The Potential of a Novel Class of EPAC-Selective Agonists to Combat Cardiovascular Inflammation

 ,
,  ,
, 
Abstract
:1. Introduction
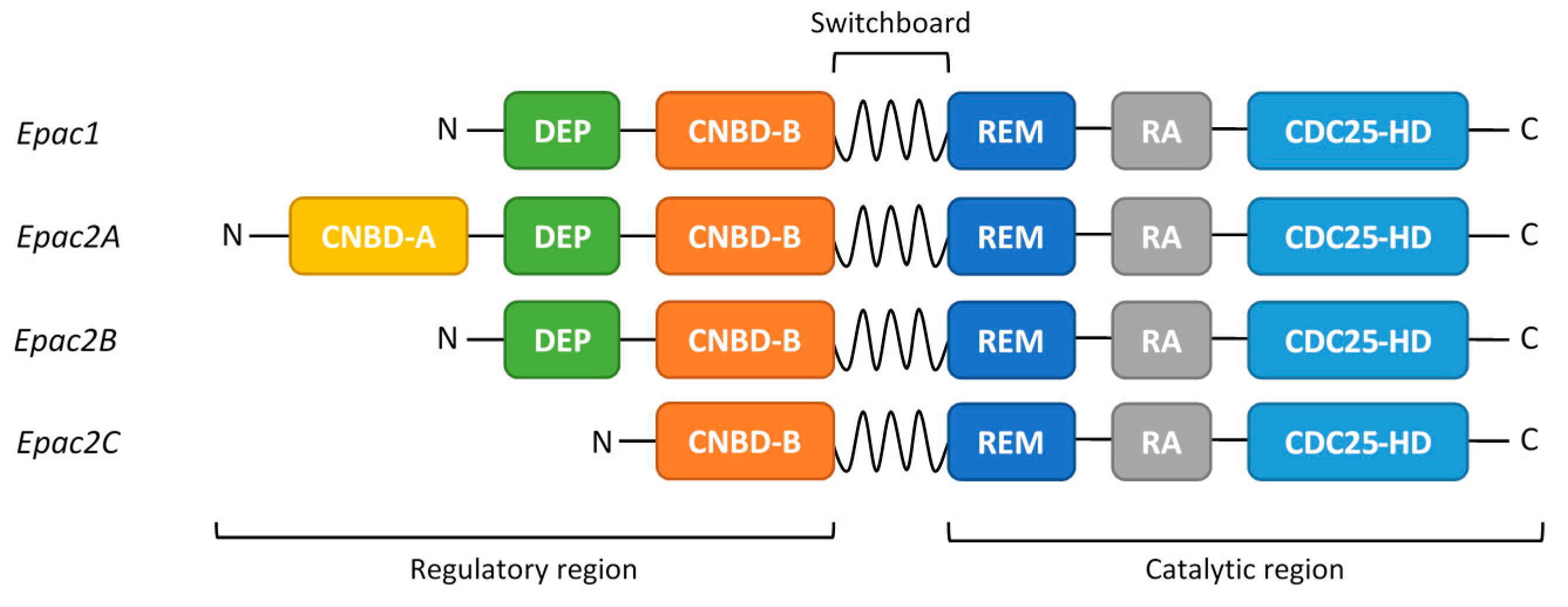
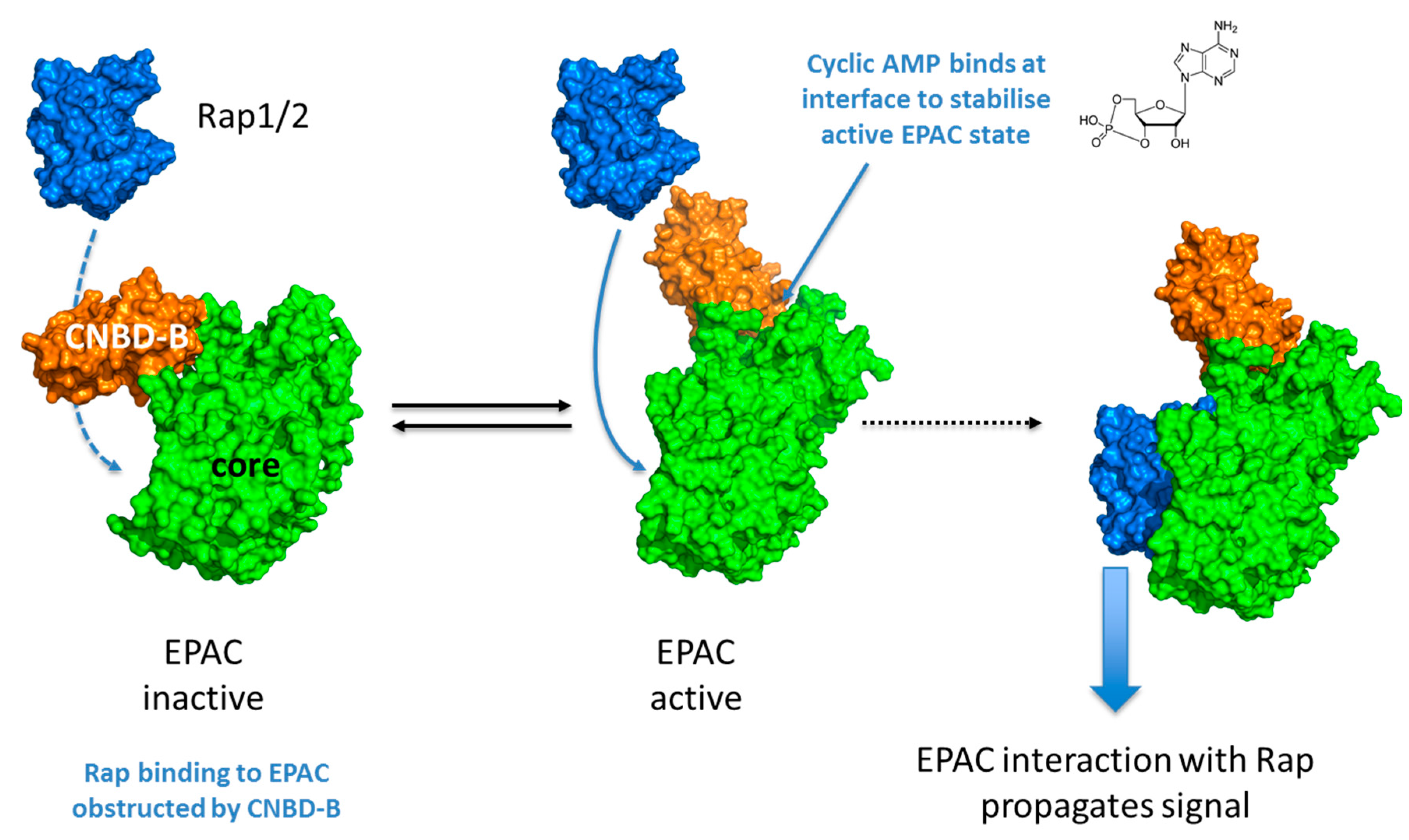
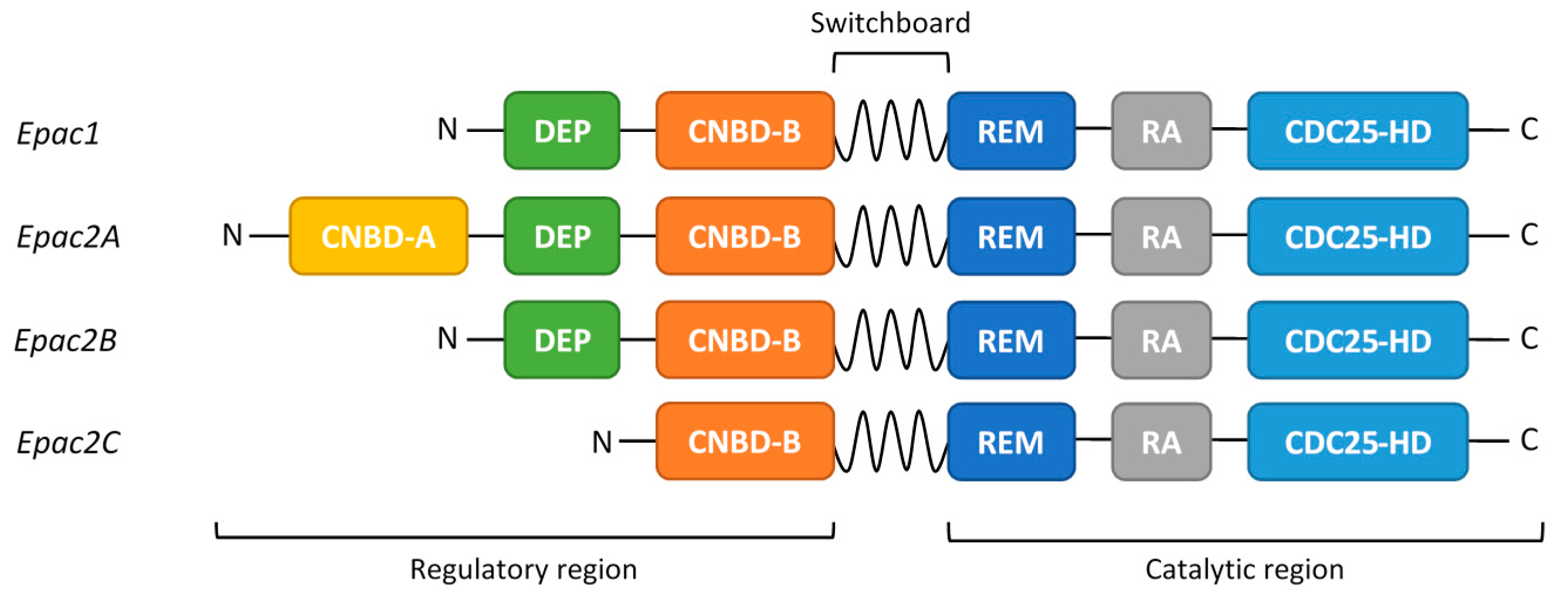
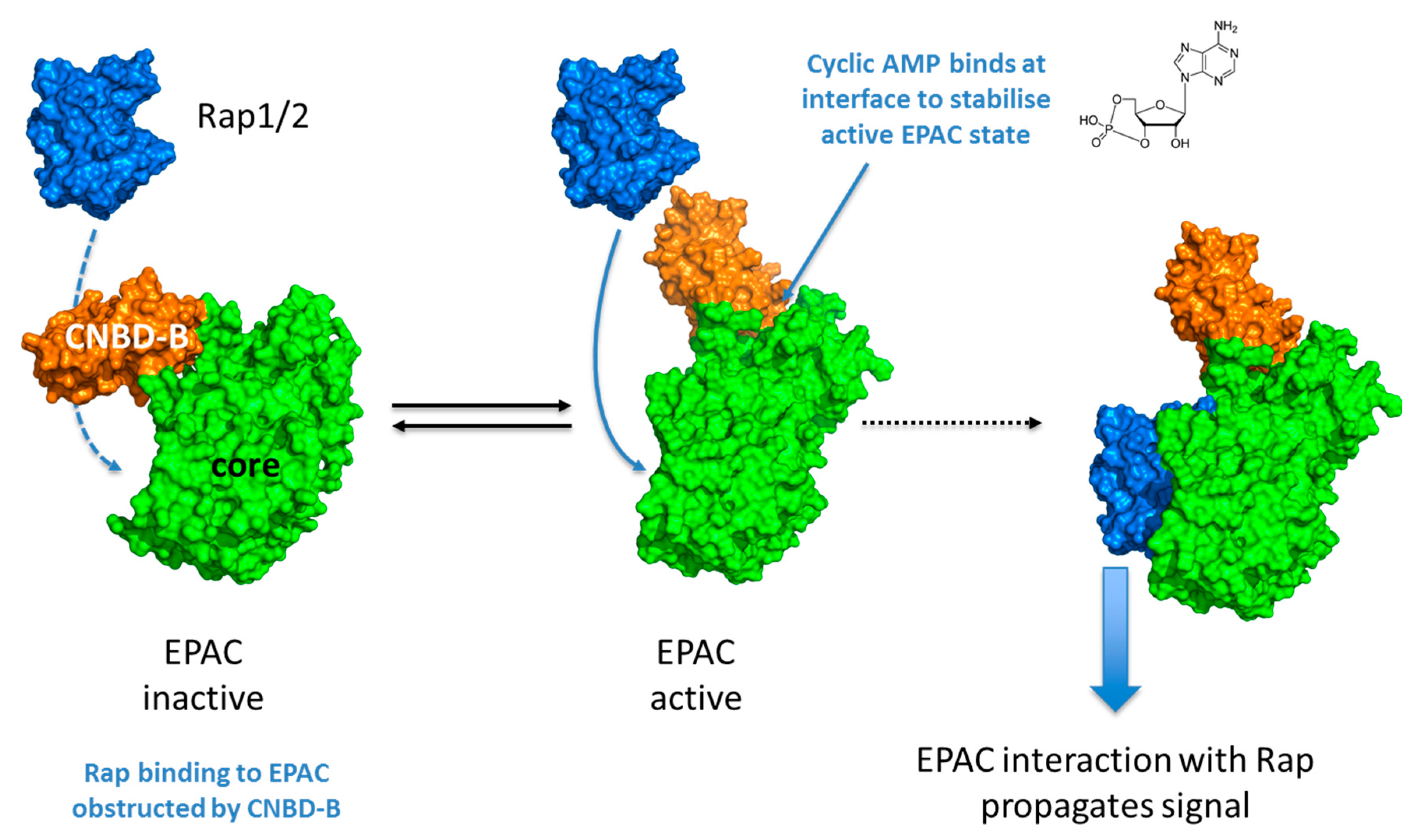
2. EPAC Proteins
3. EPAC1 Signalling and Vascular Function
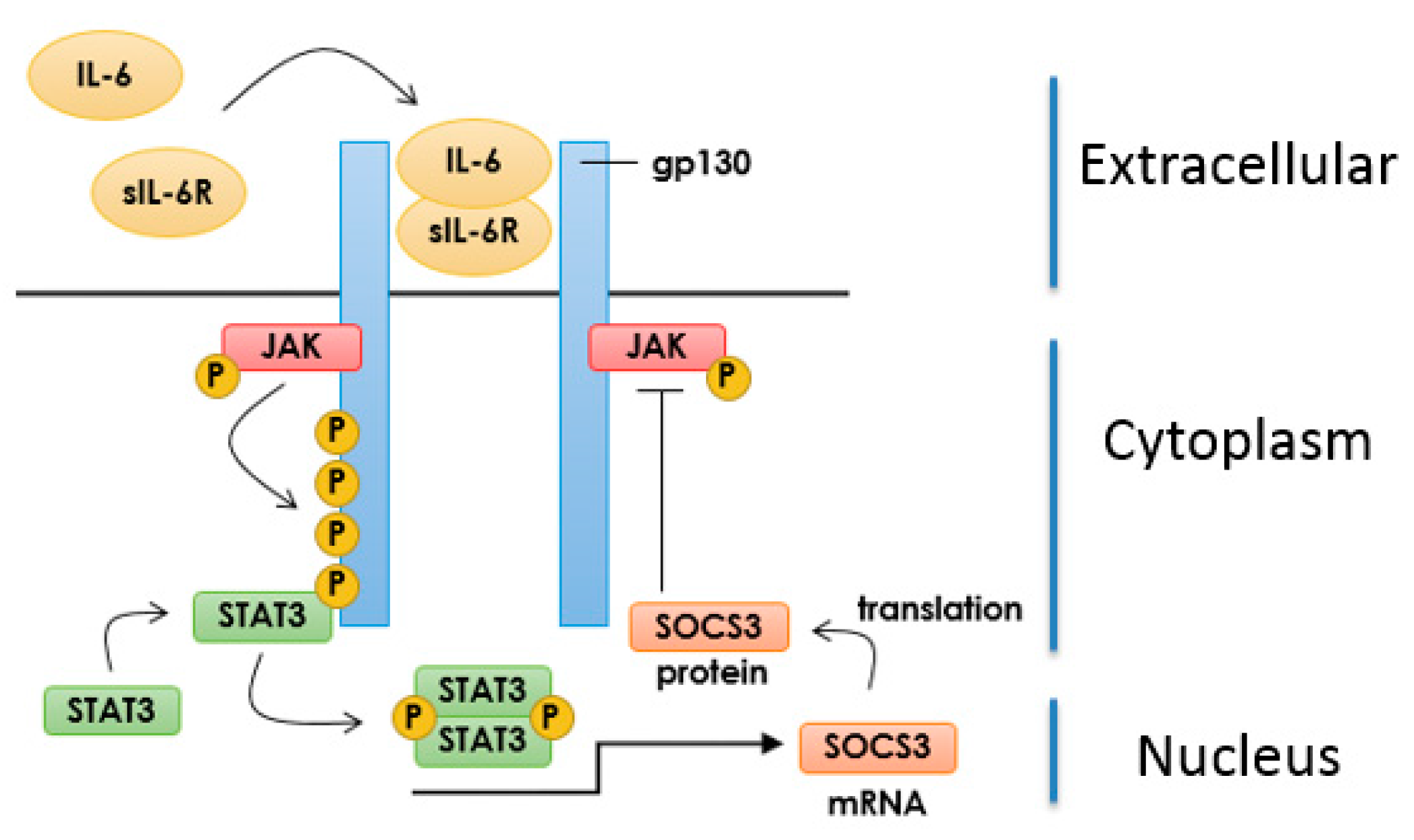
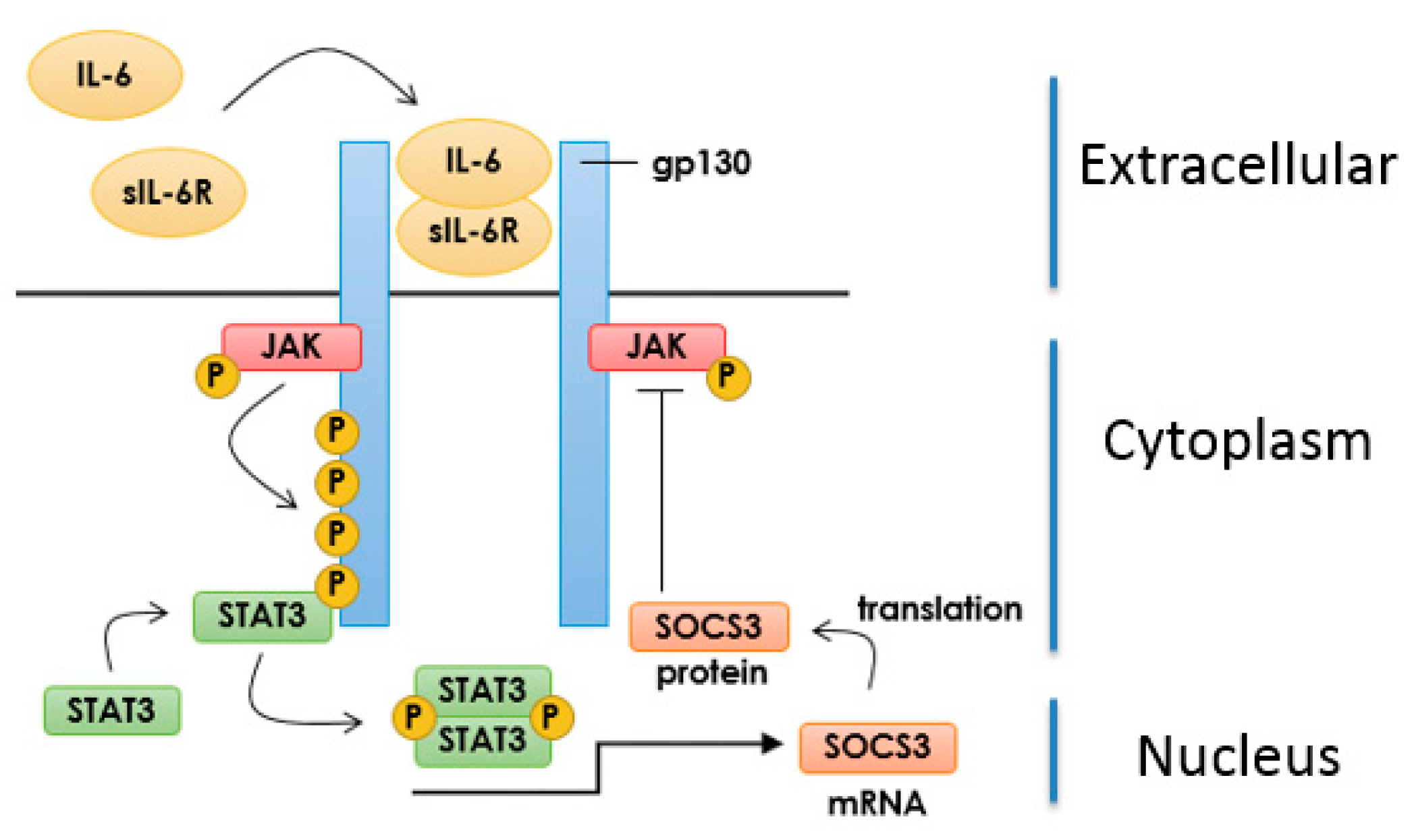
3.1. IL-6 Signalling in Vascular Endothelial Cells
3.2. Inhibition of IL-6 Signalling by SOCS3
3.3. Induction of SOCS3 by EPAC1 and Inhibition of IL-6 Signalling in VECs
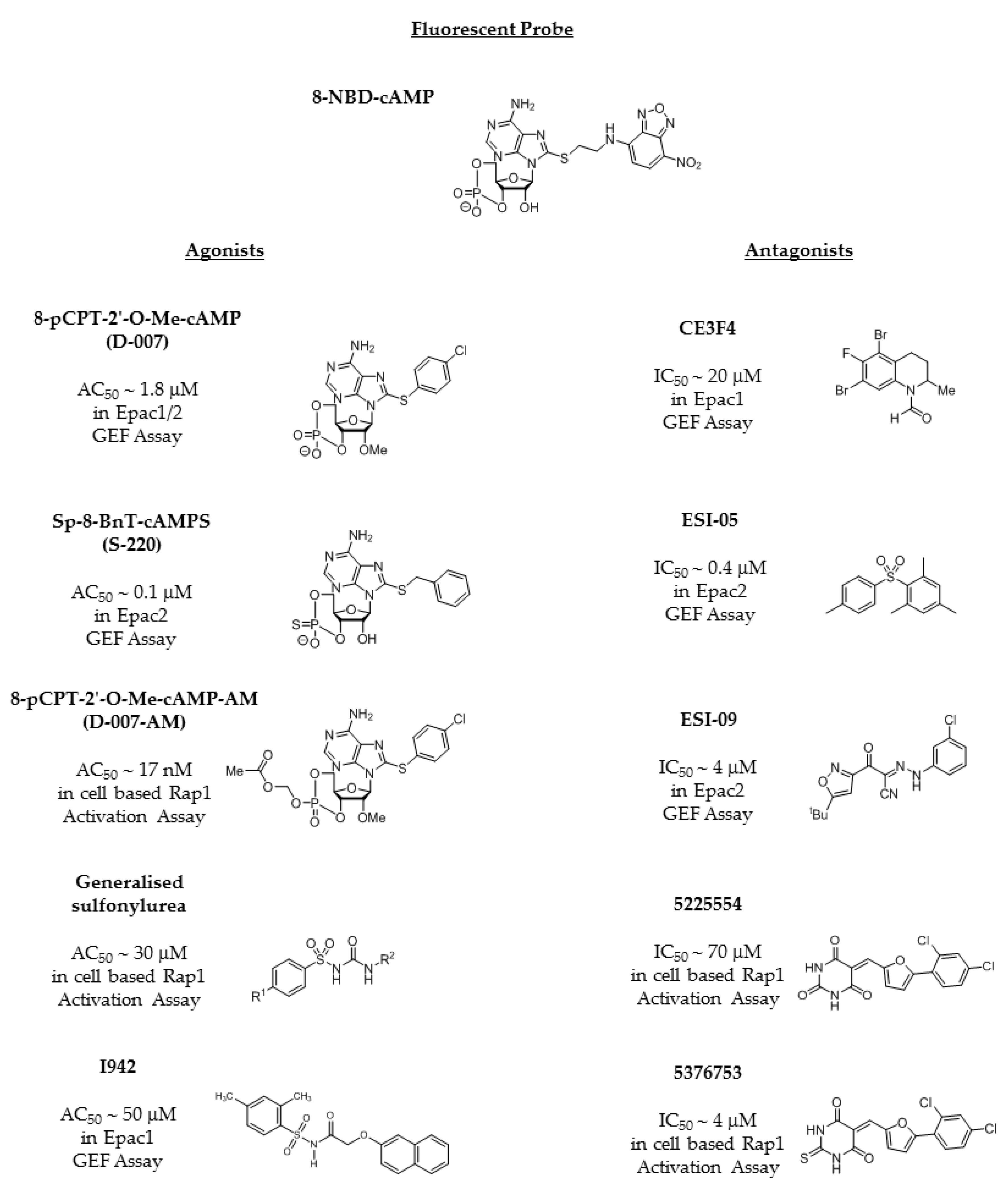
4. Development of EPAC-Selective Agonists
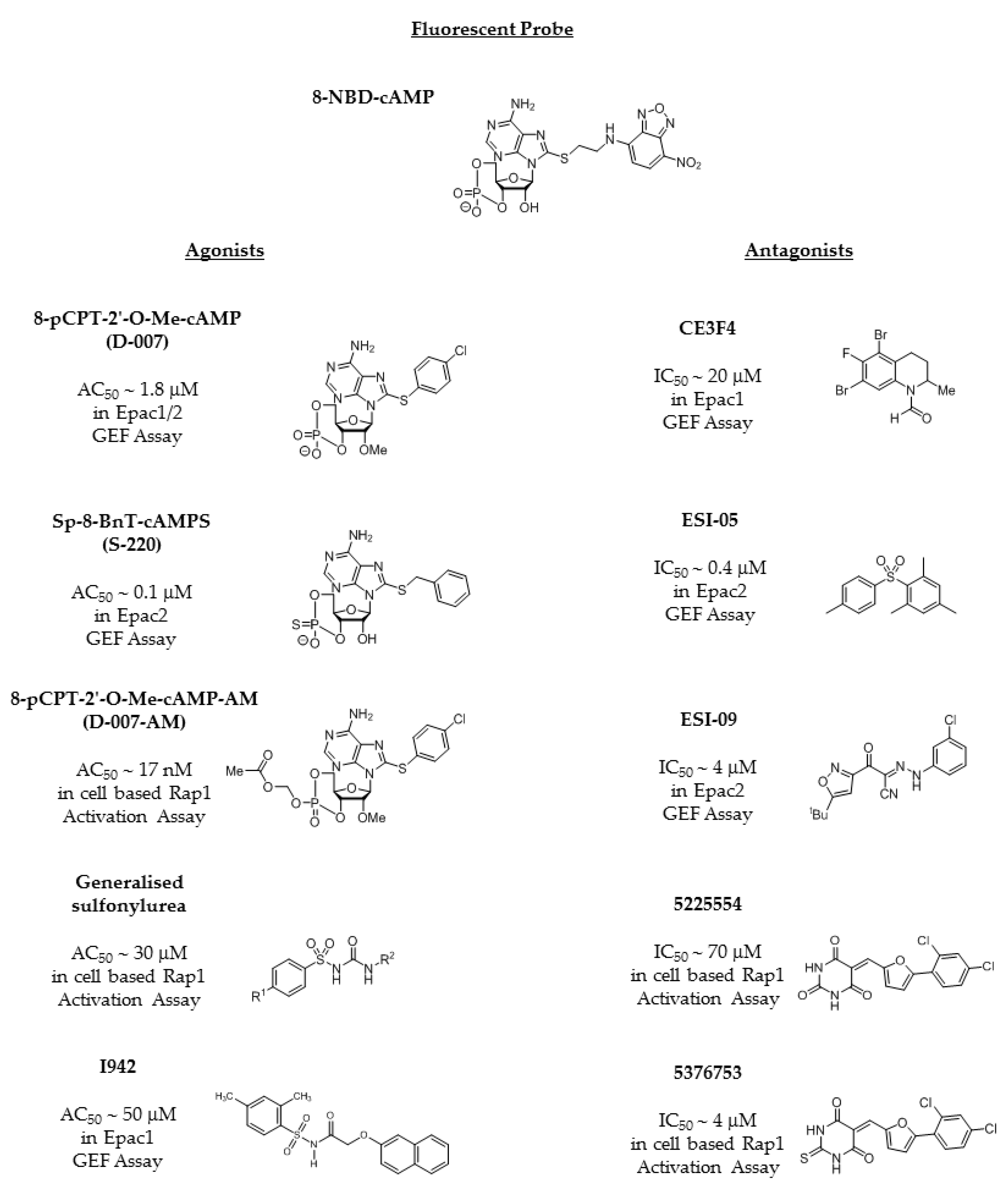
4.1. EPAC Agonists Based on Cyclic Nucleotides
4.2. The Sulfonyl Urea Family as EPAC Agonists
4.3. EPAC Antagonists
4.4. Identification of Non-Cyclic Nucleotide (NCN) EPAC Agonists
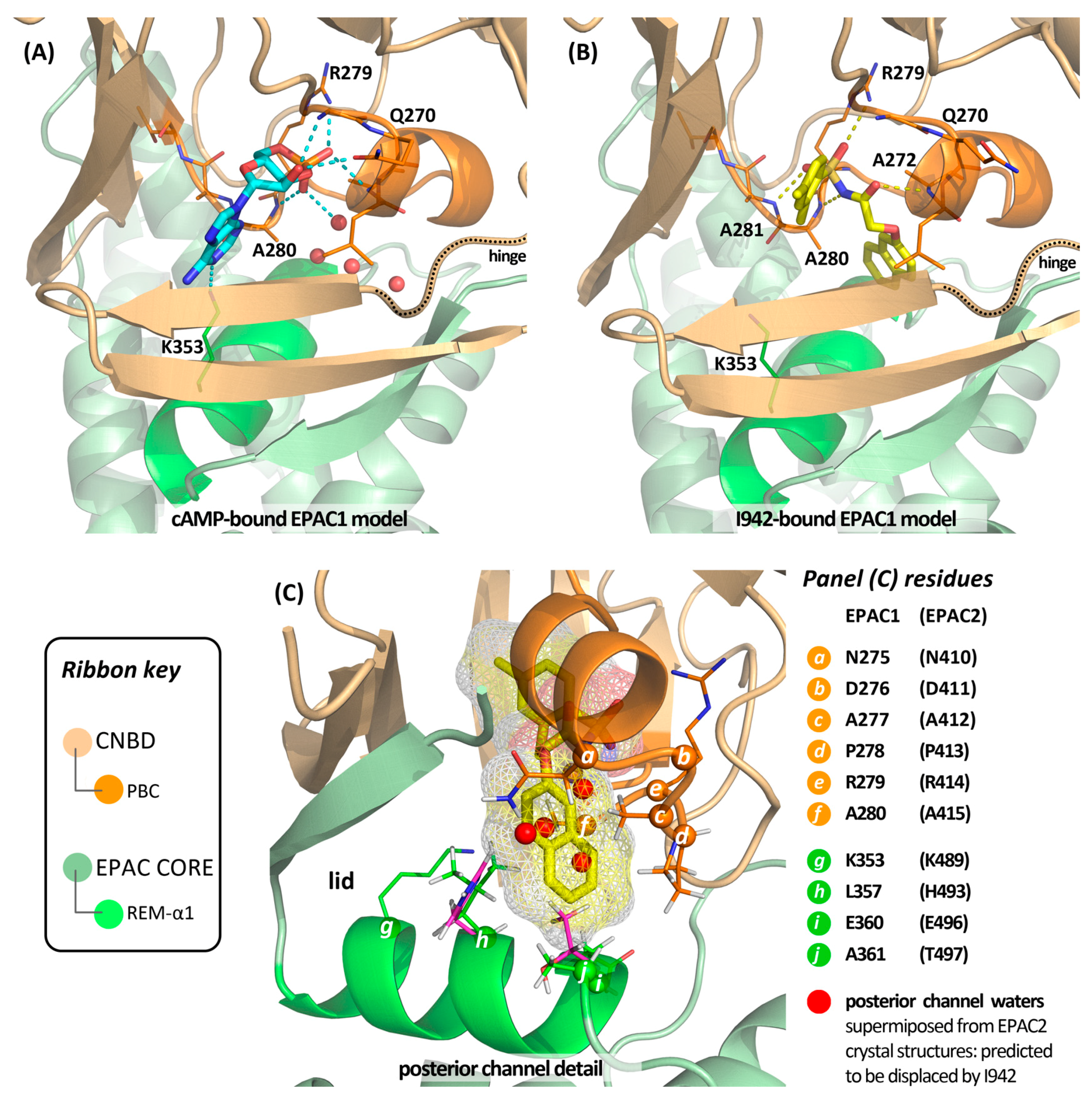
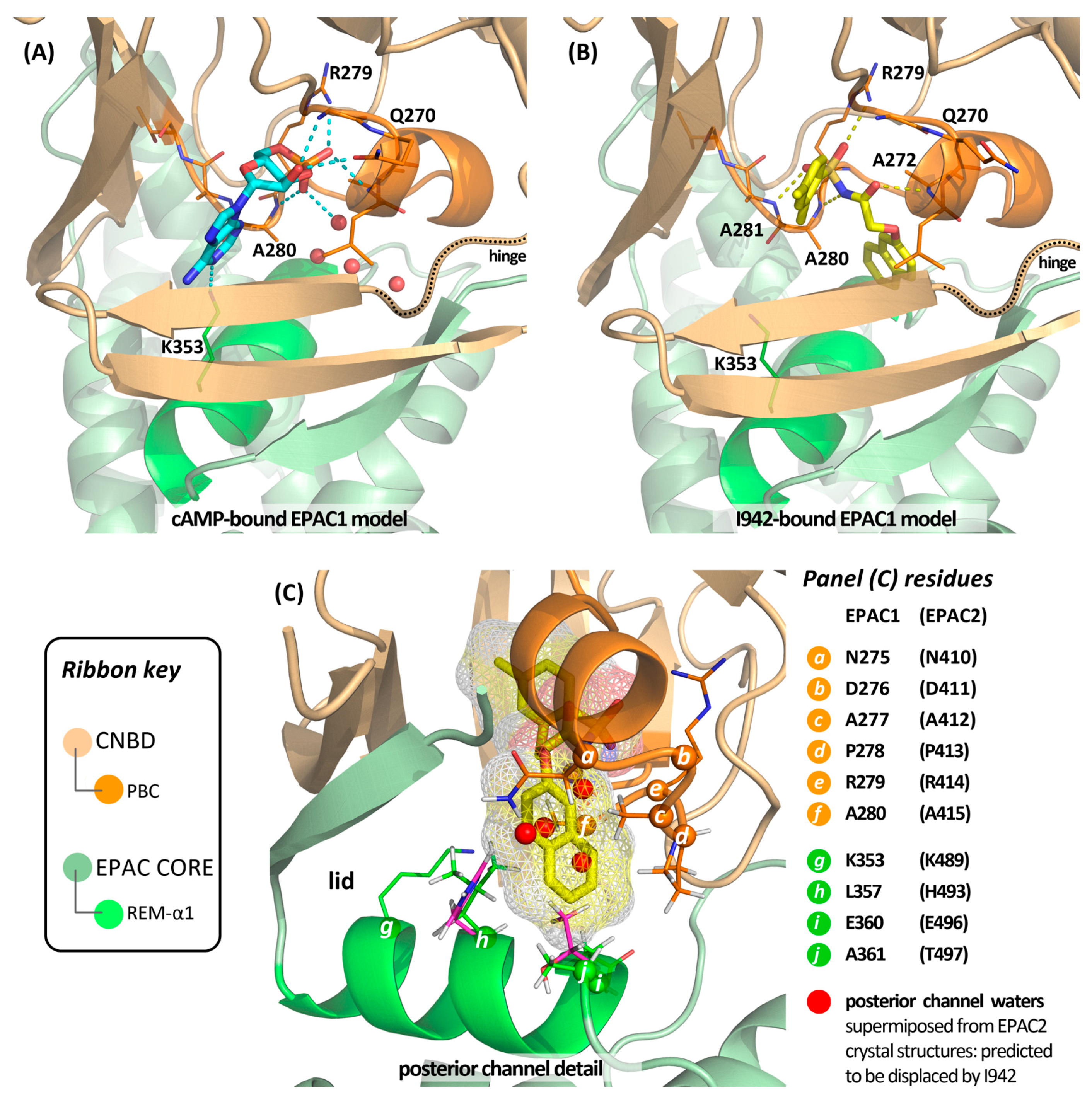
4.5. Putative Binding Mode of Novel EPAC Agonists
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brown, L.M.; Rogers, K.E.; Aroonsakool, N.; McCammon, J.A.; Insel, P.A. Allosteric inhibition of Epac: Computational modeling and experimental validation to identify allosteric sites and inhibitors. J. Biol. Chem. 2014, 289, 29148–29157. [Google Scholar] [CrossRef] [PubMed]
- Courilleau, D.; Bisserier, M.; Jullian, J.C.; Lucas, A.; Bouyssou, P.; Fischmeister, R.; Blondeau, J.P.; Lezoualc’h, F. Identification of a tetrahydroquinoline analog as a pharmacological inhibitor of the cAMP-binding protein Epac. J. Biol. Chem. 2012, 287, 44192–44202. [Google Scholar] [CrossRef] [PubMed]
- Dessauer, C.W.; Watts, V.J.; Ostrom, R.S.; Conti, M.; Dove, S.; Seifert, R. International union of basic and clinical pharmacology. CI. Structures and small molecule modulators of mammalian adenylyl cyclases. Pharmacol. Rev. 2017, 69, 93–139. [Google Scholar] [CrossRef] [PubMed]
- Halls, M.L.; Cooper, D.M. Adenylyl cyclase signalling complexes—Pharmacological challenges and opportunities. Pharmacol. Ther. 2017, 172, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Nicol, X.; Gaspar, P. Routes to camp: Shaping neuronal connectivity with distinct adenylate cyclases. Eur. J. Neurosci. 2014, 39, 1742–1751. [Google Scholar] [CrossRef] [PubMed]
- Klussmann, E. Protein-protein interactions of PDE4 family members—Functions, interactions and therapeutic value. Cell. Signal. 2016, 28, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef] [PubMed]
- Maurice, D.H.; Wilson, L.S.; Rampersad, S.N.; Hubert, F.; Truong, T.; Kaczmarek, M.; Brzezinska, P.; Freitag, S.I.; Umana, M.B.; Wudwud, A. Cyclic nucleotide phosphodiesterases (PDEs): Coincidence detectors acting to spatially and temporally integrate cyclic nucleotide and non-cyclic nucleotide signals. Biochem. Soc. Trans. 2014, 42, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Torres-Quesada, O.; Mayrhofer, J.E.; Stefan, E. The many faces of compartmentalized PKA signalosomes. Cell. Signal. 2017, 37, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Calejo, A.I.; Tasken, K. Targeting protein-protein interactions in complexes organized by a kinase anchoring proteins. Front. Pharmacol. 2015, 6, 192. [Google Scholar] [CrossRef] [PubMed]
- Dema, A.; Perets, E.; Schulz, M.S.; Deak, V.A.; Klussmann, E. Pharmacological targeting of AKAP-directed compartmentalized cAMP signalling. Cell. Signal. 2015, 27, 2474–2487. [Google Scholar] [CrossRef] [PubMed]
- Lefkimmiatis, K.; Zaccolo, M. Camp signaling in subcellular compartments. Pharmacol. Ther. 2014, 143, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, U.; Cheng, X. Exchange protein directly activated by cAMP encoded by the mammalian rapgef3 gene: Structure, function and therapeutics. Gene 2015, 570, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Parnell, E.; Palmer, T.M.; Yarwood, S.J. The future of EPAC-targeted therapies: Agonism versus antagonism. Trends Pharmacol. Sci. 2015, 36, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Liu, Z.; Chen, H.; Ye, N.; Cheng, X.; Zhou, J. Exchange proteins directly activated by cAMP (EPACs): Emerging therapeutic targets. Bioorg. Med. Chem. Lett. 2017, 27, 1633–1639. [Google Scholar] [CrossRef] [PubMed]
- Brand, T. The popeye domain-containing gene family. Cell Biochem. Biophys. 2005, 43, 95–103. [Google Scholar] [CrossRef]
- Schindler, R.F.; Brand, T. The popeye domain containing protein family—A novel class of camp effectors with important functions in multiple tissues. Prog. Biophys. Mol. Biol. 2016, 120, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Biel, M.; Michalakis, S. Cyclic nucleotide-gated channels. Handb. Exp. Pharmacol. 2009, 191, 111–136. [Google Scholar] [CrossRef]
- Ammon, H.P.; Muller, A.B. Forskolin: From an ayurvedic remedy to a modern agent. Planta Med. 1985, 51, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Murata, T.; Shimizu, K.; Degerman, E.; Maurice, D.; Manganiello, V. Cyclic nucleotide phosphodiesterases: Important signaling modulators and therapeutic targets. Oral Dis. 2015, 21, e25–e50. [Google Scholar] [CrossRef] [PubMed]
- DeNinno, M.P. Future directions in phosphodiesterase drug discovery. Bioorg. Med. Chem. Lett. 2012, 22, 6794–6800. [Google Scholar] [CrossRef] [PubMed]
- Knight, W.; Yan, C. Therapeutic potential of pde modulation in treating heart disease. Future Med. Chem. 2013, 5, 1607–1620. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.; Gil, C. cAMP-specific phosphodiesterase inhibitors: Promising drugs for inflammatory and neurological diseases. Expert Opin. Ther. Pat. 2014, 24, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Ji, Z.; Tsalkova, T.; Mei, F. EPAC and PKA: A tale of two intracellular camp receptors. Acta Biochim. Biophys. Sin. (Shanghai) 2008, 40, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Houslay, M.D.; Conti, M.; Fancis, S.H. The cyclic nucleotides. Handb. Exp. Pharmacol. 2011, 204, v–vii. [Google Scholar]
- Schlepper, M.; Thormann, J.; Mitrovic, V. Cardiovascular effects of forskolin and phosphodiesterase-III inhibitors. Basic Res. Cardiol. 1989, 84 (Suppl. 1), 197–212. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Springett, G.M.; Mochizuki, N.; Toki, S.; Nakaya, M.; Matsuda, M.; Housman, D.E.; Graybiel, A.M. A family of cAMP-binding proteins that directly activate Rap1. Science 1998, 282, 2275–2279. [Google Scholar] [CrossRef] [PubMed]
- De Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. EPAC is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Hoivik, E.A.; Witsoe, S.L.; Bergheim, I.R.; Xu, Y.; Jakobsson, I.; Tengholm, A.; Doskeland, S.O.; Bakke, M. DNA methylation of alternative promoters directs tissue specific expression of EPAC2 isoforms. PLoS ONE 2013, 8, e67925. [Google Scholar] [CrossRef] [PubMed]
- Rehmann, H.; Arias-Palomo, E.; Hadders, M.A.; Schwede, F.; Llorca, O.; Bos, J.L. Structure of EPAC2 in complex with a cyclic AMP analogue and Rap1b. Nature 2008, 27, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Rehmann, H.; Das, J.; Knipscheer, P.; Wittinghofer, A.; Bos, J.L. Structure of the cyclic-AMP-responsive exchange factor EPAC2 in its auto-inhibited state. Nature 2006, 439, 625–628. [Google Scholar] [CrossRef] [PubMed]
- Rehmann, H.; Prakash, B.; Wolf, E.; Rueppel, A.; de Rooij, J.; Bos, J.L.; Wittinghofer, A. Structure and regulation of the cAMP-binding domains of EPAC2. Nat. Struct. Biol. 2003, 10, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Parnell, E.; Smith, B.O.; Yarwood, S.J. The cAMP sensors, EPAC1 and EPAC2, display distinct subcellular distributions despite sharing a common nuclear pore localisation signal. Cell. Signal. 2015, 27, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Asuri, S.; Rebhun, J.F.; Castro, A.F.; Paranavitana, N.C.; Quilliam, L.A. The Rap1 guanine nucleotide exchange factor EPAC2 couples cyclic AMP and Ras signals at the plasma membrane. J. Biol. Chem. 2006, 281, 2506–2514. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Takahashi, M.; Li, Y.; Dillon, T.J.; Kaech, S.; Stork, P.J. The interaction of EPAC1 and Ran promotes Rap1 activation at the nuclear envelope. Mol. Cell. Biol. 2010, 30, 3956–3969. [Google Scholar] [CrossRef] [PubMed]
- Borland, G.; Smith, B.O.; Yarwood, S.J. EPAC proteins transduce diverse cellular actions of cAMP. Br. J. Pharmacol. 2009, 158, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Parnell, E.; Smith, B.O.; Palmer, T.M.; Terrin, A.; Zaccolo, M.; Yarwood, S.J. Regulation of the inflammatory response of vascular endothelial cells by EPAC1. Br. J. Pharmacol. 2012, 166, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Kirton, H.M.; Al-Owais, M.; Thireau, J.; Richard, S.; Peers, C.; Steele, D.S. EPAC2-Rap1 signaling regulates reactive oxygen species production and susceptibility to cardiac arrhythmias. Antioxid. Redox Signal. 2017, 27, 117–132. [Google Scholar] [CrossRef] [PubMed]
- Edland, F.; Wergeland, A.; Kopperud, R.; Asrud, K.S.; Hoivik, E.A.; Witso, S.L.; Æsoy, R.; Madsen, L.; Kristiansen, K.; Bakke, M.; et al. Long-term consumption of an obesogenic high fat diet prior to ischemia-reperfusion mediates cardioprotection via EPAC1-dependent signaling. Nutr. Metab. 2016, 13, 87. [Google Scholar] [CrossRef] [PubMed]
- Hwang, M.; Go, Y.; Park, J.H.; Shin, S.K.; Song, S.E.; Oh, B.C.; Im, S.S.; Hwang, I.; Jeon, Y.H.; Lee, I.K.; et al. EPAC2a-null mice exhibit obesity-prone nature more susceptible to leptin resistance. Int. J. Obes. 2017, 41, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Komai, A.M.; Musovic, S.; Peris, E.; Alrifaiy, A.; El Hachmane, M.F.; Johansson, M.; Wernstedt Asterholm, I.; Olofsson, C.S. White adipocyte adiponectin exocytosis is stimulated via beta3-adrenergic signaling and activation of EPAC1: Catecholamine resistance in obesity and type 2 diabetes. Diabetes 2016, 65, 3301–3313. [Google Scholar] [CrossRef] [PubMed]
- Lakshmikanthan, S.; Zieba, B.J.; Ge, Z.D.; Momotani, K.; Zheng, X.; Lund, H.; Artamonov, M.V.; Maas, J.E.; Szabo, A.; Zhang, D.X.; et al. Rap1b in smooth muscle and endothelium is required for maintenance of vascular tone and normal blood pressure. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1486–1494. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.P.; Fang, C.L.; Chen, H.K.; Wen, K.S.; Hseu, Y.C.; Hung, S.T.; Uen, Y.H.; Lin, K.Y. EPAC1 overexpression is a prognostic marker and its inhibition shows promising therapeutic potential for gastric cancer. Oncol. Rep. 2017, 37, 1953–1960. [Google Scholar] [CrossRef] [PubMed]
- Singhmar, P.; Huo, X.; Eijkelkamp, N.; Berciano, S.R.; Baameur, F.; Mei, F.C.; Zhu, Y.; Cheng, X.; Hawke, D.; Mayor, F., Jr.; et al. Critical role for Epac1 in inflammatory pain controlled by GRK2-mediated phosphorylation of Epac1. Proc. Natl. Acad. Sci. USA 2016, 113, 3036–3041. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, B.G.; Racke, K.; Schmidt, M. Distinct PKA and EPAC compartmentalization in airway function and plasticity. Pharmacol. Ther. 2013, 137, 248–265. [Google Scholar] [CrossRef] [PubMed]
- Oldenburger, A.; Timens, W.; Bos, S.; Smit, M.; Smrcka, A.V.; Laurent, A.C.; Cao, J.; Hylkema, M.; Meurs, H.; Maarsingh, H.; et al. Epac1 and Epac2 are differentially involved in inflammatory and remodeling processes induced by cigarette smoke. FASEB J. 2014, 28, 4617–4628. [Google Scholar] [CrossRef] [PubMed]
- Roscioni, S.S.; Prins, A.G.; Elzinga, C.R.; Menzen, M.H.; Dekkers, B.G.; Halayko, A.J.; Meurs, H.; Maarsingh, H.; Schmidt, M. Protein kinase a and the exchange protein directly activated by cAMP (Epac) modulate phenotype plasticity in human airway smooth muscle. Br. J. Pharmacol. 2011, 164, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Fanelli, C.; Aronoff, R. Restenosis following coronary angioplasty. Am. Heart J. 1990, 119, 357–368. [Google Scholar] [CrossRef]
- McKean, J.S.; Murray, F.; Gibson, G.; Shewan, D.A.; Tucker, S.J.; Nixon, G.F. The cAMP-producing agonist beraprost inhibits human vascular smooth muscle cell migration via exchange protein directly activated by camp. Cardiovasc. Res. 2015, 107, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Hewer, R.C.; Sala-Newby, G.B.; Wu, Y.J.; Newby, A.C.; Bond, M. PKA and Epac synergistically inhibit smooth muscle cell proliferation. J. Mol. Cell. Cardiol. 2011, 50, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Sands, W.A.; Woolson, H.D.; Milne, G.R.; Rutherford, C.; Palmer, T.M. Exchange protein activated by cyclic AMP (Epac)-mediated induction of suppressor of cytokine signaling 3 (SOCS-3) in vascular endothelial cells. Mol. Cell. Biol. 2006, 26, 6333–6346. [Google Scholar] [CrossRef] [PubMed]
- Bruunsgaard, H.; Pedersen, M.; Pedersen, B.K. Aging and proinflammatory cytokines. Curr. Opin. Hematol. 2001, 8, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Tieu, B.C.; Ray, S.; Recinos, I.A.; Cui, R.; Tilton, R.G.; Brasier, A.R. Roles of IL-6-gp130 signaling in vascular inflammation. Curr. Cardiol. Rev. 2008, 4, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Kleemann, R.; Zadelaar, S.; Kooistra, T. Cytokines and atherosclerosis: A comprehensive review of studies in mice. Cardiovasc. Res. 2008, 79, 360–376. [Google Scholar] [CrossRef] [PubMed]
- Schieffer, B.; Selle, T.; Hilfiker, A.; Hilfiker-Kleiner, D.; Grote, K.; Tietge, U.J.; Trautwein, C.; Luchtefeld, M.; Schmittkamp, C.; Heeneman, S.; et al. Impact of interleukin-6 on plaque development and morphology in experimental atherosclerosis. Circulation 2004, 110, 3493–3500. [Google Scholar] [CrossRef] [PubMed]
- Schuett, H.; Oestreich, R.; Waetzig, G.H.; Annema, W.; Luchtefeld, M.; Hillmer, A.; Bavendiek, U.; von Felden, J.; Divchev, D.; Kempf, T.; et al. Transsignaling of interleukin-6 crucially contributes to atherosclerosis in mice. Arterioscler 2012, 32, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Muller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Dahmen, H.; Grimm, C.; Gendo, C.; Muller-Newen, G.; Heinrich, P.C.; Schaper, F. The cytoplasmic tyrosine motifs in full-length glycoprotein 130 have different roles in IL-6 signal transduction. J. Immunol. 2000, 164, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zheng, S.G. Hall of fame among pro-inflammatory cytokines: Interleukin-6 gene and its transcriptional regulation mechanisms. Front. Immunol. 2016, 7, 604. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Robichaux, W.G.; Wang, Z.; Mei, F.C.; Cai, M.; Du, G.; Chen, J.; Cheng, X. Inhibition of Epac1 suppresses mitochondrial fission and reduces neointima formation induced by vascular injury. Sci. Rep. 2016, 6, 36552. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Yokoyama, U.; Yanai, C.; Ishige, R.; Kurotaki, D.; Umemura, M.; Fujita, T.; Kubota, T.; Okumura, S.; Sata, M.; et al. Epac1 deficiency attenuated vascular smooth muscle cell migration and neointimal formation. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2617–2625. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, U.; Minamisawa, S.; Quan, H.; Akaike, T.; Jin, M.; Otsu, K.; Ulucan, C.; Wang, X.; Baljinnyam, E.; Takaoka, M.; et al. Epac1 is upregulated during neointima formation and promotes vascular smooth muscle cell migration. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1547–H1555. [Google Scholar] [CrossRef] [PubMed]
- Adderley, S.P.; Martin, D.N.; Tulis, D.A. Exchange protein activated by cAMP (Epac) controls migration of vascular smooth muscle cells in concentration and time-dependent manner. Arch. Physiol. 2015, 2, 2. [Google Scholar] [CrossRef]
- Sprague, A.H.; Khalil, R.A. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem. Pharmacol. 2009, 78, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Babon, J.J.; Varghese, L.N.; Nicola, N.A. Inhibition of IL-6 family cytokines by SOCS3. Semin. Immunol. 2014, 26, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Yasukawa, H.; Suzuki, A.; Kamizono, S.; Syoda, T.; Kinjyo, I.; Sasaki, M.; Johnston, J.A.; Yoshimura, A. Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits janus tyrosine kinase by binding through the N-terminal kinase inhibitory region as well as SH2 domain. Genes Cells 1999, 4, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.J.; Palmer, T.M. Unbiased identification of substrates for the epac1-inducible e3 ubiquitin ligase component SOCS-3. Biochem. Soc. Trans. 2012, 40, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Recio, C.; Oguiza, A.; Mallavia, B.; Lazaro, I.; Ortiz-Munoz, G.; Lopez-Franco, O.; Egido, J.; Gomez-Guerrero, C. Gene delivery of suppressors of cytokine signaling (SOCS) inhibits inflammation and atherosclerosis development in mice. Basic Res. Cardiol. 2015, 110, 8. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.; Liu, J.; Dong, N.; Shi, J.; Xiao, Y.; Wang, Y.; Hu, X.; Gong, L.; Wang, W. Suppressor of cytokine signaling 3 is a negative regulator for neointimal hyperplasia of vein graft stenosis. J. Vasc. Res. 2014, 51, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; He, M.; Chen, T.; Liu, Y.; Tian, Y.L.; Wu, Y.L.; Zhao, Y.; Shen, Y.; Yuan, Z.Y. Multiple roles of SOCS proteins: Differential expression of SOCS1 and SOCS3 in atherosclerosis. Int. J. Mol. Med. 2013, 31, 1066–1074. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Munoz, G.; Martin-Ventura, J.L.; Hernandez-Vargas, P.; Mallavia, B.; Lopez-Parra, V.; Lopez-Franco, O.; Munoz-Garcia, B.; Fernandez-Vizarra, P.; Ortega, L.; Egido, J.; et al. Suppressors of cytokine signaling modulate JAK/STAT-mediated cell responses during atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Croker, B.A.; Kiu, H.; Pellegrini, M.; Toe, J.; Preston, S.; Metcalf, D.; O‘Donnell, J.A.; Cengia, L.H.; McArthur, K.; Nicola, N.A.; et al. IL-6 promotes acute and chronic inflammatory disease in the absence of SOCS3. Immunol. Cell Biol. 2012, 90, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Stahl, A.; Joyal, J.S.; Chen, J.; Sapieha, P.; Juan, A.M.; Hatton, C.J.; Pei, D.T.; Hurst, C.G.; Seaward, M.R.; Krah, N.M.; et al. SOCS3 is an endogenous inhibitor of pathologic angiogenesis. Blood 2012, 120, 2925–2929. [Google Scholar] [CrossRef] [PubMed]
- Jo, D.; Liu, D.; Yao, S.; Collins, R.D.; Hawiger, J. Intracellular protein therapy with SOCS3 inhibits inflammation and apoptosis. Nat. Med. 2005, 11, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Recio, C.; Oguiza, A.; Lazaro, I.; Mallavia, B.; Egido, J.; Gomez-Guerrero, C. Suppressor of cytokine signaling 1-derived peptide inhibits janus kinase/signal transducers and activators of transcription pathway and improves inflammation and atherosclerosis in diabetic mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1953–1960. [Google Scholar] [CrossRef] [PubMed]
- Yarwood, S.J.; Borland, G.; Sands, W.A.; Palmer, T.M. Identification of CCAAT/enhancer-binding proteins as exchange protein activated by cAMP-activated transcription factors that mediate the induction of the SOCS-3 gene. J. Biol. Chem. 2008, 283, 6843–6853. [Google Scholar] [CrossRef] [PubMed]
- Netherton, S.J.; Sutton, J.A.; Wilson, L.S.; Carter, R.L.; Maurice, D.H. Both protein kinase A and exchange protein activated by cAMP coordinate adhesion of human vascular endothelial cells. Circ. Res. 2007, 101, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Bogatcheva, N.V.; Garcia, J.G.; Verin, A.D. Role of tyrosine kinase signaling in endothelial cell barrier regulation. Vasc. Pharmacol. 2002, 39, 201–212. [Google Scholar] [CrossRef]
- Vouret-Craviari, V.; Grall, D.; Van Obberghen-Schilling, E. Modulation of Rho GTPase activity in endothelial cells by selective proteinase-activated receptor (PAR) agonists. J. Thromb. Haemost. 2003, 1, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Sand, C.; Jakobs, K.H.; Michel, M.C.; Weernink, P.A. Epac and the cardiovascular system. Curr. Opin. Pharmacol. 2007, 7, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Baumer, Y.; Drenckhahn, D.; Waschke, J. cAMP induced Rac 1-mediated cytoskeletal reorganization in microvascular endothelium. Histochem. Cell Biol. 2008, 129, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Birukova, A.A.; Zagranichnaya, T.; Fu, P.; Alekseeva, E.; Chen, W.; Jacobson, J.R.; Birukov, K.G. Prostaglandins PGE2 and PGI2 promote endothelial barrier enhancement via PKA- and Epac1/Rap1-dependent Rac activation. Exp. Cell Res. 2007, 313, 2504–2520. [Google Scholar] [CrossRef] [PubMed]
- Cullere, X.; Shaw, S.K.; Andersson, L.; Hirahashi, J.; Luscinskas, F.W.; Mayadas, T.N. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood 2005, 105, 1950–1955. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, S.; Sakurai, A.; Sano, H.; Yamagishi, A.; Somekawa, S.; Takakura, N.; Saito, Y.; Kangawa, K.; Mochizuki, N. Cyclic AMP potentiates vascular endothelial cadherin-mediated cell-cell contact to enhance endothelial barrier function through an Epac-Rap1 signaling pathway. Mol. Cell. Biol. 2005, 25, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Kooistra, M.R.; Corada, M.; Dejana, E.; Bos, J.L. Epac1 regulates integrity of endothelial cell junctions through VE-cadherin. FEBS Lett. 2005, 579, 4966–4972. [Google Scholar] [CrossRef] [PubMed]
- Sehrawat, S.; Cullere, X.; Patel, S.; Italiano, J., Jr.; Mayadas, T.N. Role of Epac1, an exchange factor for Rap GTPases, in endothelial microtubule dynamics and barrier function. Mol. Biol. Cell 2008, 19, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Vliem, M.J.; Ponsioen, B.; Schwede, F.; Pannekoek, W.J.; Riedl, J.; Kooistra, M.R.; Jalink, K.; Genieser, H.G.; Bos, J.L.; Rehmann, H. 8-pCPT-2′-O-Me-cAMP-AM: An improved Epac-selective cAMP analogue. Chembiochem Eur. J. Chem. Biol. 2008, 9, 2052–2054. [Google Scholar] [CrossRef] [PubMed]
- Enserink, J.M.; Christensen, A.E.; de Rooij, J.; van Triest, M.; Schwede, F.; Genieser, H.G.; Doskeland, S.O.; Blank, J.L.; Bos, J.L. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat. Cell Biol. 2002, 4, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, A.; Rehmann, H.R.; Cool, R.H.; Theiss, C.; de Rooij, J.; Bos, J.L.; Wittinghofer, A. Dynamic interaction of cAMP with the Rap guanine-nucleotide exchange factor Epac1. J. Mol. Biol. 2001, 306, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Schwede, F.; Bertinetti, D.; Langerijs, C.N.; Hadders, M.A.; Wienk, H.; Ellenbroek, J.H.; de Koning, E.J.; Bos, J.L.; Herberg, F.W.; Genieser, H.G.; et al. Structure-guided design of selective Epac1 and Epac2 agonists. PLoS Biol. 2015, 13, e1002038. [Google Scholar] [CrossRef] [PubMed]
- Metrich, M.; Berthouze, M.; Morel, E.; Crozatier, B.; Gomez, A.M.; Lezoualc’h, F. Role of the cAMP-binding protein Epac in cardiovascular physiology and pathophysiology. Pflug. Arch. Eur. J. Physiol. 2010, 459, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Hothi, S.S.; Gurung, I.S.; Heathcote, J.C.; Zhang, Y.; Booth, S.W.; Skepper, J.N.; Grace, A.A.; Huang, C.L. Epac activation, altered calcium homeostasis and ventricular arrhythmogenesis in the murine heart. Pflug. Arch. Eur. J. Physiol. 2008, 457, 253–270. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Cheng, H.; Lao, D.H.; Na, L.; van Oort, R.J.; Brown, J.H.; Wehrens, X.H.; Chen, J.; Bers, D.M. Epac2 mediates cardiac β1-adrenergic-dependent sarcoplasmic reticulum Ca2+ leak and arrhythmia. Circulation 2013, 127, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Metrich, M.; Fernandez-Velasco, M.; Lucas, A.; Leroy, J.; Perrier, R.; Morel, E.; Fischmeister, R.; Richard, S.; Benitah, J.P.; et al. The cAMP binding protein Epac modulates Ca2+ sparks by a Ca2+/calmodulin kinase signalling pathway in rat cardiac myocytes. J. Physiol. 2007, 583, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.L.; Katoh, M.; Shibasaki, T.; Minami, K.; Sunaga, Y.; Takahashi, H.; Yokoi, N.; Iwasaki, M.; Miki, T.; Seino, S. The cAMP sensor Epac2 is a direct target of antidiabetic sulfonylurea drugs. Science 2009, 325, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Herbst, K.J.; Coltharp, C.; Amzel, L.M.; Zhang, J. Direct activation of Epac by sulfonylurea is isoform selective. Chem. Biol. 2011, 18, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Rehmann, H. Epac2: A sulfonylurea receptor? Biochem. Soc. Trans. 2012, 40, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Tsalkova, T.; Gribenko, A.V.; Cheng, X. Exchange protein directly activated by cyclic AMP isoform 2 is not a direct target of sulfonylurea drugs. Assay Drug Dev. Technol. 2011, 9, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Tsalkova, T.; Mei, F.C.; Li, S.; Chepurny, O.G.; Leech, C.A.; Liu, T.; Holz, G.G.; Woods, V.L., Jr.; Cheng, X. Isoform-specific antagonists of exchange proteins directly activated by cAMP. Proc. Natl. Acad. Sci. USA 2012, 109, 18613–18618. [Google Scholar] [CrossRef] [PubMed]
- McPhee, I.; Gibson, L.C.; Kewney, J.; Darroch, C.; Stevens, P.A.; Spinks, D.; Cooreman, A.; MacKenzie, S.J. Cyclic nucleotide signalling: A molecular approach to drug discovery for alzheimer‘s disease. Biochem. Soc. Trans. 2005, 33, 1330–1332. [Google Scholar] [CrossRef] [PubMed]
- Tsalkova, T.; Mei, F.C.; Cheng, X. A fluorescence-based high-throughput assay for the discovery of exchange protein directly activated by cyclic AMP (EPAC) antagonists. PLoS ONE 2012, 7, e30441. [Google Scholar] [CrossRef] [PubMed]
- Almahariq, M.; Tsalkova, T.; Mei, F.C.; Chen, H.; Zhou, J.; Sastry, S.K.; Schwede, F.; Cheng, X. A novel EPAC-specific inhibitor suppresses pancreatic cancer cell migration and invasion. Mol. Pharmacol. 2013, 83, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Tsalkova, T.; Chepurny, O.G.; Mei, F.C.; Holz, G.G.; Cheng, X.; Zhou, J. Identification and characterization of small molecules as potent and specific EPAC2 antagonists. J. Med. Chem. 2013, 56, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Rehmann, H. Epac-inhibitors: Facts and artefacts. Sci. Rep. 2013, 3, 3032. [Google Scholar] [CrossRef] [PubMed]
- Wild, C.T.; Zhu, Y.; Na, Y.; Mei, F.; Ynalvez, M.A.; Chen, H.; Cheng, X.; Zhou, J. Functionalized N,N-diphenylamines as potent and selective EPAC2 inhibitors. ACS Med. Chem. Lett. 2016, 7, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Ye, N.; Zhu, Y.; Chen, H.; Liu, Z.; Mei, F.C.; Wild, C.; Chen, H.; Cheng, X.; Zhou, J. Structure-activity relationship studies of substituted 2-(isoxazol-3-yl)-2-oxo-N′-phenyl-acetohydrazonoyl cyanide analogues: Identification of potent exchange proteins directly activated by cAMP (EPAC) antagonists. J. Med. Chem. 2015, 58, 6033–6047. [Google Scholar] [CrossRef] [PubMed]
- Ye, N.; Zhu, Y.; Liu, Z.; Mei, F.C.; Chen, H.; Wang, P.; Cheng, X.; Zhou, J. Identification of novel 2-(benzo[d]isoxazol-3-yl)-2-oxo-N-phenylacetohydrazonoyl cyanide analoguesas potent epac antagonists. Eur. J. Med. Chem. 2017, 134, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Rehmann, H.; Wittinghofer, A.; Bos, J.L. Capturing cyclic nucleotides in action: Snapshots from crystallographic studies. Nat. Rev. Mol. Cell Biol. 2007, 8, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Rehmann, H. Characterization of the activation of the Rap-specific exchange factor Epac by cyclic nucleotides. Methods Enzymol. 2005, 407, 159–173. [Google Scholar]
- Dao, K.K.; Teigen, K.; Kopperud, R.; Hodneland, E.; Schwede, F.; Christensen, A.E.; Martinez, A.; Doskeland, S.O. Epac1 and cAMP-dependent protein kinase holoenzyme have similar cAMP affinity, but their cAMP domains have distinct structural features and cyclic nucleotide recognition. J. Biol. Chem. 2006, 281, 21500–21511. [Google Scholar] [CrossRef] [PubMed]
- Parnell, E.; McElroy, S.P.; Wiejak, J.; Baillie, G.L.; Porter, A.; Adams, D.R.; Rehmann, H.; Smith, B.O.; Yarwood, S.J. Identification of a novel, small molecule partial agonist for the cyclic AMP sensor, EPAC1. Sci. Rep. 2017, 7, 294. [Google Scholar] [CrossRef] [PubMed]
- Ammazzalorso, A.; De Filippis, B.; Giampietro, L.; Amoroso, R. N-acylsulfonamides: Synthetic routes and biological potential in medicinal chemistry. Chem. Biol. Drug Des. 2017, 90, 1094–1105. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Model | Treatments | Effects |
|---|---|---|
| Carotid arteries and vascular smooth muscle cells (VSMCs) from wild type (WT) and EPAC1 −/− mice. | Ligation of carotid arteries and pharmacological inhibition of EPAC1 | Neointima formation and VSMC proliferation were reduced in EPAC1 −/− mice. ESI09 also reduced neointima formation [60]. |
| VSMCs from thoracic aorta explants from WT and EPAC1 −/− mice. | Injury of femoral artery | Reduced neointima formation and reduced migration of VSMCs in EPAC1 −/− mice [61]. |
| Human saphenous vein VSMCs | Effects of pharmacological EPAC activation on VSMC migration | EPAC activation reduced VSMC migration and serum-induced vessel wall thickening [49]. |
| Rat VSMCs from aorta explants. | Phamacological activation of EPAC and PKA | A combination of EPAC and PKA activation inhibited serum-induced VSMC proliferation [50]. |
| VSMCs from foetal and adult rat aorta. | Pharmacological activation of EPAC and adenovirus-mediated overexpression of EPAC1 | EPAC activation and overexpression of EPAC1 enhanced intimal thickening in aorta and VSMC proliferation [62]. |
| Primary aortic VSMCs from male rats | Pharmacological activation of EPAC and PKA | PKA and EPAC work cooperatively to inhibit VSMC migration [63]. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barker, G.; Parnell, E.; Van Basten, B.; Buist, H.; Adams, D.R.; Yarwood, S.J. The Potential of a Novel Class of EPAC-Selective Agonists to Combat Cardiovascular Inflammation. J. Cardiovasc. Dev. Dis. 2017, 4, 22. https://doi.org/10.3390/jcdd4040022
Barker G, Parnell E, Van Basten B, Buist H, Adams DR, Yarwood SJ. The Potential of a Novel Class of EPAC-Selective Agonists to Combat Cardiovascular Inflammation. Journal of Cardiovascular Development and Disease. 2017; 4(4):22. https://doi.org/10.3390/jcdd4040022
Chicago/Turabian StyleBarker, Graeme, Euan Parnell, Boy Van Basten, Hanna Buist, David R. Adams, and Stephen J. Yarwood. 2017. "The Potential of a Novel Class of EPAC-Selective Agonists to Combat Cardiovascular Inflammation" Journal of Cardiovascular Development and Disease 4, no. 4: 22. https://doi.org/10.3390/jcdd4040022
APA StyleBarker, G., Parnell, E., Van Basten, B., Buist, H., Adams, D. R., & Yarwood, S. J. (2017). The Potential of a Novel Class of EPAC-Selective Agonists to Combat Cardiovascular Inflammation. Journal of Cardiovascular Development and Disease, 4(4), 22. https://doi.org/10.3390/jcdd4040022