
MAPK, PI3K/Akt Pathways, and GSK-3β Activity in Severe Acute Heart Failure in Intensive Care Patients: An Updated Review



Abstract
1. Introduction
2. MAPK Pathway Family
2.1. Canonical Architecture and Function
2.2. Upstream Activation in Acute Cardiac Stress
2.3. Downstream Effects
2.3.1. Inflammation
2.3.2. Apoptosis
2.3.3. Fibrosis
2.3.4. Crosstalk with Other Signaling Pathways
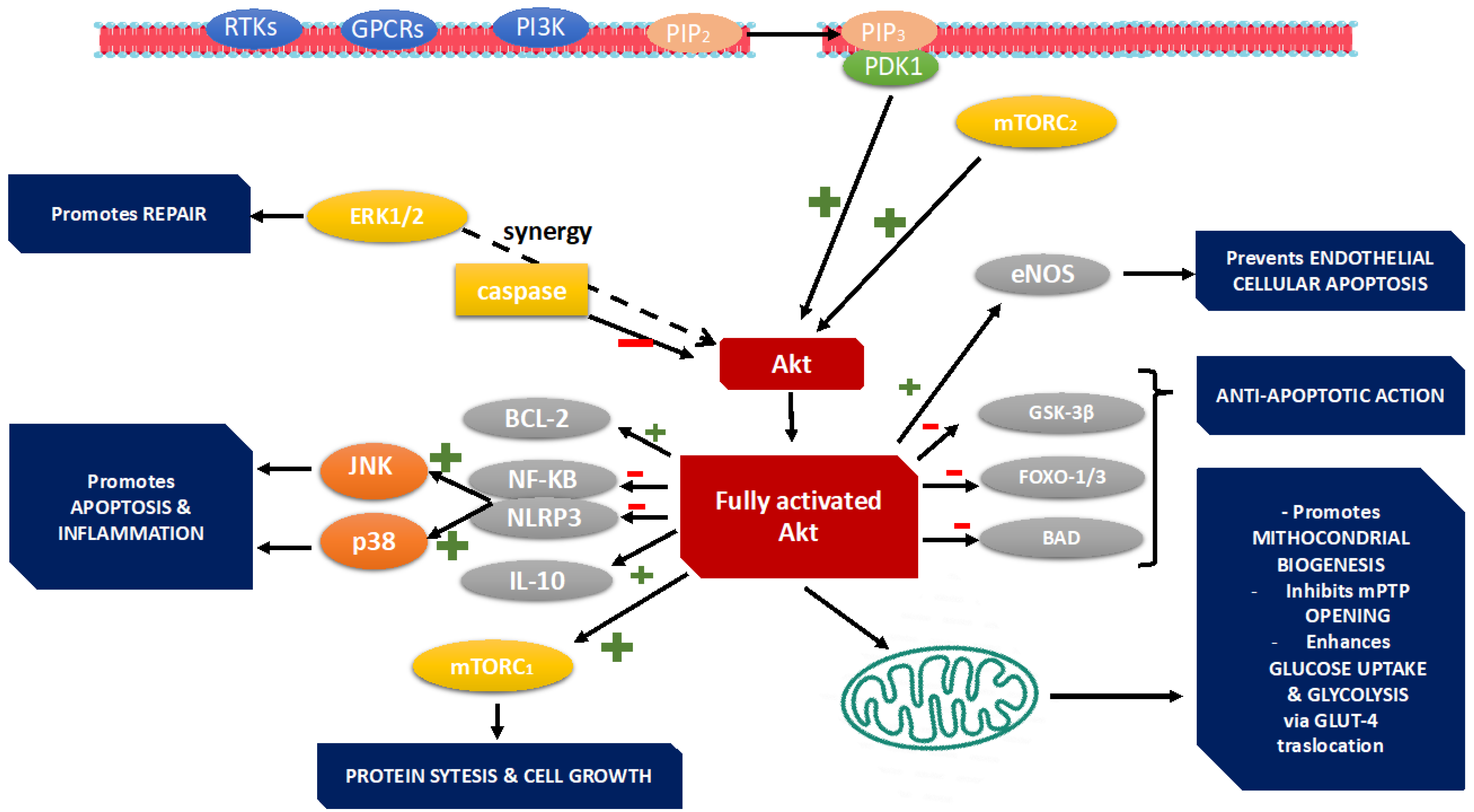
3. PI3K Pathway
3.1. Cardiomyocyte Survival and Apoptosis
3.2. Regulation of Hypertrophy
3.3. Mitochondrial Function
3.4. Inflammation, Oxidative Stress, and Autophagy
4. GSK-3β Pathway
4.1. GSK-3β in the Pathogenesis of Acute Heart Failure
4.2. GSK-3β Dysregulation in Acute Heart Failure
5. MAPK, PI3K/Akt, and GSK-3β Signaling in Acute Heart Failure in Diabetic Patients
5.1. MAPK Mechanisms and Activation
5.2. PI3K/Akt Mechanisms and Activation
5.3. GSK-3β Mechanisms and Activation
6. Therapeutic Perspectives
6.1. MAPK Pathway
6.2. PI3K/Akt/mTor Signaling Cascade
6.3. GSK-3β Pathway
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cowie, M.; Anker, S.; Cleland, J.; Felker, G.; Filippatos, G.; Jaarsma, T.; López-Sendón, J. Improving care for patients with acute heart failure: Before, during and after hospitalization. Esc. Heart Fail. 2014, 1, 110–145. [Google Scholar] [CrossRef] [PubMed]
- Jentzer, J.C.; Reddy, Y.N.; Rosenbaum, A.N.; Dunlay, S.M.; Borlang, B.A.; Hollemberg, S.M. Outcomes and predictors of mortality among cardiac intensive care uniti patients in heart failure. J. Card. Fail. 2022, 7, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Zhang, S.; Kovacs, A.; Wang, Y.; Muslni, A.J. Role of p38alfa MAPK in cardiac apoptosis and remodelling after myocaridal infarction. J. Mol. Cell Cariol. 2005, 38, 617–623. [Google Scholar] [CrossRef]
- Maulik, A.; Davidson, S.M.; Piotrowska, I.; Walker, M.; Yellon, D.M. Ischaemic preconditioning protects cariomyociyes from anthracycline-induced toxicity vie thw PI3K pathway. Cardiovasc. Drug Ther. 2018, 32, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, A.; Gruenloh, S.K.; Buonaccors, J.N.; Zhang, R.; Gross, G.J.; Falk, J.R. Multiple antiapoptotic target of the PI3K/Akt survival pathway are activated by epoxyeixosatrienoic acids to protect cardiomyocytes from hypoxia/anoxia. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H724–H735. [Google Scholar] [CrossRef] [PubMed]
- Sudgen, P.H.; Fuller, S.J.; Weiss, S.C.; Clerk, A. Glycogen syntase Kinase 3 (GSK3) in the heart: A point of integration in hypertrophic signalling and a therapeutic target? A critical analysis. Br. J. Pharmacol. 2008, 153, 137–153. [Google Scholar]
- Li, L.; Hao, J.; Jiang, X.; Li, P.; Hu, S. Cardioprotective effects of ulinastatin against isoproterenol-induced chronic heart failure through the pi3k-akt, p38 mapk and nf-κb pathways. Mol. Med. Rep. 2018, 17, 1354–1360. [Google Scholar] [CrossRef]
- Yao, H.; Han, X.; Han, X. The cardioprotection of the insulin-mediated pi3k/akt/mtor signaling pathway. Am. J. Cardiovasc. Drugs 2014, 14, 433–442. [Google Scholar] [CrossRef]
- Ferrari, R.; Bueno, H.; Chioncel, O.; Cleland, J.G.; Stough, W.G.; Lettino, M.; Metra, M.; Parissis, J.T.; Pinto, F. Acute heart failure: Lesson learned, roads haed. Eur. J. Heart Fail. 2018, 20, 842–850. [Google Scholar] [CrossRef]
- Wei, S.; Yu, Y.; Zhang, Z.; Weiss, R.; Felder, R. Mitogen-activated protein kinases mediate upregulation of hypothalamic angiotensin ii type 1 receptors in heart failure rats. Hypertension 2008, 52, 679–686. [Google Scholar] [CrossRef]
- Wang, Y. Mitogen-activated protein kinases in heart development and diseases. Circulation 2007, 116, 101161. [Google Scholar] [CrossRef]
- Avruch, J. MAP kinase pathways: The first twenty years. Biochim. Biophys. Acta 2007, 1773, 1150–1160. [Google Scholar] [CrossRef]
- Wang, X.; Liu, R. The role of the MAPK signaling pathway in cardiovascular disease: Pathophysiological mechanism and clinical therapy. Int. J. Mol. Sci. 2025, 26, 2667. [Google Scholar] [CrossRef] [PubMed]
- Kehat, I.; Molkenntin, J.D. Extracellular signal-regulated kinases 1/2 (ERK 1/2) signaling in cardiac hypertrophy. Ann. N. Y. Acad. Sci. 2010, 1188, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Yang, Y.; Zhang, Z.; Felder, R. Angiotensin II upregulates hypothalamic at1 receptor expression in rats via the mitogen-activated preotein kinase pathway. AJP Heart Circ. Physiol. 2009, 296, H1425–H1433. [Google Scholar] [CrossRef] [PubMed]
- Malathi, H.; Singhal, A.; Goyal, J. Computational docking studies of mapk1 target protein responsible for heart failure using catechin as natural inhibitor. Cardiometry 2023, 355–360. [Google Scholar] [CrossRef]
- Guitart-Mampel, M.; Urquiza, P.; Borges, J.I.; Lymperopoulos, A.; Solesso, M.E. Impact of aldosterone on failing myocardium: Insights from mitochondria and adrenergic receptors signaling and function. Cells 2021, 10, 1152. [Google Scholar] [CrossRef]
- Yocota, T.; Wang, Y. p38MAP kinases in heart. Gene 2015, 575, 369–376. [Google Scholar] [CrossRef]
- Zhang, W.; Elimban, V.; Nijjar, M.S.; Gupta, S.K.; Dhalla, N.S. Role of mitogen-activated protein kinases in cardiac hypertrophy and heart failure. Exp. Clin. Cardiol. 2003, 8, 173–183. [Google Scholar]
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of oxidative stress in cardiovascular diseases. J. Hypertens. 2000, 18, 655–673. [Google Scholar] [CrossRef]
- Sadoshima, J.; Izumo, S. Mechanical stretch rapidly activates multiple signal transduction pathways in cardiac myocytes: Potential involvement of an autocrine/paracrine mechanism. Cell 1993, 75, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, M.; Kurihara, H.; Kimura, S.; Tomobe, Y.; Kobayashi, M.; Mitsui, Y.; Yazaki, Y.; Goto, K.; Masaki, T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988, 332, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Frantz, S.; Ertl, G.; Bauersachs, J. Mechanisms of disease: Toll-like receptors in cardiovascular disease. Cardiovasc. Res. 2007, 73, 310–320. [Google Scholar] [CrossRef]
- Aplin, M.; Christensen, G.L.; Schneider, M.; Heydorn, A.; Gammeltoft, S.; Kjølbye, A.L.; Sheikh, S.P.; Hansen, J.L. Differential extracellular signal-regulated kinase 1/2 signaling and regulation of cardiac myocyte hypertrophic and apoptotic responses. J. Biol. Chem. 2007, 282, 22023–22032. [Google Scholar]
- Lijnen, P. Renin-angiotensin system, hypertrophy and gene expression in cardiac myocytes. J. Mol. Cell Cardiol. 1999, 31, 949–970. [Google Scholar] [CrossRef]
- Rose, B.A.; Force, T.; Wang, Y. Mitogen-activated protein kinase signaling in the heart: Angels vs demons in an heart-braking tale. Physiol. Rev. 2010, 90, 1507–1546. [Google Scholar] [CrossRef]
- Clerk, A.; Sudgen, P.H. Inflam my heart (by p38-MAPK). Circ. Res. 2006, 99, 455–458. [Google Scholar] [CrossRef]
- Touyz, R.M. Reactive oxygen species as mediators of calcium signaling by angiotensin II: Implications in vascular physiology and pathophysiology. Circ. Res. 2004, 94, 456–458. [Google Scholar] [CrossRef]
- Garlapati, V.; Molitor, M.; Michna, T.; Harms, G.; Finger, S.; Jung, R.; Wenzel, P. Targeting myeloid cell coagulation signaling blocks map kinase/tgf-β1–driven fibrotic remodeling in ischemic heart failure. J. Clin. Investig. 2023, 133, e156436. [Google Scholar] [CrossRef]
- Sun, H.Y.; Wang, N.P.; Halkos, M.; Kerendi, F.; Kin, H.; Guyton, R.A.; Zhao, Z.Q. Postconditioning attenuates cardiomyocytes apoptosis via inhibition of JNK and p38 mitogen-activated protein kinase signaling pathways. Apoptosis 2006, 11, 1583–1593. [Google Scholar] [CrossRef]
- Min, K.; Huang, Y.; Giordano, F.; Bajpeyi, S.; Bennett, A. Abstract 16205: Mkp-5 deficiency attenuates pressure overload-induced cardiac hypertrophy. Circulation 2020, 142 (Suppl. S3), A16205. [Google Scholar] [CrossRef]
- Nakamura, K.; Fushimi, K.; Kouchi, H.; Mihara, K.; Miyazaki, M.; Ohe, T. Inhibitory effects of an-tioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis fac-tor-alpha and angiotensin II. Circulation 1998, 98, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Schafer, S.; Viswanathan, S.; Widjaja, A.A.; Lim, W.W.; Moreno-Moral, A.; DeLaughter, D.M.; Patone, G.; Chow, K.; Khin, E.; Tan, J.; et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 2017, 552, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Ying, W.; Wang, P.; Song, L.; Chen, Q.; Zhang, M.; Zhang, Z. Reduction of myocardial fibrosis by inhibition of p38 MAPK in pressure-overloaded rats. Hypertension 2010, 55, 1101–1107. [Google Scholar]
- Czubryt, M.P. Cardiac fibroblast to myofibroblast phenotype conversion-an unexploited therapeutic target. J. Cardiovasc. Dev. Dis. 2019, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.; Law, J.P. Chronic activation of human cardiac fibriblasts in vitro attenuates the reversibility of the myofibroblasts phenotype. Sci. Rep. 2023, 13, 12137. [Google Scholar] [CrossRef]
- Liu, M.; de Juan Ababd, B.L.; Cheng, K. Cardiac fibrosis: Myofibroblast-mediated pathological regulation and drug delivery strategies. Adv. Drug Deliv. Rev. 2021, 173, 504–519. [Google Scholar] [CrossRef]
- Ma, Z.G.; Yuan, Y.P.; Wu, H.M.; Zhang, X.; Tang, Q.Z. Cardiac fibrosis: New insights into the pathogenesis. Int. J. Biol. Sci. 2018, 14, 1645–1657. [Google Scholar] [CrossRef]
- Maejima, Y.; Adachi, S.; Morikawa, K.; Ito, H.; Isobe, M. Nitric oxide inhibits vascular smooth muscle cell proliferation by modulation of the c-Src signaling pathway via activation of ERK and Akt. Cardiovasc. Res. 2005, 68, 423–431. [Google Scholar] [CrossRef]
- Duangrat, R.; Parichatikanond, W.; Chanmahasathien, W.; Mangmool, S. Adenosine a3 receptor: From molecular signaling to therapeutic strategies for heart diseases. Int. J. Mol. Sci. 2024, 25, 5763. [Google Scholar] [CrossRef]
- Liu, F.; Fan, L.M.; Geng, L.; Li, M.J. p47phox-dependent oxidant signalling through ASK1, MKK 3/6, and MAPKs in Angiotensin II-induced cardiac hypertrophy and apoptosis. Antooxidants 2021, 10, 1363. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.M.; Zhou, J. The role of PI3K/Akt signaling pathway in myocardial ischemia-reperfusion injury. Int. Immunopharmacol. 2023, 123, 110715. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri-Fard, S.; Sasi, A.; Hussen, B.; Shoorei, H.; Siddiq, A.; Taheri, M.; Ayatollahi, S. Interplay between pi3k/akt pathway and heart disorders. Mol. Biol. Rep. 2022, 49, 9767–9781. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Gao, F.; Chen, J. Role of PTEN-less in cardiac injury, hypertrophy, and regeneration. Cell Regen. 2021, 10, 25. [Google Scholar] [CrossRef]
- Meng, X.; Cui, J.; He, G. Bcl2 is involved in cardiac hypertrophy through PI3K/Akt pathway. BioMed Res. Int. 2021, 2, 6615502. [Google Scholar]
- Lin, Y.; Zhang, L.; Ding, X.; Chen, C.; Meng, M.; Ke, Y.; Wang, W. Realtionship netween the microRNAs and PIeK/Akt/mTOR axis: Focus on non-small cell lung cancer. Pathol. Res. Pract. 2022, 239, 154093. [Google Scholar] [CrossRef]
- Matsui, T.; Tao, J.; del Monte, F.; Lee, K.H.; Li, L.; Picard, M.; Force, T.L.; Franke, T.F.; Hajjar, R.J.; Rosenzweig, A. Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation 2001, 104, 330–335. [Google Scholar] [CrossRef]
- Hussini, A.M.; Quast, A.S.; Plotz, M.; Gravel, K.; Exner, T. PI3K/Akt signaling pathway is essential for survival of induced puripotent stem cells. PLoS ONE 2016, 11, e0254770. [Google Scholar]
- Ramos-Kuri, M.; Meka, S.H.; Salamanca-Buentello, F. Molecules linked to Ras signaling as therapeutic targets in cardiac pathologies. Bol. Res. 2021, 54, 23. [Google Scholar] [CrossRef]
- Wang, X.; Pan, J.; Liu, D.; Zhang, M.; Li, X.; Tian, J. Nicorandil alleviates apoptosis in diabetic cardiomyopathy through PI3K/Akt pathway. J. Cell Mol. Med. 2019, 23, 5349–5359. [Google Scholar] [CrossRef]
- Hua, Y.; Zhang, Y.; Ceylan-Isik, A.F.; Wold, L.E.; Nunn, J.M.; Ren, J. Chronic Akt activation accentuates aging-induced cardia hypertrophy and myocardial contractile dysfunction: Role of autophagy. Basic Res. Cardiol. 2011, 106, 1173–1191. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.X.; Wang, R.Y.; Guo, Y.Z.; Lv, D.Y.; Luo, M.H.; He, A.; Luo, S.X.; Xia, Y. Phosphorylation of Akt at Thr308 regulates p-eNOS Ser1177 during physiological conditions. FEBS Open Bio 2021, 11, 1953–1964. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ding, Y.; Sun, X. Carnisol suppresses microglia cell inflammation and apoptosis through PI3K/Akt/mTOR signaling pathway. Immunipharmacol. Immunotoxixol. 2022, 44, 656–662. [Google Scholar]
- Lei, Z.; Wahlquist, C.; el Azzouzi, H.; Deddens, J.C.; Kuster, D.; van Mil, A.; Rojas-Munoz, A.; Huibers, M.M. miR-132/212 impairs cariomyocytes contractility in the failing heart by suppressing SERCA2a. Front. Cardiovasc. Med. 2021, 8, 592362. [Google Scholar] [CrossRef] [PubMed]
- Abeyrathna, P.; Su, Y. The critical role of Akt in cardiovascular function. Vasc. Pharmacol. 2015, 74, 38–48. [Google Scholar] [CrossRef]
- Rolski, F.; Blyszczuk, P. Complexity of TNF-alfa signaling in heart disease. J. Clin. Med. 2020, 9, 3267. [Google Scholar] [CrossRef]
- Yarmohammadi, F.; Ebrahimian, Z.; Karimi, G. MicroRNAs target the PI3K/Akt/p53 and the Sirt1/Nrf2 signaling pathways in doxorubicin-induced cardiotoxicity. J. Biochem. Mol. Toxicol. 2023, 37, e23261. [Google Scholar] [CrossRef]
- Song, W.J.; Song, E.A.C.; Jung, M.S.; Choi, S.H.; Baik, H.H.; Jin, B.W.; Kim, J.H.; Chung, S.H. Phosphorylation and inactivation of Glycogen Sybthase Kinase 3beta by dual-specificity tyrosine phosphorylation-regulated kinase 1° (Dyrk1A). J. Biol. Chem. 2014, 290, 2321–2333. [Google Scholar] [CrossRef]
- Menon, B.; Johnson, J.N.; Ross, R.S.; Singh, M.; Singh, K. Glycogen synthase kinase-3beta plays a pro-apoptotic role in beta-adrenergic receptor-stimulated apoptosis in adult rat ventricular myocytes: Role of beta1 integrins. J. Mol. Cell Cardiol. 2006, 42, 653–661. [Google Scholar] [CrossRef]
- Wang, S.; Venkatraman, V.; Crowgey, E.; Liu, T.; Fu, Z.; Holewinski, R.; Eyk, J. Proteins S-nitrosylation controls glycogen synthase kinase 3β function independent of its phosphorylation state. Circ. Res. 2018, 122, 1517–1531. [Google Scholar] [CrossRef]
- Hirotani, S.; Zhai, P.; Tomita, H.; Galeotti, J.; Marquez, J.P.; Gao, S.; Hong, C.; Yatani, A.; Avila, J.; Sadoshima, J. Inhibition of glycogen synthase kinase 3beta during heart failure is protective. Circ. Res. 2007, 101, 1164–1174. [Google Scholar] [CrossRef] [PubMed]
- Haq, S.; Choukroun, G.; Kang, Z.B.; Ranu, H.; Matsui, T.; Rosenzweig, A.; Molketntin, J.D.; Alessandrini, A.; Woodgett, J. Glycogen Synthase kinase-3beta is a negative regulator of cardiomyocyte hypertrophy. J. Cell Biol. 2000, 151, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Tariq, U.; Uppulapu, S.; Banerjee, S. Role of gsk-3 in cardiac health: Focusing on cardiac remodeling and heart failure. Curr. Drug Targets 2021, 22, 1568–1576. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Chen, Z.; Gao, J.; Shi, W.; Li, L.; Jiang, S.; Hu, H.; Liu, Z.; Xu, D.; Wu, L. The key roles of GSK-3beta in regulating mitochondrial activity. Cell Physiol. Biochem. 2017, 44, 1445–1459. [Google Scholar] [CrossRef]
- Zhou, H.L.; Stomberski, C.T.; Stamler, J.S. Cross talk between S-nitrosylation and phosphorylation involving kinases and nitrosylases. Cric Res. 2018, 122, 1485–1487. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Deng, Y.; Yang, X.; Xue, H.; Lang, Y. The relationship between protein s-nytrosylation and human diseases: A review. Neurochem. Res. 2020, 45, 2815–2827. [Google Scholar] [CrossRef]
- Cortes-Vieyra, R.; Silva-Garcia, O.; Gomez-Garcia, A.; Gutierrez-castellanos, S.; Alvarez-Aguillar, C. Glycogen synthase kinase ebeta modulates the inflammatory response activated by bacteria, viruses, parasites. Front. Immunol. 2021, 12, 675751. [Google Scholar] [CrossRef]
- Ding, L.; Liou, G.Y.; Schmitt, D.M.; Storz, P.; Zhang, J.S.; Billadeau, P.D. Glygogen synthase kinase-3beta ablation limits pancreatitis-induced acino-ductal metaplasia. J. Pathol. 2017, 243, 65–77. [Google Scholar] [CrossRef]
- Umbakar, P.; Tousif, S.; Singh, A.P.; Anderson, J.C.; Zhang, Q.; Tallquist, N.D.; Woodgett, J.; Lal, H. Fibroblast GSK-3alfa promotes fibrosis via RAF-MEK-ERK pathway in the injured heart. Circ. Res. 2022, 131, 620–636. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.; Tang, R.; Liu, H.; Li, X.; Lei, W.; Chen, J.; Jin, Z.; Tang, J.; Wang, Z.; et al. Glycogen synthase kinase-3beta: A multifaceted player in ischemia-reperfusion injury and its therapeutic prospects. J. Cell Physiol. 2024, 239, e31335. [Google Scholar] [CrossRef]
- Nakamura, M.; Liu, T.; Husain, S.; Zhai, P.; Warren, J.S.; HSU, C.P.; Matsuda, T.; Phiel, C.J.; Cox, J.E. Glycogen synthasr kinase-3alfa promotes fatty acid uptake and lipotoxic cardiomyopathy. Cell Metab. 2019, 29, 1119–1134. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Woodgett, J.R. Emerging role of GSK-3alfa in pathophysiology: Emphasis on cardio-metabolic disorders. Biochim. Biophys. Acta Mol. Cell. Res. 2020, 1867, 118616. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Sharma, S. Mechanisms og sphingosine-1-phosphate induced cardioprotectionagainst I/R injury in diabetic rat heart: Possible involment of glygogen synthase kinase 3beta and mitochondrial permeability transition pore. Clin. Exp. Pharmacol. Physiol. 2016, 43, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Marosi, M.; Arman, P.; Acet, G.; D’Ascenzio, M.; Laezza, M. Glycogen synthase kinase-3: Ion channels, plasticity, diseases. Int. J. Mol. Sci. 2022, 23, 4413. [Google Scholar] [CrossRef]
- Popov, S.V.; Mukhomedzyanov, A.V.; Voronkov, N.S.; Derkachev, I.A.; Boshchenko, A.A. Regulation of autophagy of the heart in ischemia abd reperfusion. Apoptosis 2023, 28, 55–80. [Google Scholar] [CrossRef]
- Li, C.W.; Lim, S.O.; Xia, W.; Lee, H.H.; Chan, L.C.; Kuo, C.W.; Khoo, K.H.; Chang, S.S.; Cha, J.H. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Common. 2016, 7, 12632. [Google Scholar] [CrossRef]
- Magaye, R.M.; Savira, F.; Hua, Y.; Xiong, X.; Huang, L.; Reid, C.; Flynn, B.L.; Kaye, D.; Liew, D.; Wang, B.H. Attenuating PI3K/Akt-mTOR pathway reduces dihydrosphingosine 1 phopshate mediated collagen synthesis and hypertrophy in primary cardiac cells. Int. J. Biochem. Cell Biol. 2021, 134, 105952. [Google Scholar] [CrossRef]
- Zeng, J.; Li, Y.; Ma, Z.; Hu, M. Advances in small molecules in cellular reprogramming: Effects, structures, and mechanisms. Curr. Stem Cell Res. Ther. 2021, 16, 115–132. [Google Scholar] [CrossRef]
- Zhang, Z.; Zou, Z.; Zhang, H.; Zhang, D. Regulatory network analysis based on integrated mirna-tf reveals key genes in heart failure. Sci. Rep. 2024, 14, 13896. [Google Scholar] [CrossRef]
- Gupta, C.; Kaur, J.; Tikoo, K. Regulation of MDA-MB-231 cell proliferation by GSK-3beta involves epigenetic modifications under high glucose conditions. Exp. Cell Res. 2014, 324, 75–83. [Google Scholar] [CrossRef]
- Boudina, S.; Abel, E.D. Diabetic cardiomyopathy revisited. Circulation 2007, 115, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.P. The role of JNK in diabetes and ischemic heart disease. Diabetologia 2014, 57, 885–896. [Google Scholar]
- Liu, Q.; Sargent, M.A.; York, A.J.; Molkentin, J.D. ASK1 regulates cardiomyocyte death but not hypertrophy in transgenic mice. Circ. Res. 2009, 105, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhao, Z.; Liu, J.; Huang, N.; Long, D.; Li, X.; Liu, Y. MEK/ERK and p38 MAPK regulate chondrogenesis of rat bone marrow mesenchymal stem cells through delicate interaction with TGF-β1/Smads pathway. Cell Prolif. 2010, 43, 333–343. [Google Scholar] [CrossRef]
- Ghigo, A.; Hirsch, E. A new role for PI3Kγ in cardiac remodeling: Bridging inflammation and contractile dysfunction. Cardiovasc. Res. 2014, 102, 238–239. [Google Scholar] [CrossRef]
- Riehle, C.; Abel, E.D. Insulin signaling and heart failure. Circ. Res. 2016, 118, 1151–1169. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, X.; Wang, S.; Zhao, Y.; Liu, X.; Ding, W. Inhibition of Akt exacerbates myocardial apoptosis in diabetic rats. J. Mol. Cell Cardiol. 2010, 49, 774–782. [Google Scholar] [CrossRef]
- Cuenda, A.; Rousseau, S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 2007, 1773, 1358–1375. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: A 10-year update. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [CrossRef]
- Sedighi, S.; Liu, T.; O’Meally, R.; Cole, R.; O’Rourke, B.; Foster, D. Inhibition of cardiac p38 highlights the role of the phosphoproteome in heart failure progression. bioRxiv 2024. [Google Scholar] [CrossRef]
- Chung, E.S.; Packer, M.; Lo, K.H.; Fasanmade, A.A.; Willerson, J.T. Randomized, double-blind, placebo-controlled, pilot trial of infliximab in patients with moderate-to-severe heart failure: Results of the ATTACH trial. Circulation 2003, 107, 3133–3140. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef] [PubMed]
- Noma, T.; Lemaire, A.; Naga Prasad, S.V.; Barki-Harrington, L.; Tilley, D.G.; Chen, J.; Le Corvoisier, P.; Violin, J.D.; Wei, H.; Lefkowitz, R.J.; et al. Beta-arrestin–mediated β1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J. Clin. Investig. 2007, 117, 2445–2458. [Google Scholar] [CrossRef] [PubMed]
- Patrucco, E.; Notte, A.; Barberis, L.; Selvetella, G.; Maffei, A.; Brancaccio, M.; Marengo, S.; Russo, G.; Azzolino, O.; Rybalkin, S.D.; et al. PI3Kγ modulates the cardiac response to chronic pressure overload by regulating β-adrenergic receptor internalization. Nat. Med. 2004, 10, 600–606. [Google Scholar]
- Aoyagi, T.; Matsui, T. Pharmacological inhibition of ERK1/2 signaling prevents cardiac fibrosis and dysfunction in pressure-overloaded mice. Hypertension 2012, 60, 660–667. [Google Scholar]
- Jo, H.; Mondal, S.; Tan, D.; Nagata, E.; Takizawa, S.; Sharma, A.K.; Hou, Q.; Shanmugasundaram, K.; Prasad, A.; Tung, J.K. Small molecute-induced cytosolic activation of protein kinase Akt rescues ischemia-elicited neuronal death. Proc. Natl. Acad. Sci. USA 2012, 109, 10581–10586. [Google Scholar] [CrossRef]
- Li, Z.; Yan, S.; Attayan, N.; Ramalingam, S.; Thiele, C.J. Combination of allosteric Akt inhibitor MK-2206 with etoposide or rapamycin enhances the antitumor growth effect in neuroblastoma. Clin. Cancer Res. 2012, 18, 3603–3615. [Google Scholar] [CrossRef]
- McMullen, J.R.; Sherwood, M.C.; Tarnavski, O.; Zhang, L.; Dorfman, A.L.; Shioi, T.; Izumo, S. Inhibition of mTOR signaling with rapamycin regresses establiahed cardiac hypertrophy induced by pressure overload. Circulation 2004, 109, 3050–3055. [Google Scholar] [CrossRef]
- Morello, F.; Santulli, G.; Baillie, G.S.; Damilano, F.; Dunlop, A.J.; Pawson, C.; Walchli, T. PI3Kγ mediates the cardioprotective effects of leukocytes in heart failure. Nat. Med. 2011, 17, 610–618. [Google Scholar]
- Zhao, L.; Vogt, P.K. Class I PI3K in oncogenic cellular transformation. Oncogene 2008, 27, 5486–5496. [Google Scholar] [CrossRef]
- Shende, P.; Plaisance, I.; Morandi, C.; Pentassuglia, L.; Heim, P.; Lebboukh, S.; Berthonneche, C.; Pedrazzini, T.; Kaufmann, B.A.; Hall, M.N.; et al. Cardiac mTOR complex 2 preserves ventricular function in pressure-overload hypertrophy. Cardiovasc. Res. 2016, 109, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Juhaszova, M.; Zorov, D.B.; Yaniv, Y.; Nuss, H.B.; Wang, S.; Sollott, S.J. Role of glycogen synthase kinase-3β in cardioprotection. Circ. Res. 2009, 104, 1240–1252. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H.; Wang, D.; Rhaleb, N.E.; Yang, X.-P.; Xu, J.; Sankey, S.S.; Rudolph, A.E.; Carretero, O.A. Inhibition of p38 mitogen-activated protein kinase protects the heart against cardiac remodeling in mice with heart failure resulting from myocardial infarction. J. Card. Fail. 2005, 11, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Jope, R.S.; Johnson, G.V. Glycogen synthase kinase-3 in neural and non-neural functions: Regulation and disease. Trends Biochem. Sci. 2004, 29, 95–102. [Google Scholar] [CrossRef]
- Gross, E.R.; Hsu, A.K.; Gross, G.J. Inhibition of glycogen synthase kinase-3β during reperfusion limits myocardial infarction in vivo. J. Cardiovasc. Pharmacol. 2004, 44, 547–552. [Google Scholar]
- Das, A.; Durrant, D.; Gude, N. Glycogen synthase kinase-3β inhibition attenuates mitochondrial dysfunction and apoptosis in hypertrophied heart during ischemia-reperfusion injury. Cardiovasc. Res. 2008, 77, 626–634. [Google Scholar]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3β: Regulation, actions, and diseases. Trends Pharmacol. Sci. 2015, 36, 36–46. [Google Scholar]
- Martinez, A.; Gil, C.; Perez, D.I. Glycogen synthase kinase-3β inhibitors: A patent review (2007–2010). Curr. Pharm. Des. 2011, 17, 357–364. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
| Pathway | Targeted Molecule | Mechanism of Action | Therapeutic Agents | Potential Benefit | Limitation/Risks |
|---|---|---|---|---|---|
| MAPK | p38, ERK 1/2, JNK | Inhibition of stress-activated kinases reduces inflammation, fibrosis, hypertrophy, and cardiomyocyte apoptosis | SB203580 (p38 inhibitor), SP600125 (JNK inhibitor), U0126 (ERK1/2 inhibitor) | Decreases maladaptive cardiac remodeling, improves ventricular function | Non-specific inhibition may impair physiological adaptation to stress |
| PI3K/Akt | PI3Kγ, PI3Kα, Akt1 | Activation enhances cardiomyocyte survival, mitochondrial metabolism, and contractility; reduces apoptosis | SC79 (Akt activator), MK-2206 (Akt inhibitor), IPI-549 (PI3Kγ inhibitor) | Improves energy efficiency and cell viability; supports adaptive hypertrophy | Oncogenic potential (especially PI3Kα); context- and dose-dependent responses |
| GSK-3β | GSK-3β | Inhibition prevents mitochondrial dysfunction, apoptosis, and adverse remodeling | SB216763, Tideglusib, Lithium chloride (LiCl) | Promotes cardioprotection, facilitates post-injury recovery | Chronic inhibition may impair metabolic homeostasis and increase fibrosis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meco, M.; Giustiniano, E.; Nisi, F.; Zulli, P.; Agosteo, E. MAPK, PI3K/Akt Pathways, and GSK-3β Activity in Severe Acute Heart Failure in Intensive Care Patients: An Updated Review. J. Cardiovasc. Dev. Dis. 2025, 12, 266. https://doi.org/10.3390/jcdd12070266
Meco M, Giustiniano E, Nisi F, Zulli P, Agosteo E. MAPK, PI3K/Akt Pathways, and GSK-3β Activity in Severe Acute Heart Failure in Intensive Care Patients: An Updated Review. Journal of Cardiovascular Development and Disease. 2025; 12(7):266. https://doi.org/10.3390/jcdd12070266
Chicago/Turabian StyleMeco, Massimo, Enrico Giustiniano, Fulvio Nisi, Pierluigi Zulli, and Emiliano Agosteo. 2025. "MAPK, PI3K/Akt Pathways, and GSK-3β Activity in Severe Acute Heart Failure in Intensive Care Patients: An Updated Review" Journal of Cardiovascular Development and Disease 12, no. 7: 266. https://doi.org/10.3390/jcdd12070266
APA StyleMeco, M., Giustiniano, E., Nisi, F., Zulli, P., & Agosteo, E. (2025). MAPK, PI3K/Akt Pathways, and GSK-3β Activity in Severe Acute Heart Failure in Intensive Care Patients: An Updated Review. Journal of Cardiovascular Development and Disease, 12(7), 266. https://doi.org/10.3390/jcdd12070266