An Update on the Tissue Renin Angiotensin System and Its Role in Physiology and Pathology


Abstract
:1. Introduction
2. An Overview of RAS
3. The Concept of Tissue RAS
3.1. Angiotensin-I
3.2. Angiotensin (1–12)
3.3. Angiotensin-II
3.4. Angiotensin-(1–7)
3.5. Angiotensin-III/-IV
3.6. Angiotensin A/Alamandine/MrgD
3.7. Other Angiotensin Peptides
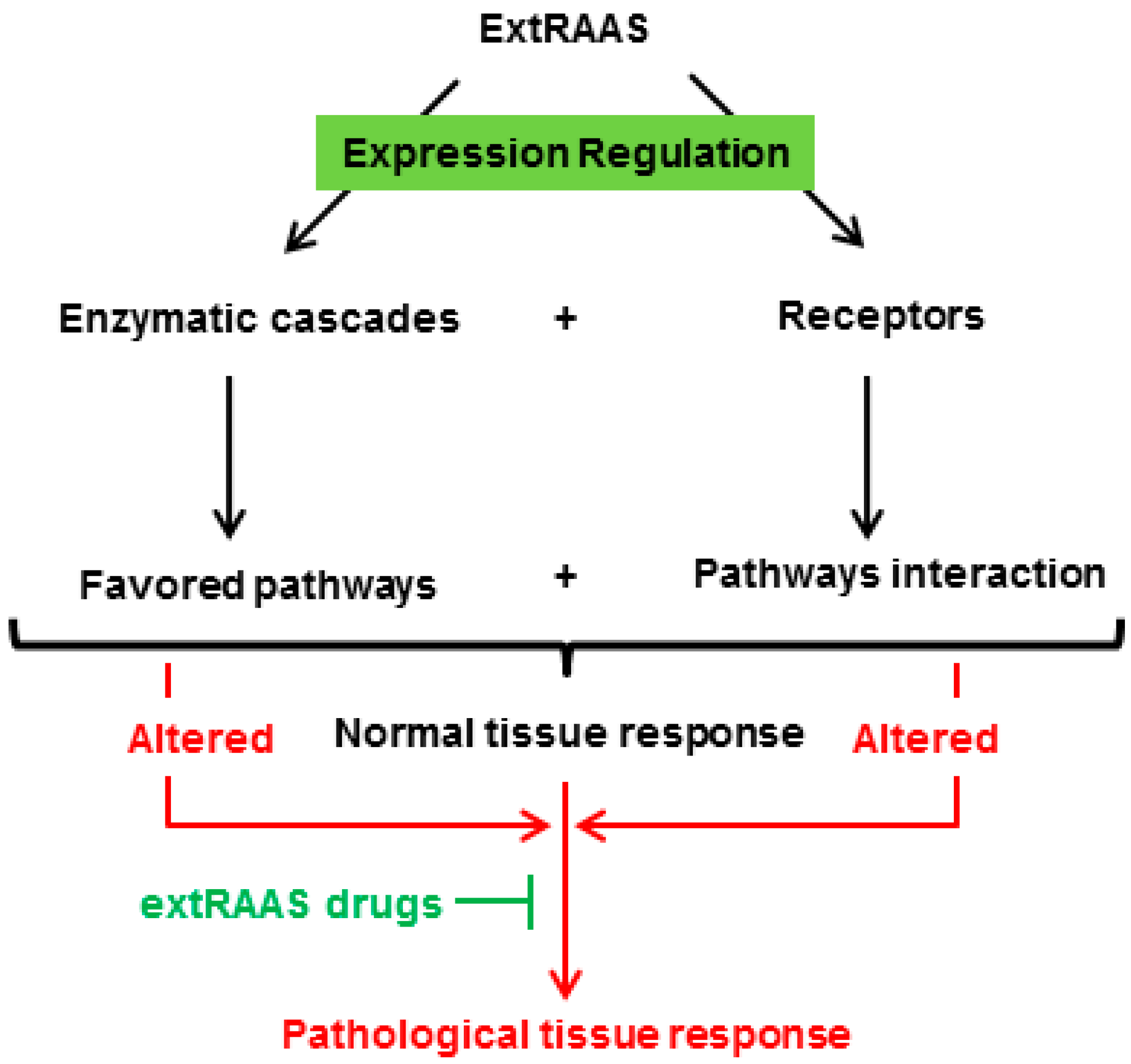
4. Conclusion and Future Directions
Funding
Conflicts of Interest
References
- Bader, M. Tissue renin-angiotensin-aldosterone systems: Targets for pharmacological therapy. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 439–465. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro-Oliveira, A.; Nogueira, A.I.; Pereira, R.M.; Boas, W.W.V.; dos Santos, R.A.S.; e Silva, A.C.S. The renin–angiotensin system and diabetes: An update. Vasc. Health Risk Manag. 2008, 4, 787–803. [Google Scholar] [PubMed]
- Nehme, A.; Zibara, K. Cellular distribution and interaction between extended renin-angiotensin-aldosterone system pathways in atheroma. Atherosclerosis 2017, 263, 334–342. [Google Scholar] [CrossRef]
- Nehme, A.; Zibara, K. Efficiency and specificity of RAAS inhibitors in cardiovascular diseases: How to achieve better end-organ protection? Hypertens. Res. 2017, 40, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Borghi, C.; SIIA Task Force; Rossi, F.; SIF Task Force. Role of the Renin-Angiotensin-Aldosterone System and Its Pharmacological Inhibitors in Cardiovascular Diseases: Complex and Critical Issues. High Blood Press Cardiovasc. Prev. 2015, 22, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Deschepper, C.F. Angiotensinogen: Hormonal regulation and relative importance in the generation of angiotensin II. Kidney Int. 1994, 46, 1561–1563. [Google Scholar] [CrossRef] [PubMed]
- Nehme, A.; Cerutti, C.; Dhaouadi, N.; Gustin, M.P.; Courand, P.-Y.; Zibara, K.; Bricca, G. Atlas of tissue renin-angiotensin-aldosterone system in human: A transcriptomic meta-analysis. Sci Rep 2015, 5, 10035. [Google Scholar] [CrossRef]
- Atlas, S.A. The renin-angiotensin aldosterone system: Pathophysiological role and pharmacologic inhibition. J. Manag. Care Pharm. 2007, 13, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Dzau, V.J.; Herrmann, H.C. Hormonal control of angiotensinogen production. Life Sci. 1982, 30, 577–584. [Google Scholar] [CrossRef]
- Hsueh, W.A. Potential effects of renin activation on the regulation of renin production. Am. J. Physiol. 1984, 247, F205–F212. [Google Scholar] [CrossRef]
- Kohlstedt, K.; Busse, R.; Fleming, I. Signaling via the angiotensin-converting enzyme enhances the expression of cyclooxygenase-2 in endothelial cells. Hypertension 2005, 45, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Gasparo, M.; de Catt, K.J.; Inagami, T.; Wright, J.W.; Unger, T. International Union of Pharmacology. XXIII. The Angiotensin II Receptors. Pharmacol. Rev. 2000, 52, 415–472. [Google Scholar]
- Kim, S.; Iwao, H. Molecular and Cellular Mechanisms of Angiotensin II-Mediated Cardiovascular and Renal Diseases. Pharmacol. Rev. 2000, 52, 11–34. [Google Scholar] [PubMed]
- Jaffe, I.Z.; Mendelsohn, M.E. Angiotensin II and aldosterone regulate gene transcription via functional mineralocortocoid receptors in human coronary artery smooth muscle cells. Circ. Res. 2005, 96, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, A.; Wang, J.; Pearce, D. Regulation of epithelial ion transport by aldosterone through changes in gene expression. Mol. Cell. Endocrinol. 2004, 217, 189–196. [Google Scholar] [CrossRef]
- Verhovez, A.; Williams, T.A.; Morello, F.; Monticone, S.; Brizzi, M.F.; Dentelli, P.; Fallo, F.; Fabris, B.; Amenta, F.; Gomez-Sanchez, C.; et al. Aldosterone does not modify gene expression in human endothelial cells. Horm. Metab. Res. 2012, 44, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.M.; Ahmad, S.; Nagata, S.; Simington, S.W.; Varagic, J.; Kon, N.; Dell’italia, L.J. An evolving story of angiotensin-II-forming pathways in rodents and humans. Clin. Sci. 2014, 126, 461–469. [Google Scholar] [CrossRef]
- Ganten, D.; Minnich, J.L.; Granger, P.; Hayduk, K.; Brecht, H.M.; Barbeau, A.; Boucher, R.; Genest, J. Angiotensin-forming enzyme in brain tissue. Science 1971, 173, 64–65. [Google Scholar] [CrossRef] [PubMed]
- Husain, A.; Bumpus, F.M.; Smeby, R.R.; Brosnihan, K.B.; Khosla, M.C.; Speth, R.C.; Ferrario, C.M. Evidence for the existence of a family of biologically active angiotensin I-like peptides in the dog central nervous system. Circ. Res. 1983, 52, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.; Delarue, F.; Burcklé, C.; Bouzhir, L.; Giller, T.; Sraer, J.-D. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J. Clin. Invest. 2002, 109, 1417–1427. [Google Scholar] [CrossRef]
- Paul, M.; Mehr, A.P.; Kreutz, R. Physiology of Local Renin-Angiotensin Systems. Physiol. Rev. 2006, 86, 747–803. [Google Scholar] [CrossRef] [PubMed]
- Lau, T.; Carlsson, P.-O.; Leung, P.S. Evidence for a local angiotensin-generating system and dose-dependent inhibition of glucose-stimulated insulin release by angiotensin II in isolated pancreatic islets. Diabetologia 2004, 47, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Dzau, V.J. Circulating versus local renin-angiotensin system in cardiovascular homeostasis. Circulation 1988, 77, I4–I13. [Google Scholar] [PubMed]
- Dzau, V.J. Tissue renin-angiotensin system in myocardial hypertrophy and failure. Arch. Intern. Med. 1993, 153, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Lu, H.; Cassis, L.A.; Daugherty, A. Molecular and Pathophysiological Features of Angiotensinogen: A Mini Review. N. Am. J. Med. Sci. (Boston) 2011, 4, 183–190. [Google Scholar] [CrossRef]
- Batenburg, W.W.; Danser, A.H.J. (Pro)renin and its receptors: Pathophysiological implications. Clin. Sci. 2012, 123, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Kaneshiro, Y.; Ichihara, A.; Sakoda, M.; Takemitsu, T.; Nabi, A.H.M.N.; Uddin, M.N.; Nakagawa, T.; Nishiyama, A.; Suzuki, F.; Inagami, T.; et al. Slowly progressive, angiotensin II-independent glomerulosclerosis in human (pro)renin receptor-transgenic rats. J. Am. Soc. Nephrol. 2007, 18, 1789–1795. [Google Scholar] [CrossRef] [PubMed]
- Lutterotti, N.; von Catanzaro, D.F.; Sealey, J.E.; Laragh, J.H. Renin is not synthesized by cardiac and extrarenal vascular tissues. A review of experimental evidence. Circulation 1994, 89, 458–470. [Google Scholar] [CrossRef]
- Boddi, M.; Poggesi, L.; Coppo, M.; Zarone, N.; Sacchi, S.; Tania, C.; Neri Serneri, G.G. Human vascular renin-angiotensin system and its functional changes in relation to different sodium intakes. Hypertension 1998, 31, 836–842. [Google Scholar] [CrossRef]
- Neri Serneri, G.G.; Boddi, M.; Coppo, M.; Chechi, T.; Zarone, N.; Moira, M.; Poggesi, L.; Margheri, M.; Simonetti, I. Evidence for the existence of a functional cardiac renin-angiotensin system in humans. Circulation 1996, 94, 1886–1893. [Google Scholar] [CrossRef]
- Cassis, L.A.; Police, S.B.; Yiannikouris, F.; Thatcher, S.E. Local adipose tissue renin-angiotensin system. Curr. Hypertens. Rep. 2008, 10, 93–98. [Google Scholar] [CrossRef]
- Santos, C.F.; Akashi, A.E.; Dionísio, T.J.; Sipert, C.R.; Didier, D.N.; Greene, A.S.; Oliveira, S.H.P.; Pereira, H.J.V.; Becari, C.; Oliveira, E.B.; et al. Characterization of a local renin-angiotensin system in rat gingival tissue. J. Periodontol. 2009, 80, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Saris, J.J.; Derkx, F.H.; De Bruin, R.J.; Dekkers, D.H.; Lamers, J.M.; Saxena, P.R.; Schalekamp, M.A.; Jan Danser, A.H. High-affinity prorenin binding to cardiac man-6-P/IGF-II receptors precedes proteolytic activation to renin. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1706–H1715. [Google Scholar] [CrossRef] [PubMed]
- Morris, B.J.; Reid, I.A. A “Renin-Like” Enzymatic Action of Cathepsin D and the Similarity in Subcellular Distributions of “Renin-Like” Activity and Cathepsin D in the Midbrain of Dogs. Endocrinology 1978, 103, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Rakoczy, P.E.; Sarks, S.H.; Daw, N.; Constable, I.J. Distribution of cathepsin D in human eyes with or without age-related maculopathy. Exp. Eye Res. 1999, 69, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Naseem, R.H.; Hedegard, W.; Henry, T.D.; Lessard, J.; Sutter, K.; Katz, S.A. Plasma cathepsin D isoforms and their active metabolites increase after myocardial infarction and contribute to plasma renin activity. Basic Res. Cardiol. 2005, 100, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Lavrentyev, E.N.; Estes, A.M.; Malik, K.U. Mechanism of high glucose induced angiotensin II production in rat vascular smooth muscle cells. Circ. Res. 2007, 101, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Belova, L.A. Angiotensin II-generating enzymes. Biochemistry Mosc. 2000, 65, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Hackenthal, E.; Hackenthal, R.; Hilgenfeldt, U. Isorenin, pseudorenin, cathepsin D and renin. A comparative enzymatic study of angiotensin-forming enzymes. Biochim. Biophys. Acta 1978, 522, 574–588. [Google Scholar] [CrossRef]
- Figueiredo, A.F.; Takii, Y.; Tsuji, H.; Kato, K.; Inagami, T. Rat kidney renin and cathepsin D: Purification and comparison of properties. Biochemistry 1983, 22, 5476–5481. [Google Scholar] [CrossRef]
- Nagata, S.; Kato, J.; Sasaki, K.; Minamino, N.; Eto, T.; Kitamura, K. Isolation and identification of proangiotensin-12, a possible component of the renin-angiotensin system. Biochem. Biophys. Res. Commun. 2006, 350, 1026–1031. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, Y.; Kida, N.; Nozaki, N.; Kuwasako, K.; Nagata, S.; Kitamura, K.; Kato, J. Effects of proangiotensin-12 infused continuously over 14 days in conscious rats. Eur. J. Pharmacol. 2012, 683, 186–189. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Von Cannon, J.; Jiao, Y.; Ahmad, S.; Bader, M.; Dell’Italia, L.J.; Groban, L.; Varagic, J. Cardiac angiotensin-(1–12) expression and systemic hypertension in rats expressing the human angiotensinogen gene. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H995–H1002. [Google Scholar] [CrossRef] [PubMed]
- Isa, K.; García-Espinosa, M.A.; Arnold, A.C.; Pirro, N.T.; Tommasi, E.N.; Ganten, D.; Chappell, M.C.; Ferrario, C.M.; Diz, D.I. Chronic immunoneutralization of brain angiotensin-(1–12) lowers blood pressure in transgenic (mRen2)27 hypertensive rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R111–R115. [Google Scholar] [CrossRef]
- Arnold, A.C.; Isa, K.; Shaltout, H.A.; Nautiyal, M.; Ferrario, C.M.; Chappell, M.C.; Diz, D.I. Angiotensin-(1–12) requires angiotensin converting enzyme and AT1 receptors for cardiovascular actions within the solitary tract nucleus. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H763–H771. [Google Scholar] [CrossRef]
- Chitravanshi, V.C.; Sapru, H.N. Cardiovascular responses elicited by a new endogenous angiotensin in the nucleus tractus solitarius of the rat. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H230–H240. [Google Scholar] [CrossRef] [PubMed]
- Chitravanshi, V.C.; Proddutur, A.; Sapru, H.N. Cardiovascular actions of angiotensin-(1–12) in the hypothalamic paraventricular nucleus of the rat are mediated via angiotensin II. Exp. Physiol. 2012, 97, 1001–1017. [Google Scholar] [CrossRef]
- Jessup, J.A.; Trask, A.J.; Chappell, M.C.; Nagata, S.; Kato, J.; Kitamura, K.; Ferrario, C.M. Localization of the novel angiotensin peptide, angiotensin-(1–12), in heart and kidney of hypertensive and normotensive rats. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2614–H2618. [Google Scholar] [CrossRef]
- Nagata, S.; Kato, J.; Kuwasako, K.; Kitamura, K. Plasma and tissue levels of proangiotensin-12 and components of the renin-angiotensin system (RAS) following low- or high-salt feeding in rats. Peptides 2010, 31, 889–892. [Google Scholar] [CrossRef] [PubMed]
- Westwood, B.M.; Chappell, M.C. Divergent pathways for the angiotensin-(1–12) metabolism in the rat circulation and kidney. Peptides 2012, 35, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Varagic, J.; Westwood, B.M.; Chappell, M.C.; Ferrario, C.M. Uptake and metabolism of the novel peptide angiotensin-(1–12) by neonatal cardiac myocytes. PLoS ONE 2011, 6, e15759. [Google Scholar] [CrossRef]
- Ahmad, S.; Simmons, T.; Varagic, J.; Moniwa, N.; Chappell, M.C.; Ferrario, C.M. Chymase-dependent generation of angiotensin II from angiotensin-(1–12) in human atrial tissue. PLoS ONE 2011, 6, e28501. [Google Scholar] [CrossRef]
- Prosser, H.C.; Richards, A.M.; Forster, M.E.; Pemberton, C.J. Regional vascular response to ProAngiotensin-12 (PA12) through the rat arterial system. Peptides 2010, 31, 1540–1545. [Google Scholar] [CrossRef] [PubMed]
- Tonnesen, M.G.; Klempner, M.S.; Austen, K.F.; Wintroub, B.U. Identification of a human neutrophil angiotension II-generating protease as cathepsin G. J. Clin. Invest. 1982, 69, 25–30. [Google Scholar] [CrossRef]
- Urata, H.; Kinoshita, A.; Misono, K.S.; Bumpus, F.M.; Husain, A. Identification of a highly specific chymase as the major angiotensin II-forming enzyme in the human heart. J. Biol. Chem. 1990, 265, 22348–22357. [Google Scholar] [PubMed]
- Park, S.; Bivona, B.J.; Kobori, H.; Seth, D.M.; Chappell, M.C.; Lazartigues, E.; Harrison-Bernard, L.M. Major role for ACE-independent intrarenal ANG II formation in type II diabetes. Am. J. Physiol. Renal Physiol. 2010, 298, F37–F48. [Google Scholar] [CrossRef] [PubMed]
- Nouet, S.; Nahmias, C. Signal transduction from the angiotensin II AT2 receptor. Trends Endocrinol. Metab. 2000, 11, 1–6. [Google Scholar] [CrossRef]
- Fei, D.T.; Coghlan, J.P.; Fernley, R.T.; Scoggins, B.A.; Tregear, G.W. Peripheral production of angiotensin II and III in sheep. Circ. Res. 1980, 46, I135–I137. [Google Scholar]
- Van Kats, J.P.; van Meegen, J.R.; Verdouw, P.D.; Duncker, D.J.; Schalekamp, M.A.; Danser, A.H. Subcellular localization of angiotensin II in kidney and adrenal. J. Hypertens. 2001, 19, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Van Kats, J.P.; Danser, A.H.; van Meegen, J.R.; Sassen, L.M.; Verdouw, P.D.; Schalekamp, M.A. Angiotensin production by the heart: A quantitative study in pigs with the use of radiolabeled angiotensin infusions. Circulation 1998, 98, 73–81. [Google Scholar] [CrossRef]
- Danser, A.H.; van Kats, J.P.; Verdouw, P.D.; Schalekamp, M.A. Evidence for the existence of a functional cardiac renin-angiotensin system in humans. Circulation 1997, 96, 3795–3796. [Google Scholar] [PubMed]
- Sealey, J.E. Evidence for cardiovascular effects of prorenin. J. Hum. Hypertens. 1995, 9, 381–384. [Google Scholar]
- Esther, C.R.; Marino, E.M.; Howard, T.E.; Machaud, A.; Corvol, P.; Capecchi, M.R.; Bernstein, K.E. The critical role of tissue angiotensin-converting enzyme as revealed by gene targeting in mice. J. Clin. Invest. 1997, 99, 2375–2385. [Google Scholar] [CrossRef]
- Arakawa, K.; Ikeda, M.; Fukuyama, J.; Sakai, T. A pressor formation by trypsin from renin-denatured human plasma protein. J. Clin. Endocrinol. Metab. 1976, 42, 599–602. [Google Scholar] [CrossRef]
- Arakawa, K.; Maruta, H. Ability of kallikrein to generate angiotensin II-like pressor substance and a proposed ‘kinin-tensin enzyme system’. Nature 1980, 288, 705–706. [Google Scholar] [CrossRef] [PubMed]
- Miura, S.; Ideishi, M.; Sakai, T.; Motoyama, M.; Kinoshita, A.; Sasaguri, M.; Tanaka, H.; Shindo, M.; Arakawa, K. Angiotensin II formation by an alternative pathway during exercise in humans. J. Hypertens. 1994, 12, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, K.; Urata, H. Hypothesis regarding the pathophysiological role of alternative pathways of angiotensin II formation in atherosclerosis. Hypertension 2000, 36, 638–641. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, X.-H.; Yuan, H. Angiotensin II type-2 receptor-specific effects on the cardiovascular system. Cardiovasc. Diagn. Ther. 2012, 2, 56–62. [Google Scholar] [PubMed]
- Carey, R.M.; Wang, Z.Q.; Siragy, H.M. Update: Role of the angiotensin type-2 (AT(2)) receptor in blood pressure regulation. Curr. Hypertens. Rep. 2000, 2, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.H.; Wang, Z.Q.; Siragy, H.M.; Guerrant, R.L.; Carey, R.M. Regulation of jejunal sodium and water absorption by angiotensin subtype receptors. Am. J. Physiol. 1998, 275, R515–R523. [Google Scholar] [CrossRef]
- Strauss, M.H.; Hall, A.S. Angiotensin Receptor Blockers May Increase Risk of Myocardial Infarction. Circulation 2006, 114, 838–854. [Google Scholar] [CrossRef]
- Reudelhuber, T.L. The continuing saga of the AT2 receptor: A case of the good, the bad, and the innocuous. Hypertension 2005, 46, 1261–1262. [Google Scholar] [CrossRef]
- Pahlavani, M.; Kalupahana, N.S.; Ramalingam, L.; Moustaid-Moussa, N. Regulation and Functions of the Renin-Angiotensin System in White and Brown Adipose Tissue. Compr Physiol 2017, 7, 1137–1150. [Google Scholar]
- Tonnaer, J.A.; Engels, G.M.; Wiegant, V.M.; Burbach, J.P.; De Jong, W.; De Wied, D. Proteolytic conversion of angiotensins in rat brain tissue. Eur. J. Biochem. 1983, 131, 415–421. [Google Scholar] [CrossRef]
- Santos, R.A.; Brosnihan, K.B.; Chappell, M.C.; Pesquero, J.; Chernicky, C.L.; Greene, L.J.; Ferrario, C.M. Converting enzyme activity and angiotensin metabolism in the dog brainstem. Hypertension 1988, 11, I153–I157. [Google Scholar] [CrossRef]
- Tom, B.; Dendorfer, A.; Danser, A.H.J. Bradykinin, angiotensin-(1–7), and ACE inhibitors: How do they interact? Int. J. Biochem. Cell Biol. 2003, 35, 792–801. [Google Scholar] [CrossRef]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A Human Homolog of Angiotensin-converting Enzyme CLONING AND FUNCTIONAL EXPRESSION AS A CAPTOPRIL-INSENSITIVE CARBOXYPEPTIDASE. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef]
- Rice, G.I.; Thomas, D.A.; Grant, P.J.; Turner, A.J.; Hooper, N.M. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem. J. 2004, 383, 45–51. [Google Scholar] [CrossRef]
- Jackman, H.L.; Massad, M.G.; Sekosan, M.; Tan, F.; Brovkovych, V.; Marcic, B.M.; Erdös, E.G. Angiotensin 1–9 and 1–7 release in human heart: Role of cathepsin A. Hypertension 2002, 39, 976–981. [Google Scholar] [CrossRef]
- Pereira, M.G.A.G.; Souza, L.L.; Becari, C.; Duarte, D.A.; Camacho, F.R.B.; Oliveira, J.A.C.; Gomes, M.D.; Oliveira, E.B.; Salgado, M.C.O.; Garcia-Cairasco, N.; et al. Angiotensin II-independent angiotensin-(1–7) formation in rat hippocampus: Involvement of thimet oligopeptidase. Hypertension 2013, 62, 879–885. [Google Scholar] [CrossRef]
- Douglas, G.C.; O’Bryan, M.K.; Hedger, M.P.; Lee, D.K.L.; Yarski, M.A.; Smith, A.I.; Lew, R.A. The novel angiotensin-converting enzyme (ACE) homolog, ACE2, is selectively expressed by adult Leydig cells of the testis. Endocrinology 2004, 145, 4703–4711. [Google Scholar] [CrossRef]
- Santos, R.A.S.; Simoes e Silva, A.C.; Maric, C.; Silva, D.M.R.; Machado, R.P.; de Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.B.; Lopes, M.T.; Bader, M.; et al. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263. [Google Scholar] [CrossRef] [PubMed]
- Simões e Silva, A.C.; Silveira, K.D.; Ferreira, A.J.; Teixeira, M.M. ACE2, angiotensin-(1–7) and Mas receptor axis in inflammation and fibrosis. Br. J. Pharmacol. 2013, 169, 477–492. [Google Scholar] [CrossRef] [PubMed]
- Kostenis, E.; Milligan, G.; Christopoulos, A.; Sanchez-Ferrer, C.F.; Heringer-Walther, S.; Sexton, P.M.; Gembardt, F.; Kellett, E.; Martini, L.; Vanderheyden, P.; et al. G-protein-coupled receptor Mas is a physiological antagonist of the angiotensin II type 1 receptor. Circulation 2005, 111, 1806–1813. [Google Scholar] [CrossRef] [PubMed]
- Villela, D.; Leonhardt, J.; Patel, N.; Joseph, J.; Kirsch, S.; Hallberg, A.; Unger, T.; Bader, M.; Santos, R.A.; Sumners, C.; et al. Angiotensin type 2 receptor (AT2R) and receptor Mas: A complex liaison. Clin. Sci. 2015, 128, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Karnik, S.S.; Khuraijam, D.; Tirupula, K.; Unal, H. Significance of Ang(1–7) coupling with MAS1 and other GPCRs to the Renin-Angiotensin System: IUPHAR Review “X”. Br. J. Pharmacol. 2017. [Google Scholar] [CrossRef]
- Chappell, M.C. Emerging evidence for a functional angiotensin-converting enzyme 2-angiotensin-(1–7)-MAS receptor axis: More than regulation of blood pressure? Hypertension 2007, 50, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Oudit, G.Y.; Kassiri, Z.; Patel, M.P.; Chappell, M.; Butany, J.; Backx, P.H.; Tsushima, R.G.; Scholey, J.W.; Khokha, R.; Penninger, J.M. Angiotensin II-mediated oxidative stress and inflammation mediate the age-dependent cardiomyopathy in ACE2 null mice. Cardiovasc. Res. 2007, 75, 29–39. [Google Scholar] [CrossRef]
- Liu, R.; Qi, H.; Wang, J.; Wang, Y.; Cui, L.; Wen, Y.; Yin, C. Angiotensin-converting enzyme (ACE and ACE2) imbalance correlates with the severity of cerulein-induced acute pancreatitis in mice. Exp. Physiol. 2014, 99, 651–663. [Google Scholar] [CrossRef]
- Wösten-van Asperen, R.M.; Lutter, R.; Specht, P.A.; Moll, G.N.; van Woensel, J.B.; van der Loos, C.M.; van Goor, H.; Kamilic, J.; Florquin, S.; Bos, A.P. Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1–7) or an angiotensin II receptor antagonist. J. Pathol. 2011, 225, 618–627. [Google Scholar] [CrossRef]
- Simões e Silva, A.C.; Miranda, A.S.; Rocha, N.P.; Teixeira, A.L. Renin angiotensin system in liver diseases: Friend or foe? World J. Gastroenterol. 2017, 23, 3396–3406. [Google Scholar] [CrossRef]
- Moreira de Macêdo, S.; Guimarães, T.A.; Feltenberger, J.D.; Sousa Santos, S.H. The role of renin-angiotensin system modulation on treatment and prevention of liver diseases. Peptides 2014, 62, 189–196. [Google Scholar] [CrossRef]
- Shim, K.Y.; Eom, Y.W.; Kim, M.Y.; Kang, S.H.; Baik, S.K. Role of the renin-angiotensin system in hepatic fibrosis and portal hypertension. Korean J. Intern. Med. 2018, 33, 453–461. [Google Scholar] [CrossRef]
- Mak, K.Y.; Chin, R.; Cunningham, S.C.; Habib, M.R.; Torresi, J.; Sharland, A.F.; Alexander, I.E.; Angus, P.W.; Herath, C.B. ACE2 Therapy Using Adeno-associated Viral Vector Inhibits Liver Fibrosis in Mice. Mol. Ther. 2015, 23, 1434–1443. [Google Scholar] [CrossRef]
- Osterreicher, C.H.; Taura, K.; De Minicis, S.; Seki, E.; Penz-Osterreicher, M.; Kodama, Y.; Kluwe, J.; Schuster, M.; Oudit, G.Y.; Penninger, J.M.; et al. Angiotensin-converting-enzyme 2 inhibits liver fibrosis in mice. Hepatology 2009, 50, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Saldanha da Silva, A.A.; Rodrigues Prestes, T.R.; Lauar, A.O.; Finotti, B.B.; Simoes, E.; Silva, A.C. Renin Angiotensin System and Cytokines in Chronic Kidney Disease: Clinical and Experimental Evidence. Protein Pept. Lett. 2017, 24, 799–808. [Google Scholar] [CrossRef]
- Caruso-Neves, C.; Lara, L.S.; Rangel, L.B.; Grossi, A.L.; Lopes, A.G. Angiotensin-(1–7) modulates the ouabain-insensitive Na+-ATPase activity from basolateral membrane of the proximal tubule. Biochim. Biophys. Acta 2000, 1467, 189–197. [Google Scholar] [CrossRef]
- Lara, L.S.; Bica, R.B.S.; Sena, S.L.F.; Correa, J.S.; Marques-Fernandes, M.F.; Lopes, A.G.; Caruso-Neves, C. Angiotensin-(1–7) reverts the stimulatory effect of angiotensin II on the proximal tubule Na(+)-ATPase activity via a A779-sensitive receptor. Regul. Pept. 2002, 103, 17–22. [Google Scholar] [CrossRef]
- Esteban, V.; Heringer-Walther, S.; Sterner-Kock, A.; de Bruin, R.; van den Engel, S.; Wang, Y.; Mezzano, S.; Egido, J.; Schultheiss, H.-P.; Ruiz-Ortega, M.; et al. Angiotensin-(1–7) and the g protein-coupled receptor MAS are key players in renal inflammation. PLoS ONE 2009, 4, e5406. [Google Scholar] [CrossRef]
- Jiang, T.; Gao, L.; Guo, J.; Lu, J.; Wang, Y.; Zhang, Y. Suppressing inflammation by inhibiting the NF-κB pathway contributes to the neuroprotective effect of angiotensin-(1–7) in rats with permanent cerebral ischaemia. Br. J. Pharmacol. 2012, 167, 1520–1532. [Google Scholar] [CrossRef]
- Chiu, A.T.; Ryan, J.W.; Stewart, J.M.; Dorer, F.E. Formation of angiotensin III by angiotensin-converting enzyme. Biochem. J. 1976, 155, 189–192. [Google Scholar] [CrossRef]
- Cesari, M.; Rossi, G.P.; Pessina, A.C. Biological properties of the angiotensin peptides other than angiotensin II: Implications for hypertension and cardiovascular diseases. J. Hypertens. 2002, 20, 793–799. [Google Scholar] [CrossRef]
- Padia, S.H.; Kemp, B.A.; Howell, N.L.; Siragy, H.M.; Fournie-Zaluski, M.-C.; Roques, B.P.; Carey, R.M. Intrarenal aminopeptidase N inhibition augments natriuretic responses to angiotensin III in angiotensin type 1 receptor-blocked rats. Hypertension 2007, 49, 625–630. [Google Scholar] [CrossRef]
- Kotlo, K.; Hughes, D.E.; Herrera, V.L.M.; Ruiz-Opazo, N.; Costa, R.H.; Robey, R.B.; Danziger, R.S. Functional polymorphism of the Anpep gene increases promoter activity in the Dahl salt-resistant rat. Hypertension 2007, 49, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Carrera, M.P.; Ramírez-Expósito, M.J.; Valenzuela, M.T.; Dueñas, B.; García, M.J.; Mayas, M.D.; Martínez-Martos, J.M. Renin-angiotensin system-regulating aminopeptidase activities are modified in the pineal gland of rats with breast cancer induced by N-methyl-nitrosourea. Cancer Invest. 2006, 24, 149–153. [Google Scholar] [CrossRef]
- Chai, S.Y.; Fernando, R.; Peck, G.; Ye, S.-Y.; Mendelsohn, F.A.O.; Jenkins, T.A.; Albiston, A.L. The angiotensin IV/AT4 receptor. Cell. Mol. Life Sci. 2004, 61, 2728–2737. [Google Scholar] [CrossRef] [PubMed]
- Appenrodt, E.; Brattström, A. Effects of central angiotensin II and angiotensin III on baroreflex regulation. Neuropeptides 1994, 26, 175–180. [Google Scholar] [CrossRef]
- Handa, R.K. Biphasic actions of angiotensin IV on renal blood flow in the rat. Regul. Pept. 2006, 136, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Albiston, A.L.; McDowall, S.G.; Matsacos, D.; Sim, P.; Clune, E.; Mustafa, T.; Lee, J.; Mendelsohn, F.A.; Simpson, R.J.; Connolly, L.M.; et al. Evidence that the angiotensin IV (AT(4)) receptor is the enzyme insulin-regulated aminopeptidase. J. Biol. Chem. 2001, 276, 48623–48626. [Google Scholar] [CrossRef] [PubMed]
- Lochard, N.; Thibault, G.; Silversides, D.W.; Touyz, R.M.; Reudelhuber, T.L. Chronic production of angiotensin IV in the brain leads to hypertension that is reversible with an angiotensin II AT1 receptor antagonist. Circ. Res. 2004, 94, 1451–1457. [Google Scholar] [CrossRef]
- Li, X.C.; Campbell, D.J.; Ohishi, M.; Yuan, S.; Zhuo, J.L. AT1 receptor-activated signaling mediates angiotensin IV-induced renal cortical vasoconstriction in rats. Am. J. Physiol. Renal Physiol. 2006, 290, F1024–F1033. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, V.; Vanholder, R.; van der Giet, M.; Tölle, M.; Karadogan, S.; Gobom, J.; Furkert, J.; Oksche, A.; Krause, E.; Tran, T.N.A.; et al. Mass-spectrometric identification of a novel angiotensin peptide in human plasma. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 297–302. [Google Scholar] [CrossRef]
- Yang, R.; Smolders, I.; Vanderheyden, P.; Demaegdt, H.; Van Eeckhaut, A.; Vauquelin, G.; Lukaszuk, A.; Tourwé, D.; Chai, S.Y.; Albiston, A.L.; et al. Pressor and renal hemodynamic effects of the novel angiotensin A peptide are angiotensin II type 1A receptor dependent. Hypertension 2011, 57, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, D.C.O.; Foureaux, G.; Rodrigues, K.D.L.; Salles, R.L.A.; Moraes, P.L.; Murça, T.M.; De Maria, M.L.A.; Gomes, E.R.M.; Santos, R.A.S.; Guatimosim, S.; et al. Cardiovascular effects of angiotensin A: A novel peptide of the renin-angiotensin system. J. Renin. Angiotensin Aldosterone Syst. 2014, 15, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Lautner, R.Q.; Villela, D.C.; Fraga-Silva, R.A.; Silva, N.; Verano-Braga, T.; Costa-Fraga, F.; Jankowski, J.; Jankowski, V.; Sousa, F.; Alzamora, A.; et al. Discovery and characterization of alamandine: A novel component of the renin-angiotensin system. Circ. Res. 2013, 112, 1104–1111. [Google Scholar] [CrossRef]
- Tetzner, A.; Gebolys, K.; Meinert, C.; Klein, S.; Uhlich, A.; Trebicka, J.; Villacañas, Ó.; Walther, T. G-Protein-Coupled Receptor MrgD Is a Receptor for Angiotensin-(1–7) Involving Adenylyl Cyclase, cAMP, and Phosphokinase, A. Hypertension 2016, 68, 185–194. [Google Scholar] [CrossRef]
- Habiyakare, B.; Alsaadon, H.; Mathai, M.L.; Hayes, A.; Zulli, A. Reduction of angiotensin A and alamandine vasoactivity in the rabbit model of atherogenesis: Differential effects of alamandine and Ang(1–7). Int. J. Exp. Pathol. 2014, 95, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Mogielnicki, A.; Kramkowski, K.; Hermanowicz, J.M.; Leszczynska, A.; Przyborowski, K.; Buczko, W. Angiotensin-(1–9) enhances stasis-induced venous thrombosis in the rat because of the impairment of fibrinolysis. J Renin. Angiotensin Aldosterone Syst. 2014, 15, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Karwowska-Polecka, W.; Kułakowska, A.; Wiśniewski, K.; Braszko, J.J. Losartan influences behavioural effects of angiotensin II(3–7) in rats. Pharmacol. Res. 1997, 36, 275–283. [Google Scholar] [CrossRef]
- Handa, R.K. Metabolism alters the selectivity of angiotensin-(1–7) receptor ligands for angiotensin receptors. J. Am. Soc. Nephrol. 2000, 11, 1377–1386. [Google Scholar] [PubMed]
- Nehme, A.; Marcelo, P.; Nasser, R.; Kobeissy, F.; Bricca, G.; Zibara, K. The kinetics of angiotensin-I metabolism in human carotid atheroma: An emerging role for angiotensin (1–7). Vascul. Pharmacol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Nehme, A.; Cerutti, C.; Zibara, K. Transcriptomic analysis reveals novel transcription factors associated with renin-angiotensin-aldosterone system in human atheroma. Hypertension 2016, 68, 1375–1384. [Google Scholar] [CrossRef]
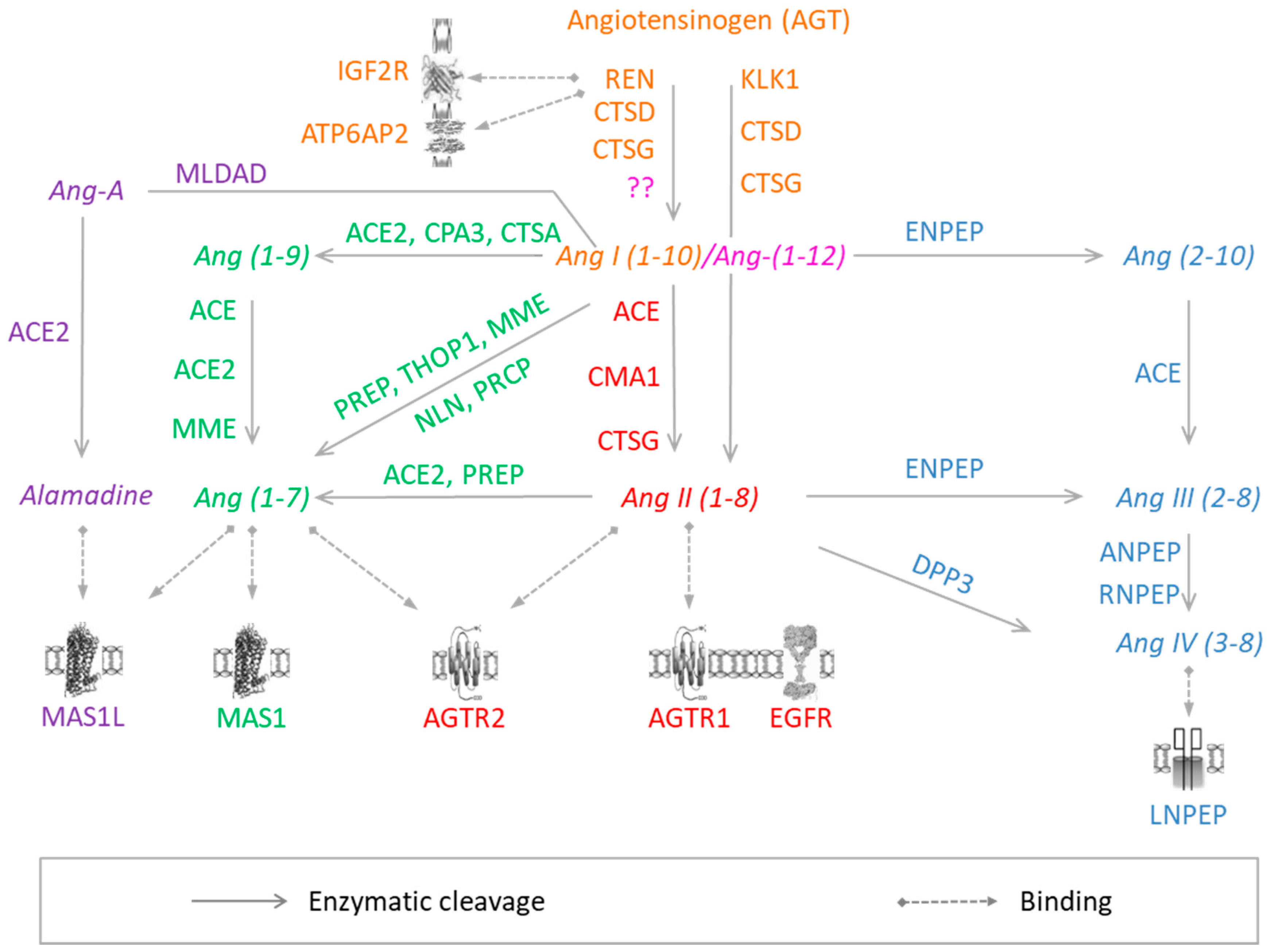

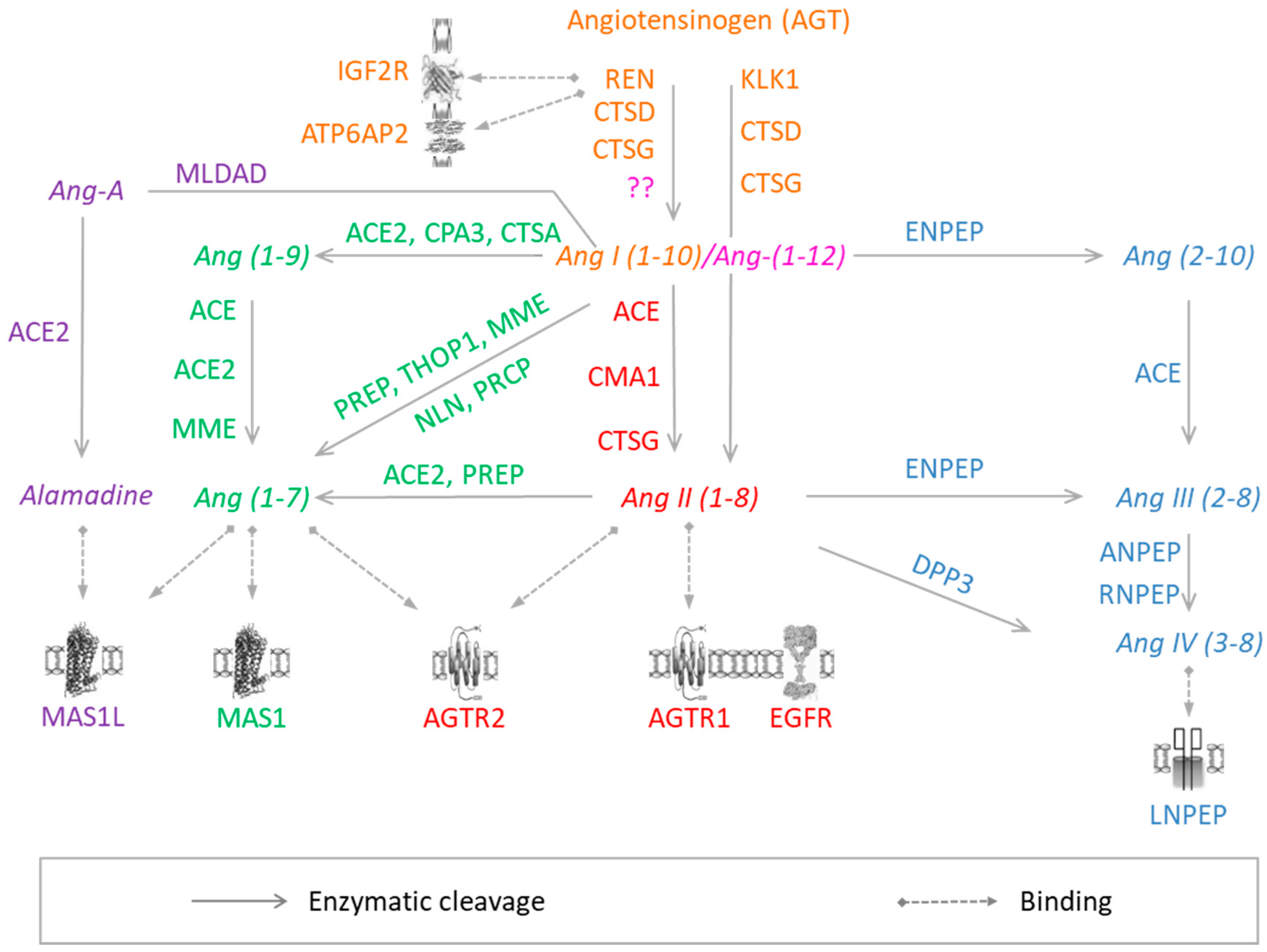{kind=link}
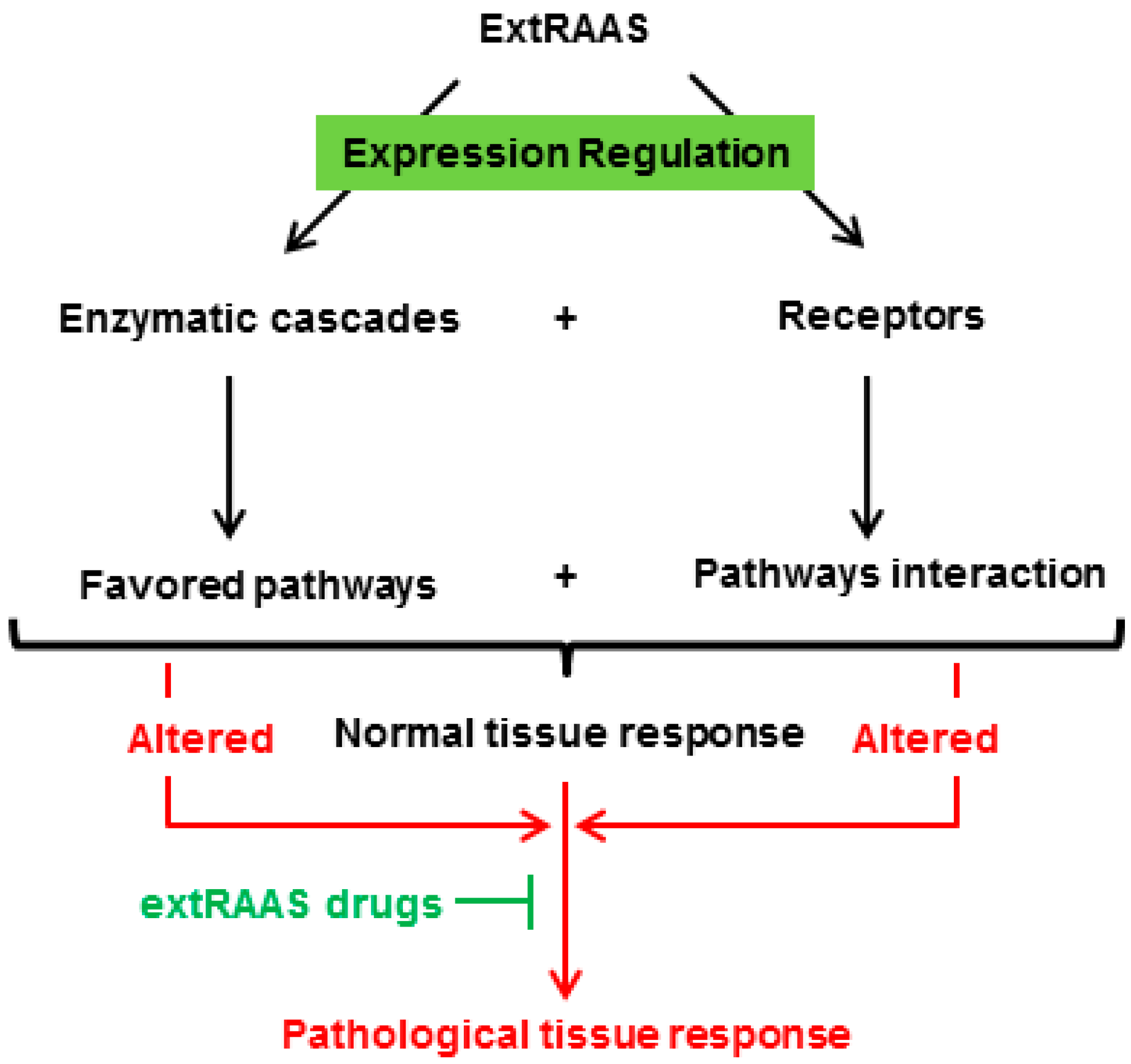{kind=link}
| Gene Symbol | Gene Description | Gene ID |
|---|---|---|
| ACE * | angiotensin I converting enzyme (peptidyl-dipeptidase A) 1 | 1636 |
| ACE2 | angiotensin I converting enzyme (peptidyl-dipeptidase A) 2 | 59272 |
| AGT * | angiotensinogen (serpin peptidase inhibitor, clade A, member 8) | 183 |
| AGTR1 * | angiotensin II receptor, type 1 | 185 |
| AGTR2 | angiotensin II receptor, type 2 | 186 |
| ANPEP | alanyl (membrane) aminopeptidase | 290 |
| ATP6AP2 | ATPase, H+ transporting, lysosomal accessory protein 2 | 10159 |
| CMA1 | chymase 1, mast cell | 1215 |
| CPA3 | carboxypeptidase A3 (mast cell) | 1359 |
| CTSA | cathepsin A | 5476 |
| CTSD | cathepsin D | 1509 |
| CTSG | cathepsin G | 1511 |
| DPP3 | dipeptidyl-peptidase 3 | 10072 |
| EGFR | epidermal growth factor receptor | 1956 |
| ENPEP | glutamyl aminopeptidase (aminopeptidase A) | 2028 |
| IGF2R | insulin-like growth factor 2 receptor | 3482 |
| KLK1 | kallikrein 1 | 3816 |
| LNPEP | leucyl/cystinyl aminopeptidase | 4012 |
| MAS1 | MAS1 oncogene | 4142 |
| MME | membrane metallo-endopeptidase | 4311 |
| NLN | neurolysin (metallopeptidase M3 family) | 57486 |
| PREP | prolyl endopeptidase | 5550 |
| REN * | renin | 5972 |
| RNPEP | arginyl aminopeptidase (aminopeptidase B) | 6051 |
| THOP1 | thimet oligopeptidase 1 | 7064 |
| AKR1C4 | aldo-keto reductase family 1, member C4 | 1109 |
| AKR1D1 | aldo-keto reductase family 1, member D1 | 6718 |
| CYP11A1 | cytochrome P450, family 11, subfamily A, polypeptide 1 | 1583 |
| CYP11B1 | cytochrome P450, family 11, subfamily B, polypeptide 1 | 1584 |
| CYP11B2 * | cytochrome P450, family 11, subfamily B, polypeptide 2 | 1585 |
| CYP17A1 | cytochrome P450, family 17, subfamily A, polypeptide 1 | 1586 |
| CYP21A2 | cytochrome P450, family 21, subfamily A, polypeptide 2 | 1589 |
| GPER | G protein-coupled estrogen receptor 1 | 2852 |
| HSD11B1 | hydroxysteroid (11-beta) dehydrogenase 1 | 3290 |
| HSD11B2 * | hydroxysteroid (11-beta) dehydrogenase 2 | 3291 |
| NR3C1 | nuclear receptor subfamily 3, group C, member 1 (glucocorticoid receptor) | 2908 |
| NR3C2 * | nuclear receptor subfamily 3, group C, member 2 (Mineralocorticoid receptor) | 4306 |
| Tissue | Physiological Role of RAS | Associated Diseases |
|---|---|---|
| Blood vessel | Vasomotor regulation, oxidative metabolism | Hypertension, atherosclerosis |
| Heart | Vasomotor tone, fibrotic regulation, oxidative metabolism | Heart failure, cardiac hypertrophy and fibrosis |
| Kidney | Blood pressure regulation | Chronic kidney disease |
| CNS | Sympathetic regulation of blood pressure | Hypertension |
| Adipose tissue | Adipogenesis | Insulin resistance and obesity |
| Eye | Aqueous humor dynamics | Glaucoma and diabetic retinopathy |
| Liver | Glucose metabolism | Glucose intolerance and fibrosis |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nehme, A.; Zouein, F.A.; Deris Zayeri, Z.; Zibara, K. An Update on the Tissue Renin Angiotensin System and Its Role in Physiology and Pathology. J. Cardiovasc. Dev. Dis. 2019, 6, 14. https://doi.org/10.3390/jcdd6020014
Nehme A, Zouein FA, Deris Zayeri Z, Zibara K. An Update on the Tissue Renin Angiotensin System and Its Role in Physiology and Pathology. Journal of Cardiovascular Development and Disease. 2019; 6(2):14. https://doi.org/10.3390/jcdd6020014
Chicago/Turabian StyleNehme, Ali, Fouad A. Zouein, Zeinab Deris Zayeri, and Kazem Zibara. 2019. "An Update on the Tissue Renin Angiotensin System and Its Role in Physiology and Pathology" Journal of Cardiovascular Development and Disease 6, no. 2: 14. https://doi.org/10.3390/jcdd6020014
APA StyleNehme, A., Zouein, F. A., Deris Zayeri, Z., & Zibara, K. (2019). An Update on the Tissue Renin Angiotensin System and Its Role in Physiology and Pathology. Journal of Cardiovascular Development and Disease, 6(2), 14. https://doi.org/10.3390/jcdd6020014