.H.; Goldstein, R.H.; Bing, O.H.L. Transition from compensated hypertrophy to systolic heart failure in the spontaneously hypertensive rat: Structure, function, and transcript analysis. Genomics 2010, 95, 84–92. [Google Scholar] [CrossRef]
- Esko, J.D.; Kimata, K.; Lindahl, U. Proteoglycans and Sulfated Glycosaminoglycans. Essentials of Glycobiology. A.; Varki, R.D., Cummings, J.D., Eds.; Cold Spring Harbor: New York, NY, USA, 2009. [Google Scholar]
- Kammerer, R.A.; Schulthess, T.; Landwehr, R.; Schumacher, B.; Lustig, A.; Yurchenco, P.D.; Ruegg, M.A.; Engel, J.; Denzer, A.J. Interaction of agrin with laminin requires a coiled-coil conformation of the agrin-binding site within the laminin gamma1 chain. EMBO J. 1999, 18, 6762–6770. [Google Scholar] [CrossRef]
- Yamada, H.; Denzer, A.J.; Hori, H.; Tanaka, T.; Anderson, L.V.; Fujita, S.; Fukuta-Ohi, H.; Shimizu, T.; Ruegg, M.A.; Matsumura, K. Dystroglycan is a dual receptor for agrin and laminin-2 in Schwann cell membrane. J. Biol. Chem. 1996, 271, 23418–23423. [Google Scholar] [CrossRef]
- Burgess, M.I.; Atkinson, P.; Ray, S.G. Restrictive left ventricular filling pattern after myocardial infarction: Significance of concomitant preserved systolic function. Echocardiography 2000, 17, 659–664. [Google Scholar] [CrossRef][Green Version]
- Denzer, A.J.; Brandenberger, R.; Gesemann, M.; Chiquet, M.; Ruegg, M.A. Agrin binds to the nerve-muscle basal lamina via laminin. J. Cell Biol. 1997, 137, 671–683. [Google Scholar] [CrossRef]
- Noonan, D.M.; Fulle, A.; Valente, P.; Cai, S.; Horigan, E.; Sasaki, M.; Yamada, Y.; Hassell, J.R. The complete sequence of perlecan, a basement membrane heparan sulfate proteoglycan, reveals extensive similarity with laminin A chain, low density lipoprotein-receptor, and the neural cell adhesion molecule. J. Biol. Chem. 1991, 266, 22939–22947. [Google Scholar]
- Murdoch, A.D.; Dodge, G.R.; Cohen, I.; Tuan, R.S.; Iozzo, R.V. Primary structure of the human heparan sulfate proteoglycan from basement membrane (HSPG2/perlecan). A chimeric molecule with multiple domains homologous to the low density lipoprotein receptor, laminin, neural cell adhesion molecules, and epidermal growth factor. J. Biol. Chem. 1992, 267, 8544–8557. [Google Scholar]
- Henry, M.D.; Satz, J.S.; Brakebusch, C.; Costell, M.; Gustafsson, E.; Fassler, R.; Campbell, K.P. Distinct roles for dystroglycan, beta1 integrin and perlecan in cell surface laminin organization. J. Cell Sci. 2001, 114, 1137–1144. [Google Scholar]
- Sasse, P.; Malan, D.; Fleischmann, M.; Roell, W.; Gustafsson, E.; Bostani, T.; Fan, Y.; Kolbe, T.; Breitbach, M.; Addicks, K.; et al. Perlecan is critical for heart stability. Cardiovasc. Res. 2008, 80, 435–444. [Google Scholar] [CrossRef]
- Rider, C.C.; Mulloy, B. Heparin, Heparan Sulphate and the TGF-beta Cytokine Superfamily. Molecules 2017, 22. [Google Scholar]
- Schaefer, L.; Iozzo, R.V. Biological functions of the small leucine-rich proteoglycans: From genetics to signal transduction. J. Biol. Chem. 2008, 283, 21305–21309. [Google Scholar] [CrossRef]
- Frey, H.; Schroeder, N.; Manon-Jensen, T.; Iozzo, R.V.; Schaefer, L. Biological interplay between proteoglycans and their innate immune receptors in inflammation. FEBS J. 2013, 280, 2165–2179. [Google Scholar] [CrossRef]
- Jahanyar, J.; Joyce, D.L.; Southard, R.E.; Loebe, M.; Noon, G.P.; Koerner, M.M.; Torre-Amione, G.; Youker, K.A. Decorin-mediated Transforming Growth Factor-β Inhibition Ameliorates Adverse Cardiac Remodeling. J. Heart Lung Transplantation 2007, 26, 34–40. [Google Scholar] [CrossRef]
- Chakravarti, S. Functions of lumican and fibromodulin: Lessons from knockout mice. Glycoconjugate J. 2002, 19, 287–293. [Google Scholar] [CrossRef]
- Dupuis, L.E.; Kern, C.B. Small leucine-rich proteoglycans exhibit unique spatiotemporal expression profiles during cardiac valve development. Dev. Dyn 2014, 243, 601–611. [Google Scholar] [CrossRef]
- Van Aelst, L.N.; Voss, S.; Carai, P.; Van Leeuwen, R.; Vanhoutte, D.; Sanders-van Wijk, S.; Eurlings, L.; Swinnen, M.; Verheyen, F.K.; Verbeken, E.; et al. Osteoglycin prevents cardiac dilatation and dysfunction after myocardial infarction through infarct collagen strengthening. Circ. Res. 2015, 116, 425–436. [Google Scholar] [CrossRef]
- Deckx, S.; Heggermont, W.; Carai, P.; Rienks, M.; Dresselaers, T.; Himmelreich, U.; van Leeuwen, R.; Lommen, W.; van der Velden, J.; Gonzalez, A.; et al. Osteoglycin prevents the development of age-related diastolic dysfunction during pressure overload by reducing cardiac fibrosis and inflammation. Matrix Biol. 2018, 66, 110–124. [Google Scholar] [CrossRef]
- Rienks, M.; Papageorgiou, A.; Wouters, K.; Verhesen, W.; Leeuwen, R.V.; Carai, P.; Summer, G.; Westermann, D.; Heymans, S. A novel 72-kDa leukocyte-derived osteoglycin enhances the activation of toll-like receptor 4 and exacerbates cardiac inflammation during viral myocarditis. Cell Mol. Life Sci. 2017, 74, 1511–1525. [Google Scholar] [CrossRef]
- Belmadani, S.; Bernal, J.; Wei, C.C.; Pallero, M.A.; Dell’Italia, L.; Murphy-Ullrich, J.E.; Berecek, K.H. A thrombospondin-1 antagonist of transforming growth factor-beta activation blocks cardiomyopathy in rats with diabetes and elevated angiotensin II. Am. J. Pathol. 2007, 171, 777–789. [Google Scholar] [CrossRef]
- Xia, Y.; Dobaczewski, M.; Gonzalez-Quesada, C.; Chen, W.; Biernacka, A.; Li, N.; Lee, D.W.; Frangogiannis, N.G. Endogenous thrombospondin 1 protects the pressure-overloaded myocardium by modulating fibroblast phenotype and matrix metabolism. Hypertension 2011, 58, 902–911. [Google Scholar] [CrossRef]
- Ramos-DeSimone, N.; Hahn-Dantona, E.; Sipley, J.; Nagase, H.; French, D.L.; Quigley, J.P. Activation of matrix metalloproteinase-9 (MMP-9) via a converging plasmin/stromelysin-1 cascade enhances tumor cell invasion. J. Biol Chem. 1999, 274, 13066–13076. [Google Scholar] [CrossRef]
- Schellings, M.W.; Vanhoutte, D.; Swinnen, M.; Cleutjens, J.P.; Debets, J.; van Leeuwen, R.E.; d’Hooge, J.; Van de Werf, F.; Carmeliet, P.; Pinto, Y.M.; et al. Absence of SPARC results in increased cardiac rupture and dysfunction after acute myocardial infarction. J. Exp. Med. 2009, 206, 113–123. [Google Scholar] [CrossRef]
- Takayama, G.; Arima, K.; Kanaji, T.; Toda, S.; Tanaka, H.; Shoji, S.; McKenzie, A.N.; Nagai, H.; Hotokebuchi, T.; Izuhara, K. Periostin: A novel component of subepithelial fibrosis of bronchial asthma downstream of IL-4 and IL-13 signals. J. Allergy Clin. Immunol. 2006, 118, 98–104. [Google Scholar] [CrossRef]
- Peng, H.; Sarwar, Z.; Yang, X.-P.; Peterson, E.L.; Xu, J.; Janic, B.; Rhaleb, N.; Carretero, O.A.; Rhaleb, N.-E. Profibrotic Role for Interleukin-4 in Cardiac Remodeling and Dysfunction. Hypertension 2015, 66, 582–589. [Google Scholar] [CrossRef]
- Yang, H.; Chen, Y.; Gao, C. Interleukin-13 reduces cardiac injury and prevents heart dysfunction in viral myocarditis via enhanced M2 macrophage polarization. Oncotarget 2017, 8, 99495–99503. [Google Scholar] [CrossRef]
- Spinale, F.G. Myocardial matrix remodeling and the matrix metalloproteinases: Influence on cardiac form and function. Physiol. Rev. 2007, 87, 1285–1342. [Google Scholar] [CrossRef]
- Lindsey, M.L. Assigning matrix metalloproteinase roles in ischaemic cardiac remodelling. Nat. Rev. Cardiol. 2018, 15, 471–479. [Google Scholar] [CrossRef]
- Davis, G.E.; Bayless, K.J.; Davis, M.J.; Meininger, G.A. Regulation of tissue injury responses by the exposure of matricryptic sites within extracellular matrix molecules. Am. J. Pathol. 2000, 156, 1489–1498. [Google Scholar] [CrossRef]
- Gruszka, A.; Kunert-Radek, J.; Pawlikowski, M.; Stepien, H. Serum endostatin levels are elevated and correlate with serum vascular endothelial growth factor levels in patients with pituitary adenomas. Pituitary 2005, 8, 163–168. [Google Scholar] [CrossRef]
- Okada, M.; Imoto, K.; Sugiyama, A.; Yasuda, J.; Yamawaki, H. New Insights into the Role of Basement Membrane-Derived Matricryptins in the Heart. Biol. Pharm. Bull. 2017, 40, 2050–2060. [Google Scholar] [CrossRef]
- Isobe, K.; Kuba, K.; Maejima, Y.; Suzuki, J.; Kubota, S.; Isobe, M. Inhibition of endostatin/collagen XVIII deteriorates left ventricular remodeling and heart failure in rat myocardial infarction model. Circ. J. 2010, 74, 109–119. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Takihara, T.; Chambers, R.A.; Veraldi, K.L.; Larregina, A.T.; Feghali-Bostwick, C.A. A peptide derived from endostatin ameliorates organ fibrosis. Sci Transl. Med. 2012, 4, 136ra171. [Google Scholar] [CrossRef]
- Okada, M.; Oba, Y.; Yamawaki, H. Endostatin stimulates proliferation and migration of adult rat cardiac fibroblasts through PI3K/Akt pathway. Eur. J. Pharmacol. 2015, 750, 20–26. [Google Scholar] [CrossRef]
- Lindsey, M.L.; Iyer, R.P.; Zamilpa, R.; Yabluchanskiy, A.; DeLeon-Pennell, K.Y.; Hall, M.E.; Kaplan, A.; Zouein, F.A.; Bratton, D.; Flynn, E.R.; et al. A Novel Collagen Matricryptin Reduces Left Ventricular Dilation Post-Myocardial Infarction by Promoting Scar Formation and Angiogenesis. J. Am. Coll Cardiol 2015, 66, 1364–1374. [Google Scholar] [CrossRef]
- Okada, M.; Murata, N.; Yamawaki, H. Canstatin stimulates migration of rat cardiac fibroblasts via secretion of matrix metalloproteinase-2. Am. J. Physiol Cell Physiol. 2017, 312, C199–C208. [Google Scholar] [CrossRef]
- Sugiyama, A.; Okada, M.; Yamawaki, H. Pathophysiological roles of canstatin on myofibroblasts after myocardial infarction in rats. Eur. J. Pharmacol. 2017, 807, 32–43. [Google Scholar] [CrossRef]
- Yasuda, J.; Fukui, K.; Okada, M.; Yamawaki, H. T3 peptide, a fragment of tumstatin, stimulates proliferation and migration of cardiac fibroblasts through activation of Akt signaling pathway. Naunyn Schmiedebergs Arch. Pharmacol. 2017, 390, 1135–1144. [Google Scholar] [CrossRef]
- Sugiyama, A.; Mitsui, A.; Okada, M.; Yamawaki, H. Cathepsin S degrades arresten and canstatin in infarcted area after myocardial infarction in rats. J. Veterinary Med. Sci. 2019, 81, 522–531. [Google Scholar] [CrossRef]
- Lauten, A.; Gerhard-Garcia, A.; Suhr, F.; Fischer, J.H.; Figulla, H.R.; Bloch, W. Impact of ischemia-reperfusion on extracellular matrix processing and structure of the basement membrane of the heart. PLoS ONE 2014, 9, e92833. [Google Scholar] [CrossRef]
- Sudhakar, A.; Nyberg, P.; Keshamouni, V.G.; Mannam, A.P.; Li, J.; Sugimoto, H.; Cosgrove, D.; Kalluri, R. Human alpha1 type IV collagen NC1 domain exhibits distinct antiangiogenic activity mediated by alpha1beta1 integrin. J. Clin. Invest. 2005, 115, 2801–2810. [Google Scholar] [CrossRef]
- Nyberg, P.; Xie, L.; Sugimoto, H.; Colorado, P.; Sund, M.; Holthaus, K.; Sudhakar, A.; Salo, T.; Kalluri, R. Characterization of the anti-angiogenic properties of arresten, an alpha1beta1 integrin-dependent collagen-derived tumor suppressor. Exp. Cell Res. 2008, 314, 3292–3305. [Google Scholar] [CrossRef]
- Cailhier, J.F.; Sirois, I.; Laplante, P.; Lepage, S.; Raymond, M.A.; Brassard, N.; Prat, A.; Iozzo, R.V.; Pshezhetsky, A.V.; Hebert, M.J. Caspase-3 activation triggers extracellular cathepsin L release and endorepellin proteolysis. J. Biol. Chem. 2008, 283, 27220–27229. [Google Scholar] [CrossRef]
- Bix, G.; Iozzo, R.V. Matrix revolutions: "tails" of basement-membrane components with angiostatic functions. Trends Cell Biol. 2005, 15, 52–60. [Google Scholar] [CrossRef]
- Hornebeck, W.; Emonard, H.; Monboisse, J.C.; Bellon, G. Matrix-directed regulation of pericellular proteolysis and tumor progression. Semin Cancer Biol. 2002, 12, 231–241. [Google Scholar] [CrossRef]
- Rebustini, I.T.; Myers, C.; Lassiter, K.S.; Surmak, A.; Szabova, L.; Holmbeck, K.; Pedchenko, V.; Hudson, B.G.; Hoffman, M.P. MT2-MMP-dependent release of collagen IV NC1 domains regulates submandibular gland branching morphogenesis. Dev. Cell 2009, 17, 482–493. [Google Scholar] [CrossRef]
- Wang, B.; Sun, J.; Kitamoto, S.; Yang, M.; Grubb, A.; Chapman, H.A.; Kalluri, R.; Shi, G.P. Cathepsin S controls angiogenesis and tumor growth via matrix-derived angiogenic factors. J. Biol Chem 2006, 281, 6020–6029. [Google Scholar] [CrossRef]
- Gonzalez, E.M.; Reed, C.C.; Bix, G.; Fu, J.; Zhang, Y.; Gopalakrishnan, B.; Greenspan, D.S.; Iozzo, R.V. BMP-1/Tolloid-like metalloproteases process endorepellin, the angiostatic C-terminal fragment of perlecan. J. Biol. Chem. 2005, 280, 7080–7087. [Google Scholar] [CrossRef] [PubMed]
- Eberlein, M.; Scheibner, K.A.; Black, K.E.; Collins, S.L.; Chan-Li, Y.; Powell, J.D.; Horton, M.R. Anti-oxidant inhibition of hyaluronan fragment-induced inflammatory gene expression. J. Inflamm (Lond) 2008, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Colorado, P.C.; Torre, A.; Kamphaus, G.; Maeshima, Y.; Hopfer, H.; Takahashi, K.; Volk, R.; Zamborsky, E.D.; Herman, S.; Sarkar, P.K.; et al. Anti-angiogenic cues from vascular basement membrane collagen. Cancer Res. 2000, 60, 2520–2526. [Google Scholar] [PubMed]
- Eckes, B.; Zweers, M.C.; Zhang, Z.G.; Hallinger, R.; Mauch, C.; Aumailley, M.; Krieg, T. Mechanical tension and integrin alpha 2 beta 1 regulate fibroblast functions. J. Investig Dermatol. Symp. Proc. 2006, 11, 66–72. [Google Scholar] [CrossRef] [PubMed]


| Fibrillar Proteins | Expression in Fibrotic Heart Diseases | Role in Fibrotic Heart Disease |
|---|---|---|
| Collagen type I | Expression is increased in dilated myocardiopathy, ischemic heart disease and myocardial infarction, and in response to hypertension and pressure overload in both humans and animal models [5,6,7,8]. | Deposited by activated fibroblasts [6]. Excessive collagen I deposition reduces heart compliance and result in increased dilation of the heart and can lead to heart failure [5,6]. In the ischemic heart, collagen I replaces necrotic cardiomyocytes to form the infart scar [6]. |
| Collagen III | Expression is increased in all forms of fibrotic heart disease [6,7,8,9]. | Deposited by activated fibroblasts [6]. Increased collagen III can reduce heart compliance and result in increased dilation of the heart [6,9]. |
| Elastin | Degraded in rats and patients with acute myocardial ischemia [10,11]. During the compensatory phase of pressure overload, elastin expression is increased [12]. | Degradation or mutation of elastin can lead to fibrotic heart disease (and aortic wall stiffening) [12,13]. Overexpression of elastin the infarct of ischemic hearts reduced LV dilation and infarct size, suggesting that preserving elastin is beneficial in fibrotic heart disease [14]. |
| Expression in Fibrotic Heart Disease | Role in Fibrotic Heart Disease | |
|---|---|---|
| Fibronectin (FN) | Expression is increased in hypertrophic rat hearts, and post-MI [15,16]. | Enables the differentiation of cardiac fibroblasts to myofibroblasts to promote fibrosis [17]. Fibronectin promotes the maturation, stabilization and deposition of collagen I [18]. |
| Syndecan-1 | Increased expression in fibrotic areas of angiotensin II induced cardiac fibrosis [19]. Increased expression post-MI [20]. | Promotes collagen upregulation, cross-linking and matrix formation [21]. |
| Syndecan-4 | Increased expression post-MI [20]. Found at focal adhesions, the site of mechanotransduction signaling [22]. | Promotes myofibroblast differentiation and collagen expression [23,24]. Facilitates LOX collagen cross-linking activity [25]. |
| Glypican-6 (GPC6) | Produced by cardiac fibroblasts and cardiomyocytes; upregulated with pressure overload and angiotensin II in mice [26]. | In vitro experiments could not show that GPC6 has an effect on cardiac fibroblast collagen expression, proliferation, or migration [26]. |
| Versican | Produced by cardiac fibroblasts and cardiomyocytes [27,28]. Upregulated in the hearts of pressure-overloaded rats [29]. | Versican expression and cleavage was increased following stimulation of cardiac cells with cytokines associated with heart failure, indicating that versican is under inflammatory control [30]. |
| Agrin and Perlecan | Found in the infarct and border regions post-MI [31]. | Perlecan and agrin can bind to and accumulate growth factors (FGF, TGFβ, BMP, VEGF, HB-EGF) in the ECM through their heparin sulfate groups [32,33,34,35,36,37]. |
| Biglycan | Expression is increased following MI or pressure overload [38]. | A pro-fibrotic proteoglycan that co-localizes with and stabilizes collagen; necessary in wound healing following MI, but is detrimental following pressure overload through increased cardiac fibrosis [39,40]. |
| Decorin | Expression is increased following MI or pressure overload [38]. | Inhibits TGFβ1 activity by sequestering TGFβ1 (in latent form) and decreasing its expression [41]. Antifibrotic by inhibiting the TGFβ1-Smad pathway in hypertensive rats [42]. |
| Lumican | Lumican is abundant in fibrotic tissues including the ischemic hearts [43]. Expression is increased in the hearts of HFrEF patients and pressure overloaded mice [38]. Expression is increased following mechanical stretch, IL-1β and LPS [38]. | Addition of recombinant lumican in vitro increased collagen I and lysyl oxidase expression [38]. |
| Secreted protein acidic and rich in cysteine (SPARC) | Secreted by fibroblasts and macrophages [44,45]. Expression is increased in response to pressure overload; is necessary for the development of myocardial fibrosis [46]. Expression is increased after MI and is spatially; temporally related to scar formation [47,48]. | Necessary for processing soluble procollagen into insoluble fibrillar collagen [46]. Macrophage-derived SPARC contributes to development of fibrosis in a murine pressure overload model [49]. |
| Thrombospondin-1 (TSP1) | Expression is strongly correlated to collagen expression in human explanted hearts [50]. | Can activate TGFβ1 during the inflammatory stage of MI, allows fibroblasts to differentiate into myofibroblasts [51]. Binds to pro-LOX, inhibiting its activation by BMP1 [52]. TSP-1 can clear MMP2 and MMP9 via endocytosis to promote fibrosis [53,54,55]. |
| Thrombospondin-2 (TSP2) | Expression is strongly correlated to collagen expression in human explanted hearts [50]. | Promotes fibrotic deposition following angiotensin II infusion [56]. |
| Thrombospondin-3 (TSP3) | Upregulated in cardiac disease [56,57,58,59]. | Promotes greater hypertrophy, exacerbated ventricular remodeling and dilation, and greater cardiac fibrosis in a pressure overload model [60]. |
| Thrombospondin-4 (TSP4) | Upregulated in cardiac disease [56,57,58,59]. | TSP-4 acts in opposition to TSP-1 and -2 by inhibiting profibrotic mechanisms [61,62,63]. |
| Periostin (PN) | Upregulated in fibrotic hearts and found in activated myofibroblasts and the interstitial matrix [64]. | Contributes to recruitment of fibroblasts and collagen fibrillogenesis [64,65]. Addition of periostin in vitro to cultured fibroblasts increased connective tissue growth factor and LOX mRNA expression [66]. |
| Tenascin-C (TNC) | Upregulated in fibrotic hearts and is localized in areas with activated myofibroblasts [67,68,69,70,71]. | TNC negatively impacts post-MI remodeling, but is beneficial in attenuating fibrosis in a pressure overload model [72,73]. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chute, M.; Aujla, P.; Jana, S.; Kassiri, Z. The Non-Fibrillar Side of Fibrosis: Contribution of the Basement Membrane, Proteoglycans, and Glycoproteins to Myocardial Fibrosis. J. Cardiovasc. Dev. Dis. 2019, 6, 35. https://doi.org/10.3390/jcdd6040035
Chute M, Aujla P, Jana S, Kassiri Z. The Non-Fibrillar Side of Fibrosis: Contribution of the Basement Membrane, Proteoglycans, and Glycoproteins to Myocardial Fibrosis. Journal of Cardiovascular Development and Disease. 2019; 6(4):35. https://doi.org/10.3390/jcdd6040035
Chicago/Turabian StyleChute, Michael, Preetinder Aujla, Sayantan Jana, and Zamaneh Kassiri. 2019. "The Non-Fibrillar Side of Fibrosis: Contribution of the Basement Membrane, Proteoglycans, and Glycoproteins to Myocardial Fibrosis" Journal of Cardiovascular Development and Disease 6, no. 4: 35. https://doi.org/10.3390/jcdd6040035
APA StyleChute, M., Aujla, P., Jana, S., & Kassiri, Z. (2019). The Non-Fibrillar Side of Fibrosis: Contribution of the Basement Membrane, Proteoglycans, and Glycoproteins to Myocardial Fibrosis. Journal of Cardiovascular Development and Disease, 6(4), 35. https://doi.org/10.3390/jcdd6040035