The Functions of Long Non-Coding RNA during Embryonic Cardiovascular Development and Its Potential for Diagnosis and Treatment of Congenital Heart Disease


Abstract
1. Introduction
2. Early Heart Development: Embryonic Specification of Cardiovascular Lineages
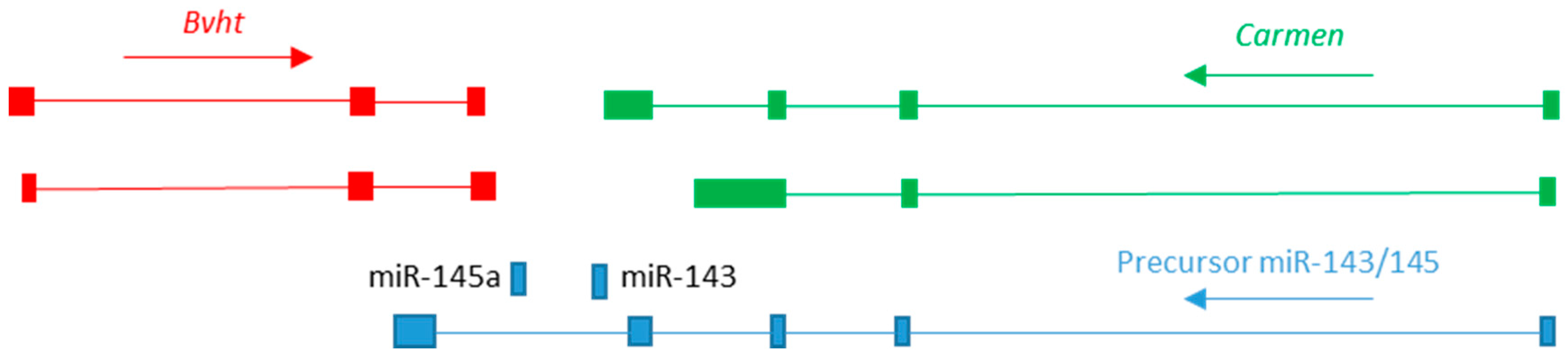
3. The Role of lncRNA in Cardiogenic Lineage Specification
3.1. Roles in Specification of Cardiogenic Mesoderm
3.2. Regulation of the Mesoderm-Endoderm Choice Point
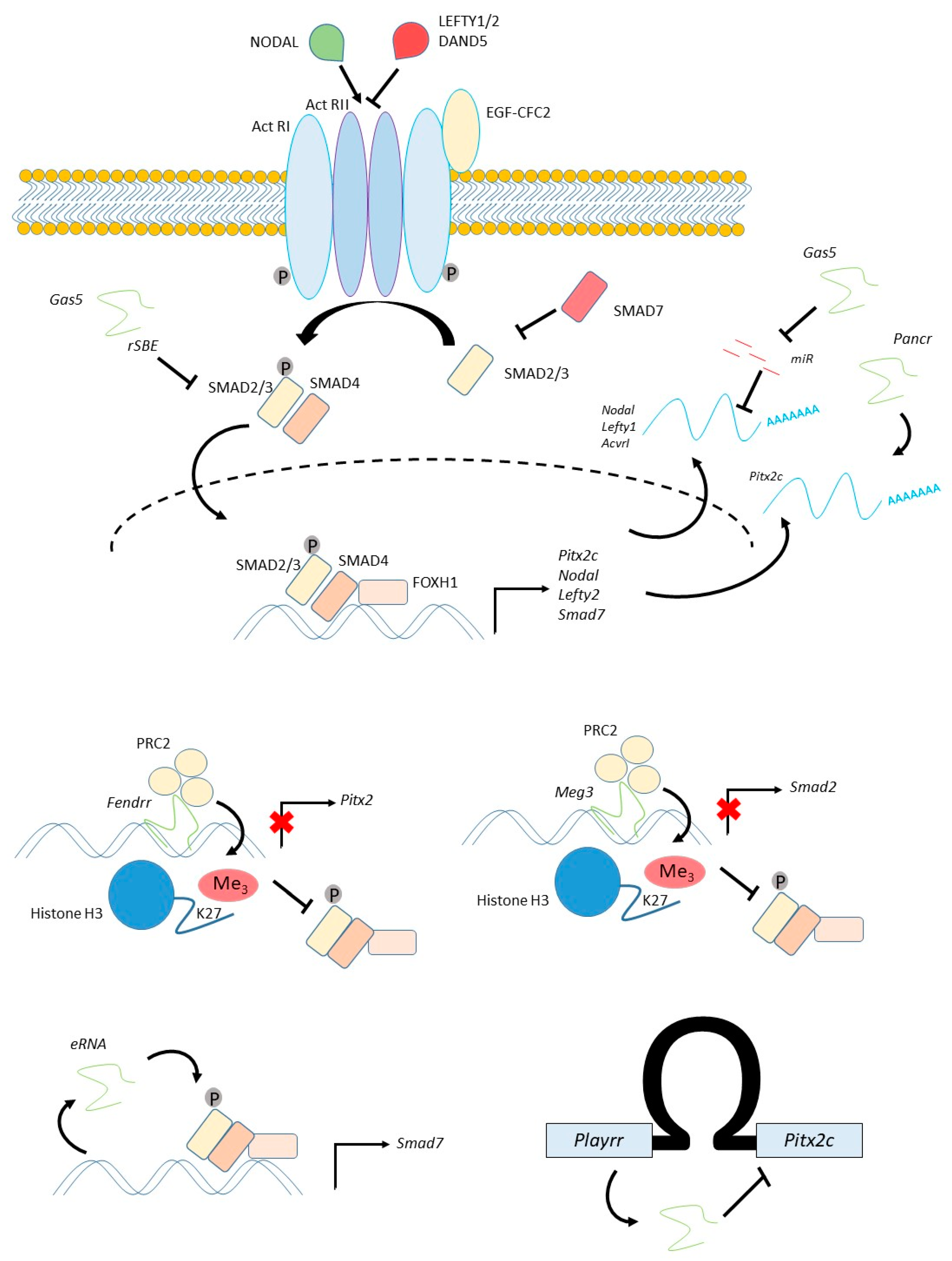
4. Patterning the Embryo: The NODAL Pathway
4.1. The NODAL Pathway Regulates Left-Right Patterning of the Cardiogenic Mesoderm
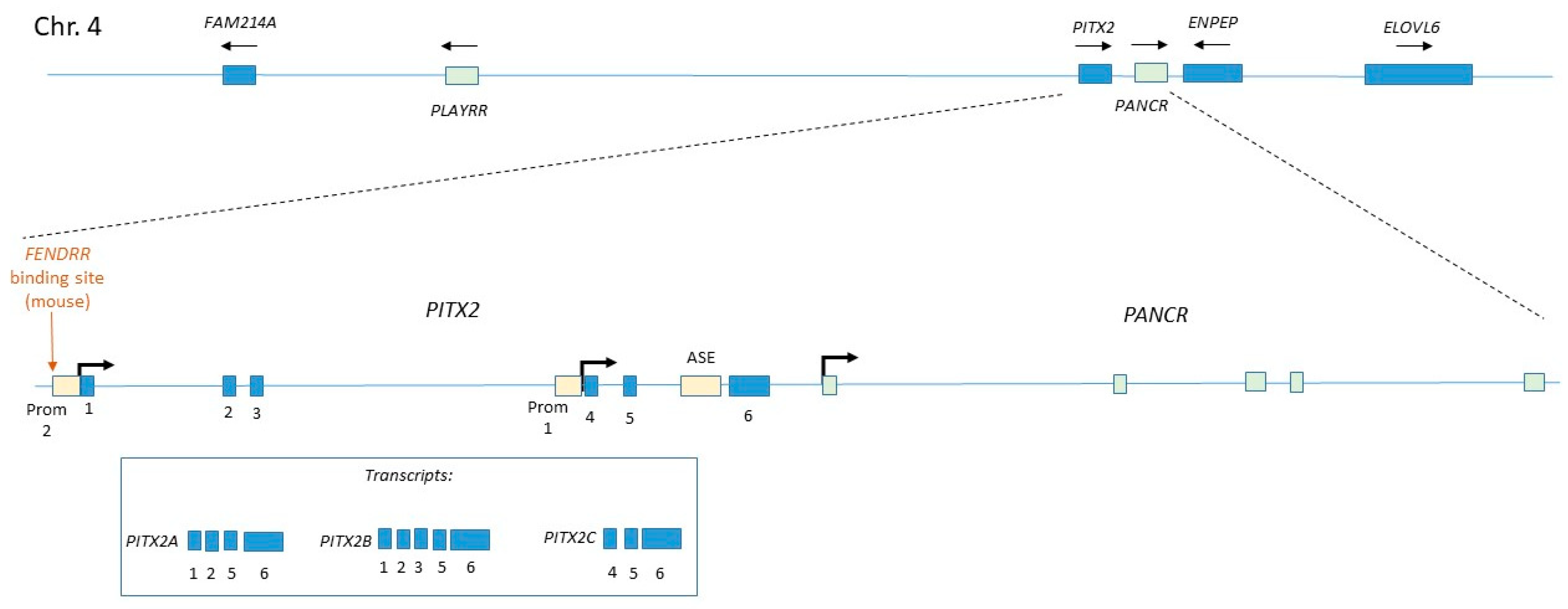
4.2. Regulation of Pitx2c
4.3. Regulation of NODAL and SMAD Signalling
5. lncRNA and Congenital Heart Disease: Challenges and Prospects
5.1. Lack of Evidence Pointing Towards A Role in CHD
5.2. Challenges in lncRNA Research
5.2.1. The Problem of Low Cross-Species Sequence Conservation
5.2.2. The Problem of Targeting Nuclear RNA Expression
5.2.3. The Problem of Low Endogenous Expression of lncRNA
5.2.4. The Problem of Identifying lncRNA Binding Partners
6. Diagnostics Applications of lncRNA
7. Therapeutics Applications of lncRNA
8. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Hoffman, J.I.E.; Kaplan, S. The incidence of congenital heart disease. J. Am. Coll. Cardiol. 2002, 39, 1890–1900. [Google Scholar] [CrossRef]
- Pandya, B.; Cullen, S.; Walker, F. Congenital heart disease in adults. BMJ 2016, 354, i3905. [Google Scholar] [CrossRef] [PubMed]
- Tsilimigras, D.I.; Oikonomou, E.K.; Moris, D.; Schizas, D.; Economopoulos, K.P.; Mylonas, K.S. Stem Cell Therapy for Congenital Heart Disease. Circulation 2017, 136, 2373–2385. [Google Scholar] [CrossRef] [PubMed]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed]
- de Hoon, M.; Shin, J.W.; Carninci, P. Paradigm shifts in genomics through the FANTOM projects. Mamm. Genome 2015, 26, 391–402. [Google Scholar] [CrossRef]
- Dykes, I.M.; Emanueli, C. Transcriptional and Post-Transcriptional Gene Regulation by Long Non-Coding RNA. Genom. Proteom. Bioinform. 2017, 15, 177–186. [Google Scholar] [CrossRef]
- Deng, X.; Zhou, J.; Li, F.F.; Yan, P.; Zhao, E.Y.; Hao, L.; Yu, K.J.; Liu, S.L. Characterization of nodal/TGF-lefty signaling pathway gene variants for possible roles in congenital heart diseases. PLoS ONE 2014, 9, e104535. [Google Scholar] [CrossRef]
- Ramsdell, A.F. Left-right asymmetry and congenital cardiac defects: Getting to the heart of the matter in vertebrate left-right axis determination. Dev. Biol 2005, 288, 1–20. [Google Scholar] [CrossRef]
- Van Vliet, P.; Wu, S.M.; Zaffran, S.; Puceat, M. Early cardiac development: A view from stem cells to embryos. Cardiovasc. Res. 2012, 96, 352–362. [Google Scholar] [CrossRef]
- Loh, K.M.; Chen, A.; Koh, P.W.; Deng, T.Z.; Sinha, R.; Tsai, J.M.; Barkal, A.A.; Shen, K.Y.; Jain, R.; Morganti, R.M.; et al. Mapping the Pairwise Choices Leading from Pluripotency to Human Bone, Heart, and Other Mesoderm Cell Types. Cell 2016, 166, 451–467. [Google Scholar] [CrossRef]
- Chabab, S.; Lescroart, F.; Rulands, S.; Mathiah, N.; Simons, B.D.; Blanpain, C. Uncovering the Number and Clonal Dynamics of Mesp1 Progenitors during Heart Morphogenesis. Cell Rep. 2016, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.N. Gene Regulatory Networks in the Evolution and Development of the Heart. Science 2006, 313, 1922–1927. [Google Scholar] [CrossRef] [PubMed]
- Bruneau, B.G. Signaling and transcriptional networks in heart development and regeneration. Cold Spring Harb. Perspect. Biol. 2013, 5, a008292. [Google Scholar] [CrossRef] [PubMed]
- Ieda, M.; Fu, J.D.; Delgado-Olguin, P.; Vedantham, V.; Hayashi, Y.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 2010, 142, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Nam, Y.J.; Luo, X.; Qi, X.; Tan, W.; Huang, G.N.; Acharya, A.; Smith, C.L.; Tallquist, M.D.; Neilson, E.G.; et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 2012, 485, 599–604. [Google Scholar] [CrossRef]
- Bodmer, R. The gene tinman is required for specification of the heart and visceral muscles in Drosophila. Development 1993, 118, 719–729. [Google Scholar] [PubMed]
- Costello, I.; Pimeisl, I.-M.; Dräger, S.; Bikoff, E.K.; Robertson, E.J.; Arnold, S.J. The T-box transcription factor Eomesodermin acts upstream of Mesp1 to specify cardiac mesoderm during mouse gastrulation. Nat. Cell Biol. 2011, 13, 1084. [Google Scholar] [CrossRef]
- Bondue, A.; Lapouge, G.; Paulissen, C.; Semeraro, C.; Iacovino, M.; Kyba, M.; Blanpain, C. Mesp1 Acts as a Master Regulator of Multipotent Cardiovascular Progenitor Specification. Cell Stem Cell 2008, 3, 69–84. [Google Scholar] [CrossRef]
- Saga, Y.; Miyagawa-Tomita, S.; Takagi, A.; Kitajima, S.; Miyazaki, J.; Inoue, T. MesP1 is expressed in the heart precursor cells and required for the formation of a single heart tube. Development 1999, 126, 3437–3447. [Google Scholar]
- David, R.; Brenner, C.; Stieber, J.; Schwarz, F.; Brunner, S.; Vollmer, M.; Mentele, E.; Müller-Höcker, J.; Kitajima, S.; Lickert, H.; et al. MesP1 drives vertebrate cardiovascular differentiation through Dkk-1-mediated blockade of Wnt-signalling. Nat. Cell Biol. 2008, 10, 338. [Google Scholar] [CrossRef]
- Islas, J.F.; Liu, Y.; Weng, K.C.; Robertson, M.J.; Zhang, S.; Prejusa, A.; Harger, J.; Tikhomirova, D.; Chopra, M.; Iyer, D.; et al. Transcription factors ETS2 and MESP1 transdifferentiate human dermal fibroblasts into cardiac progenitors. Proc. Natl. Acad. Sci. USA 2012, 109, 13016–13021. [Google Scholar] [CrossRef] [PubMed]
- Lescroart, F.; Wang, X.; Lin, X.; Swedlund, B.; Gargouri, S.; Sanchez-Danes, A.; Moignard, V.; Dubois, C.; Paulissen, C.; Kinston, S.; et al. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science 2018, 359, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Lescroart, F.; Chabab, S.; Lin, X.; Rulands, S.; Paulissen, C.; Rodolosse, A.; Auer, H.; Achouri, Y.; Dubois, C.; Bondue, A. Early lineage restriction in temporally distinct populations of Mesp1 progenitors during mammalian heart development. Nat. Cell Biol. 2014, 16, 829. [Google Scholar] [CrossRef] [PubMed]
- Klattenhoff, C.A.; Scheuermann, J.C.; Surface, L.E.; Bradley, R.K.; Fields, P.A.; Steinhauser, M.L.; Ding, H.; Butty, V.L.; Torrey, L.; Haas, S.; et al. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell 2013, 152, 570–583. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343. [Google Scholar] [CrossRef]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef]
- Davidovich, C.; Cech, T.R. The recruitment of chromatin modifiers by long noncoding RNAs: Lessons from PRC2. RNA 2015, 21, 2007–2022. [Google Scholar] [CrossRef]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Morales, D.R.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef]
- Xue, Z.; Hennelly, S.; Doyle, B.; Gulati, A.A.; Novikova, I.V.; Sanbonmatsu, K.Y.; Boyer, L.A. A G-Rich Motif in the lncRNA Braveheart Interacts with a Zinc-Finger Transcription Factor to Specify the Cardiovascular Lineage. Mol. Cell 2016, 64, 37–50. [Google Scholar] [CrossRef]
- Benhalevy, D.; Gupta, S.K.; Danan, C.H.; Ghosal, S.; Sun, H.W.; Kazemier, H.G.; Paeschke, K.; Hafner, M.; Juranek, S.A. The Human CCHC-Type Zinc Finger Nucleic Acid-Binding Protein Binds G-Rich Elements in Target mRNA Coding Sequences and Promotes Translation. Cell Rep. 2017, 18, 2979–2990. [Google Scholar] [CrossRef]
- Chen, W.; Liang, Y.; Deng, W.; Shimizu, K.; Ashique, A.M.; Li, E.; Li, Y.-P. The zinc-finger protein CNBP is required for forebrain formation in the mouse. Development 2003, 130, 1367–1379. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Zhang, P.; Li, J.; Wu, M. LAST, a c-Myc-inducible long noncoding RNA, cooperates with CNBP to promote CCND1 mRNA stability in human cells. eLife 2017, 6, e30433. [Google Scholar] [CrossRef] [PubMed]
- Kent, O.A.; Chivukula, R.R.; Mullendore, M.; Wentzel, E.A.; Feldmann, G.; Lee, K.H.; Liu, S.; Leach, S.D.; Maitra, A.; Mendell, J.T. Repression of the miR-143/145 cluster by oncogenic Ras initiates a tumor-promoting feed-forward pathway. Genes Dev. 2010, 24, 2754–2759. [Google Scholar] [CrossRef] [PubMed]
- Cordes, K.R.; Sheehy, N.T.; White, M.P.; Berry, E.C.; Morton, S.U.; Muth, A.N.; Lee, T.H.; Miano, J.M.; Ivey, K.N.; Srivastava, D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 2009, 460, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Elia, L.; Quintavalle, M.; Zhang, J.; Contu, R.; Cossu, L.; Latronico, M.V.G.; Peterson, K.L.; Indolfi, C.; Catalucci, D.; Chen, J.; et al. The knockout of miR-143 and -145 alters smooth muscle cell maintenance and vascular homeostasis in mice: Correlates with human disease. Cell Death Differ. 2009, 16, 1590. [Google Scholar] [CrossRef]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef]
- Mousavi, K.; Zare, H.; Dell’Orso, S.; Grontved, L.; Gutierrez-Cruz, G.; Derfoul, A.; Hager, G.L.; Sartorelli, V. eRNAs promote transcription by establishing chromatin accessibility at defined genomic loci. Mol. Cell 2013, 51, 606–617. [Google Scholar] [CrossRef]
- Ounzain, S.; Pezzuto, I.; Micheletti, R.; Burdet, F.; Sheta, R.; Nemir, M.; Gonzales, C.; Sarre, A.; Alexanian, M.; Blow, M.J.; et al. Functional importance of cardiac enhancer-associated noncoding RNAs in heart development and disease. J. Mol. Cell. Cardiol. 2014, 76, 55–70. [Google Scholar] [CrossRef]
- Ounzain, S.; Micheletti, R.; Arnan, C.; Plaisance, I.; Cecchi, D.; Schroen, B.; Reverter, F.; Alexanian, M.; Gonzales, C.; Ng, S.Y.; et al. CARMEN, a human super enhancer-associated long noncoding RNA controlling cardiac specification, differentiation and homeostasis. J. Mol. Cell. Cardiol. 2015, 89, 98–112. [Google Scholar] [CrossRef]
- Guttman, M.; Donaghey, J.; Carey, B.W.; Garber, M.; Grenier, J.K.; Munson, G.; Young, G.; Lucas, A.B.; Ach, R.; Bruhn, L.; et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2011, 477, 295. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Xu, Y.; Wang, Z.; Wu, Y.; Chen, J.; Wang, G.; Lu, C.; Jia, W.; Xi, J.; Zhu, S.; et al. A Linc1405/Eomes Complex Promotes Cardiac Mesoderm Specification and Cardiogenesis. Cell Stem Cell 2018, 22, 893–908. [Google Scholar] [CrossRef] [PubMed]
- Schuettengruber, B.; Martinez, A.-M.; Iovino, N.; Cavalli, G. Trithorax group proteins: Switching genes on and keeping them active. Nat. Rev. Mol. Cell Biol. 2011, 12, 799. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, Y.G. Signaling Control of Differentiation of Embryonic Stem Cells toward Mesendoderm. J. Mol. Biol. 2016, 428, 1409–1422. [Google Scholar] [CrossRef]
- Alexanian, M.; Maric, D.; Jenkinson, S.P.; Mina, M.; Friedman, C.E.; Ting, C.C.; Micheletti, R.; Plaisance, I.; Nemir, M.; Maison, D.; et al. A transcribed enhancer dictates mesendoderm specification in pluripotency. Nat. Commun. 2017, 8, 1806. [Google Scholar] [CrossRef]
- Daneshvar, K.; Pondick, J.V.; Kim, B.M.; Zhou, C.; York, S.R.; Macklin, J.A.; Abualteen, A.; Tan, B.; Sigova, A.A.; Marcho, C.; et al. DIGIT Is a Conserved Long Noncoding RNA that Regulates GSC Expression to Control Definitive Endoderm Differentiation of Embryonic Stem Cells. Cell Rep. 2016, 17, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Barnes, R.M.; Black, B.L. Nodal Signaling and Congenital Heart Defects. In Etiology and Morphogenesis of Congenital Heart Disease: from Gene Function and Cellular Interaction to Morphology; Nakanishi, T., Markwald, R.R., Baldwin, H.S., Keller, B.B., Srivastava, D., Yamagishi, H., Eds.; Springer: Tokyo, Japan, 2016; pp. 183–192. [Google Scholar] [CrossRef]
- Dykes, I.M. Left Right Patterning, Evolution and Cardiac Development. J. Cardiovasc. Dev. Dis. 2014, 1, 52–72. [Google Scholar] [CrossRef]
- Marques, S.; Borges, A.C.; Silva, A.C.; Freitas, S.; Cordenonsi, M.; Belo, J.A. The activity of the Nodal antagonist Cerl-2 in the mouse node is required for correct L/R body axis. Genes Dev. 2004, 18, 2342–2347. [Google Scholar] [CrossRef]
- Schier, A.F. Nodal morphogens. Cold Spring Harb. Perspect. Biol. 2009, 1, a003459. [Google Scholar] [CrossRef]
- Pauklin, S.; Vallier, L. Activin/Nodal signalling in stem cells. Development 2015, 142, 607–619. [Google Scholar] [CrossRef]
- Yamamoto, M.; Meno, C.; Sakai, Y.; Shiratori, H.; Mochida, K.; Ikawa, Y.; Saijoh, Y.; Hamada, H. The transcription factor FoxH1 (FAST) mediates Nodal signaling during anterior-posterior patterning and node formation in the mouse. Genes Dev. 2001, 15, 1242–1256. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.; Hill, C.S. How the Smads regulate transcription. Int. J. Biochem. Cell Biol. 2008, 40, 383–408. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.S. Transcriptional Control by the SMADs. Cold Spring Harb. Perspect. Biol. 2016, 8, a022079. [Google Scholar] [CrossRef]
- Dahle, O.; Kuehn, M.R. Polycomb determines responses to smad2/3 signaling in embryonic stem cell differentiation and in reprogramming. Stem Cells 2013, 31, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Dahle, O.; Kumar, A.; Kuehn, M.R. Nodal signaling recruits the histone demethylase Jmjd3 to counteract polycomb-mediated repression at target genes. Sci. Signal. 2010, 3, ra48. [Google Scholar] [CrossRef]
- Muller, P.; Rogers, K.W.; Jordan, B.M.; Lee, J.S.; Robson, D.; Ramanathan, S.; Schier, A.F. Differential diffusivity of Nodal and Lefty underlies a reaction-diffusion patterning system. Science 2012, 336, 721–724. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Abdollah, S.; Qiu, Y.; Cai, J.; Xu, Y.-Y.; Grinnell, B.W.; Richardson, M.A.; Topper, J.N.; Gimbrone, M.A., Jr.; Wrana, J.L. The MAD-related protein Smad7 associates with the TGFβ receptor and functions as an antagonist of TGFβ signaling. Cell 1997, 89, 1165–1173. [Google Scholar] [CrossRef]
- Shiratori, H.; Sakuma, R.; Watanabe, M.; Hashiguchi, H.; Mochida, K.; Sakai, Y.; Nishino, J.; Saijoh, Y.; Whitman, M.; Hamada, H. Two-step regulation of left-right asymmetric expression of Pitx2: Initiation by nodal signaling and maintenance by Nkx2. Mol. Cell 2001, 7, 137–149. [Google Scholar] [CrossRef]
- Franco, D.; Christoffels, V.M.; Campione, M. Homeobox transcription factor Pitx2: The rise of an asymmetry gene in cardiogenesis and arrhythmogenesis. Trends Cardiovasc. Med. 2013. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Miura, H.; Miyagawa-Tomita, S.; Yanazawa, M.; Katoh-Fukui, Y.; Suzuki, R.; Ohuchi, H.; Suehiro, A.; Motegi, Y.; Nakahara, Y.; et al. Mouse Pitx2 deficiency leads to anomalies of the ventral body wall, heart, extra- and periocular mesoderm and right pulmonary isomerism. Development 1999, 126, 5749–5758. [Google Scholar]
- Hernandez-Torres, F.; Franco, D.; Aranega, A.E.; Navarro, F. Expression patterns and immunohistochemical localization of PITX2B transcription factor in the developing mouse heart. Int. J. Dev. Biol. 2015, 59, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Gore-Panter, S.R.; Hsu, J.; Barnard, J.; Moravec, C.S.; Van Wagoner, D.R.; Chung, M.K.; Smith, J.D. PANCR, the PITX2 Adjacent Noncoding RNA, Is Expressed in Human Left Atria and Regulates PITX2c Expression. Circ. Arrhythm. Electrophysiol. 2016, 9, e003197. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Surendran, H.; Bharti, K.; Morishita, K.; Varshney, A.; Pal, R. Long Noncoding RNA RP11-380D23.2 Drives Distal-Proximal Patterning of the Lung by Regulating PITX2 Expression. Stem Cells 2018, 36, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Tulin, A.V. The roles of PARP1 in gene control and cell differentiation. Curr. Opin. Genet. Dev. 2010, 20, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Welsh, I.C.; Kwak, H.; Chen, F.L.; Werner, M.; Shopland, L.S.; Danko, C.G.; Lis, J.T.; Zhang, M.; Martin, J.F.; Kurpios, N.A. Chromatin Architecture of the Pitx2 Locus Requires CTCF- and Pitx2-Dependent Asymmetry that Mirrors Embryonic Gut Laterality. Cell Rep. 2015, 13, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Grote, P.; Wittler, L.; Hendrix, D.; Koch, F.; Wahrisch, S.; Beisaw, A.; Macura, K.; Blass, G.; Kellis, M.; Werber, M.; et al. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev. Cell 2013, 24, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Sauvageau, M.; Goff, L.A.; Lodato, S.; Bonev, B.; Groff, A.F.; Gerhardinger, C.; Sanchez-Gomez, D.B.; Hacisuleyman, E.; Li, E.; Spence, M.; et al. Multiple knockout mouse models reveal lincRNAs are required for life and brain development. eLife 2013, 2, e01749. [Google Scholar] [CrossRef]
- Wu, L.; Murat, P.; Matak-Vinkovic, D.; Murrell, A.; Balasubramanian, S. Binding interactions between long noncoding RNA HOTAIR and PRC2 proteins. Biochemistry 2013, 52, 9519–9527. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.J.; Ji, Y.X.; Zhang, P.; Deng, K.Q.; Gong, J.; Ren, S.; Wang, X.; Chen, I.; Wang, H.; et al. The long noncoding RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nat. Med. 2016, 22, 1131–1139. [Google Scholar] [CrossRef]
- Gage, P.J.; Suh, H.; Camper, S.A. Dosage requirement of Pitx2 for development of multiple organs. Development 1999, 126, 4643–4651. [Google Scholar]
- Mahlapuu, M.; Enerbäck, S.; Carlsson, P. Haploinsufficiency of the forkhead gene Foxf1, a target for sonic hedgehog signaling, causes lung and foregut malformations. Development 2001, 128, 2397–2406. [Google Scholar] [PubMed]
- Xu, C.; Zhang, Y.; Wang, Q.; Xu, Z.; Jiang, J.; Gao, Y.; Gao, M.; Kang, J.; Wu, M.; Xiong, J.; et al. Long non-coding RNA GAS5 controls human embryonic stem cell self-renewal by maintaining NODAL signalling. Nat. Commun. 2016, 7, 13287. [Google Scholar] [CrossRef]
- Tang, R.; Zhang, G.; Wang, Y.C.; Mei, X.; Chen, S.Y. The long non-coding RNA GAS5 regulates transforming growth factor beta (TGF-beta)-induced smooth muscle cell differentiation via RNA Smad-binding elements. J. Biol. Chem. 2017, 292, 14270–14278. [Google Scholar] [CrossRef] [PubMed]
- Kino, T.; Hurt, D.E.; Ichijo, T.; Nader, N.; Chrousos, G.P. Noncoding RNA Gas5 Is a Growth Arrest- and Starvation-Associated Repressor of the Glucocorticoid Receptor. Sci. Signal. 2010, 3, ra8. [Google Scholar] [CrossRef] [PubMed]
- Mondal, T.; Subhash, S.; Vaid, R.; Enroth, S.; Uday, S.; Reinius, B.; Mitra, S.; Mohammed, A.; James, A.R.; Hoberg, E.; et al. MEG3 long noncoding RNA regulates the TGF-beta pathway genes through formation of RNA-DNA triplex structures. Nat. Commun. 2015, 6, 7743. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kumari, P.; Gilligan, P.; Quach, H.N.; Mathavan, S.; Sampath, K. Dorsal activity of maternal squint is mediated by a non-coding function of the RNA. Development 2012, 139, 2903–2915. [Google Scholar] [CrossRef] [PubMed]
- Sampath, K.; Ephrussi, A. CncRNAs: RNAs with both coding and non-coding roles in development. Development 2016, 143, 1234–1241. [Google Scholar] [CrossRef]
- Goudarzi, M.; Berg, K.; Pieper, L.M.; Schier, A.F. Individual long non-coding RNAs have no overt functions in zebrafish embryogenesis, viability and fertility. eLife 2019, 8, e40815. [Google Scholar] [CrossRef]
- Nakamura, T.; Saito, D.; Kawasumi, A.; Shinohara, K.; Asai, Y.; Takaoka, K.; Dong, F.; Takamatsu, A.; Belo, J.A.; Mochizuki, A.; et al. Fluid flow and interlinked feedback loops establish left-right asymmetric decay of Cerl2 mRNA. Nat. Commun. 2012, 3, 1322. [Google Scholar] [CrossRef]
- Denissova, N.G.; Pouponnot, C.; Long, J.; He, D.; Liu, F. Transforming growth factor β-inducible independent binding of SMAD to the Smad7 promoter. Proc. Natl. Acad. Sci. USA 2000, 97, 6397–6402. [Google Scholar] [CrossRef]
- Kutter, C.; Watt, S.; Stefflova, K.; Wilson, M.D.; Goncalves, A.; Ponting, C.P.; Odom, D.T.; Marques, A.C. Rapid Turnover of Long Noncoding RNAs and the Evolution of Gene Expression. PLoS Genet. 2012, 8, e1002841. [Google Scholar] [CrossRef] [PubMed]
- Necsulea, A.; Soumillon, M.; Warnefors, M.; Liechti, A.; Daish, T.; Zeller, U.; Baker, J.C.; Grützner, F.; Kaessmann, H. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 2014, 505, 635. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.R.; Oakes, B.L.; Sternberg, S.H.; East-Seletsky, A.; Kaplan, M.; Doudna, J.A. Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature 2014, 516, 263. [Google Scholar] [CrossRef] [PubMed]
- Ploner, A.; Ploner, C.; Lukasser, M.; Niederegger, H.; Hüttenhofer, A. Methodological obstacles in knocking down small noncoding RNAs. RNA 2009, 15, 1797–1804. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, K.T.; Li, L.; Chu, Y.; Janowski, B.A.; Corey, D.R. RNAi factors are present and active in human cell nuclei. Cell Rep. 2014, 6, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Notani, D.; Ma, Q.; Tanasa, B.; Nunez, E.; Chen, A.Y.; Merkurjev, D.; Zhang, J.; Ohgi, K.; Song, X.; et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 2013, 498, 516. [Google Scholar] [CrossRef]
- Lam, M.T.Y.; Cho, H.; Lesch, H.P.; Gosselin, D.; Heinz, S.; Tanaka-Oishi, Y.; Benner, C.; Kaikkonen, M.U.; Kim, A.S.; Kosaka, M.; et al. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature 2013, 498, 511. [Google Scholar] [CrossRef]
- Robb, G.B.; Brown, K.M.; Khurana, J.; Rana, T.M. Specific and potent RNAi in the nucleus of human cells. Nat. Struct. Mol. Biol. 2005, 12, 133–137. [Google Scholar] [CrossRef]
- Wahlestedt, C.; Salmi, P.; Good, L.; Kela, J.; Johnsson, T.; Hokfelt, T.; Broberger, C.; Porreca, F.; Lai, J.; Ren, K.; et al. Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proc. Natl. Acad. Sci. USA 2000, 97, 5633–5638. [Google Scholar] [CrossRef]
- Roux, B.T.; Lindsay, M.A.; Heward, J.A. Knockdown of Nuclear-Located Enhancer RNAs and Long ncRNAs Using Locked Nucleic Acid GapmeRs. In Enhancer RNAs: Methods and Protocols; Ørom, U.A., Ed.; Springer: New York, NY, USA, 2017; pp. 11–18. [Google Scholar] [CrossRef]
- Ilott, N.E.; Heward, J.A.; Roux, B.; Tsitsiou, E.; Fenwick, P.S.; Lenzi, L.; Goodhead, I.; Hertz-Fowler, C.; Heger, A.; Hall, N.; et al. Long non-coding RNAs and enhancer RNAs regulate the lipopolysaccharide-induced inflammatory response in human monocytes. Nat. Commun. 2014, 5, 3979. [Google Scholar] [CrossRef]
- Lennox, K.A.; Behlke, M.A. Cellular localization of long non-coding RNAs affects silencing by RNAi more than by antisense oligonucleotides. Nucleic Acids Res. 2016, 44, 863–877. [Google Scholar] [CrossRef] [PubMed]
- Dykes, I.M. Exosomes in Cardiovascular Medicine. Cardiol. Ther. 2017, 6, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.S.; Tong, H.S.; Zhao, Y.; Hong, C.Y.; Bin, J.P.; Su, L. Differential Expression Pattern of Exosome Long Non-Coding RNAs (lncRNAs) and MicroRNAs (miRNAs) in Vascular Endothelial Cells Under Heat Stroke. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 7965–7974. [Google Scholar] [CrossRef] [PubMed]
- Kumarswamy, R.; Bauters, C.; Volkmann, I.; Maury, F.; Fetisch, J.; Holzmann, A.; Lemesle, G.; de Groote, P.; Pinet, F.; Thum, T. Circulating long noncoding RNA, LIPCAR, predicts survival in patients with heart failure. Circ. Res. 2014, 114, 1569–1575. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Gao, W.; Long, Q.Q.; Zhang, J.; Li, Y.F.; Liu, D.C.; Yan, J.J.; Yang, Z.J.; Wang, L.S. Increased plasma levels of lncRNA H19 and LIPCAR are associated with increased risk of coronary artery disease in a Chinese population. Sci. Rep. 2017, 7, 7491. [Google Scholar] [CrossRef] [PubMed]
- McKiernan, J.; Donovan, M.J.; O’Neill, V.; Bentink, S.; Noerholm, M.; Belzer, S.; Skog, J.; Kattan, M.W.; Partin, A.; Andriole, G.; et al. A Novel Urine Exosome Gene Expression Assay to Predict High-Grade Prostate Cancer at Initial BiopsyUrine Exosome Signature to Predict High-Grade Prostate CancerUrine Exosome Signature to Predict High-Grade Prostate Cancer. JAMA Oncol. 2016, 2, 882–889. [Google Scholar] [CrossRef]
- Gu, M.; Zheng, A.; Tu, W.; Zhao, J.; Li, L.; Li, M.; Han, S.; Hu, X.; Zhu, J.; Pan, Y.; et al. Circulating LncRNAs as Novel, Non-Invasive Biomarkers for Prenatal Detection of Fetal Congenital Heart Defects. Cell. Physiol. Biochem. 2016, 38, 1459–1471. [Google Scholar] [CrossRef]
- Song, G.; Shen, Y.; Zhu, J.; Liu, H.; Liu, M.; Shen, Y.Q.; Zhu, S.; Kong, X.; Yu, Z.; Qian, L. Integrated analysis of dysregulated lncRNA expression in fetal cardiac tissues with ventricular septal defect. PLoS ONE 2013, 8, e77492. [Google Scholar] [CrossRef]
- Wang, J.; Greene, S.B.; Martin, J.F. BMP signaling in congenital heart disease: New developments and future directions. Birth Defects Res. Part A Clin. Mol. Teratol. 2011, 91, 441–448. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, Z.; Wu, C.; Pan, Z.; Xiang, L.; Liu, H.; Jin, X.; Tong, K.; Fan, S.; Jin, X. Potential association of long noncoding RNA HA117 with tetralogy of Fallot. Genes Dis. 2018, 5, 185–190. [Google Scholar] [CrossRef]
- Jiang, Y.; Mo, H.; Luo, J.; Zhao, S.; Liang, S.; Zhang, M.; Yuan, J. HOTAIR Is a Potential Novel Biomarker in Patients with Congenital Heart Diseases. BioMed Res. Int. 2018, 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhu, W.; Zhang, B.; Wu, Y.; Yan, S.; Yuan, Y.; Zhang, H.; Li, J.; Sun, K.; Wang, H.; et al. The MALAT1 gene polymorphism and its relationship with the onset of congenital heart disease in Chinese. Biosci. Rep. 2018, 38, BSR20171381. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Long, H.; Zhou, C.; Zheng, S.; Wu, H.; Guo, T.; Wu, Q.; Zhong, T.; Wang, T. Long noncoding RNA Braveheart promotes cardiogenic differentiation of mesenchymal stem cells In Vitro. Stem Cell Res. Ther. 2017, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Viereck, J.; Kumarswamy, R.; Foinquinos, A.; Xiao, K.; Avramopoulos, P.; Kunz, M.; Dittrich, M.; Maetzig, T.; Zimmer, K.; Remke, J.; et al. Long noncoding RNA Chast promotes cardiac remodeling. Sci. Transl. Med. 2016, 8, 326ra22. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Hu, Y.; Li, X.; Jin, G.; Chen, X.; Chen, G.; Chen, Y.; Huang, S.; Liao, W.; Liao, Y.; et al. Sirt1 Antisense Long Noncoding RNA Promotes Cardiomyocyte Proliferation by Enhancing the Stability of Sirt1. J. Am. Heart Assoc. 2018, 7, e009700. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, X.; Shen, D.; Ge, D.; Chen, J.; Pei, J.; Li, Y.; Yue, Z.; Feng, J.; Chu, M.; et al. A long noncoding RNA NR_045363 controls cardiomyocyte proliferation and cardiac repair. J. Mol. Cell Cardiol. 2019, 127, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Ponnusamy, M.; Liu, F.; Zhang, Y.H.; Li, R.B.; Zhai, M.; Liu, F.; Zhou, L.Y.; Liu, C.Y.; Yan, K.W.; Dong, Y.H.; et al. The Long Non-Coding RNA CPR Regulates Cardiomyocyte Proliferation and Cardiac Repair. Circulation 2019. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Li, J.; Li, W.; Wang, Y.; Wu, F.; Xi, Y.; Zhang, L.; Ding, C.; Luo, H.; Li, Y.; et al. MicroRNAs activate gene transcription epigenetically as an enhancer trigger. RNA Biol. 2017, 14, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Fahed, A.C.; Gelb, B.D.; Seidman, J.G.; Seidman, C.E. Genetics of congenital heart disease: The glass half empty. Circ. Res. 2013, 112, 707–720. [Google Scholar] [CrossRef]
- Xu, T.P.; Huang, M.D.; Xia, R.; Liu, X.X.; Sun, M.; Yin, L.; Chen, W.M.; Han, L.; Zhang, E.B.; Kong, R.; et al. Decreased expression of the long non-coding RNA FENDRR is associated with poor prognosis in gastric cancer and FENDRR regulates gastric cancer cell metastasis by affecting fibronectin1 expression. J. Hematol. Oncol. 2014, 7, 63. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| lncRNA | Model System(s) Used | Expression Profile | Nuclear/Cytoplasmic | Loss of Function Methods | Loss of Function Phenotype | Target Genes | Protein Interactions | Evidence for lncRNA Expression in Humans | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Bvht | Mouse (ESC/EB) | ESC, Mesoderm, Cardiogenic mesoderm, Adult heart | Both | shRNA | Loss of beating CMs in EB culture | Mesp1 | PRC2 CNBP | RNA-seq from adult heart indicates lack of expression in human and rat, suggesting Bvht is mouse-specific [24] | [24,105] |
| Carmen | Mouse PC19CL6, Human foetal ventricle CPC | Cardiogenic mesoderm, Early heart tube, Adult heart, VSMC | Nuclear | shRNA | Bvht Eomes Oct4 Nanog | PRC2 | RNA-seq from adult heart indicates expression conserved in human and rat. Expressed in ventricle of week 12 foetus | [24,40] | |
| Linc1405 | Mouse (ESC/EB) | Cardiogenic mesoderm, Heart (E10.5, E16.5) | Nuclear | shRNA | Loss of beating CMs in EB culture | Mesp1 | TrxG EOMES | ND | [42] |
| Meteor | Mouse (ESC/EB) | ESCs | Nuclear | CRISPR | Lack of mesoderm specification, Gain of ectodermal specification | Eomes | ND | ChIP-seq data suggests transcription is conserved | [45] |
| Digit | Mouse/Human (ESC/EB) | Endoderm | Nuclear | shRNA LNA CRISPR | Lack of endoderm | Gsc | ND | Expressed in hESC derived endoderm | [46] |
| PANCR | Human (ESC/EB) | Cardiogenic mesoderm, Lung epithelium, Adult left atrium, Adult eye | Cytoplasmic | siRNA | Loss of Pitx2 expression | Pitx2 | PARP1 | Human-specific gene. No evidence of mouse homologue | [63,64] |
| PLAYRR | Mouse/chick (embryo) | Right-sided dorsal mesentery | Nuclear | CRISPR | Gain of Pitx2 expression | Pitx2 | CTCF | Conserved in human | [66] |
| FENDRR | Mouse (embryo) | Cardiogenic mesoderm, Lateral plate mesoderm (caudal, bilateral), Adult lung | Nuclear | Transgenic inducible shRNA | No phenotype | Pitx2 Foxf1 | PRC2 TrxG | Expression of human homologue confirmed in numerous studies | [67,68,112] |
| Null allele 1 | Embryonic lethal, Myocardial hypoplasia | ||||||||
| Null allele 2 | Perinatal lethality, VSD lung maturation and vascularization defect | ||||||||
| GAS5 | Human (ESC) | ESC Mesenchymal stem cells | Both | shRNA | Differentiation of ESCs, Differentiation of VSMCs | TGFβ/Nodal pathway | SMAD3 | Expressed in human ESCs | [73,74,75] |
| MEG3 | Human (BT-549 cells) | Breast cancer | Nuclear | siRNA | Gain of TGFβ expression | TGFβ/Nodal pathway | PRC2 | Expressed in human BT-549 | [76] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turton, N.; Swan, R.; Mahenthiralingam, T.; Pitts, D.; Dykes, I.M. The Functions of Long Non-Coding RNA during Embryonic Cardiovascular Development and Its Potential for Diagnosis and Treatment of Congenital Heart Disease. J. Cardiovasc. Dev. Dis. 2019, 6, 21. https://doi.org/10.3390/jcdd6020021
Turton N, Swan R, Mahenthiralingam T, Pitts D, Dykes IM. The Functions of Long Non-Coding RNA during Embryonic Cardiovascular Development and Its Potential for Diagnosis and Treatment of Congenital Heart Disease. Journal of Cardiovascular Development and Disease. 2019; 6(2):21. https://doi.org/10.3390/jcdd6020021
Chicago/Turabian StyleTurton, Nadia, Ross Swan, Thanujan Mahenthiralingam, Dominic Pitts, and Iain M. Dykes. 2019. "The Functions of Long Non-Coding RNA during Embryonic Cardiovascular Development and Its Potential for Diagnosis and Treatment of Congenital Heart Disease" Journal of Cardiovascular Development and Disease 6, no. 2: 21. https://doi.org/10.3390/jcdd6020021
APA StyleTurton, N., Swan, R., Mahenthiralingam, T., Pitts, D., & Dykes, I. M. (2019). The Functions of Long Non-Coding RNA during Embryonic Cardiovascular Development and Its Potential for Diagnosis and Treatment of Congenital Heart Disease. Journal of Cardiovascular Development and Disease, 6(2), 21. https://doi.org/10.3390/jcdd6020021