
Dextran Sulphate Sodium Acute Colitis Rat Model: A Suitable Tool for Advancing Our Understanding of Immune and Microbial Mechanisms in the Pathogenesis of Inflammatory Bowel Disease

 , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Design
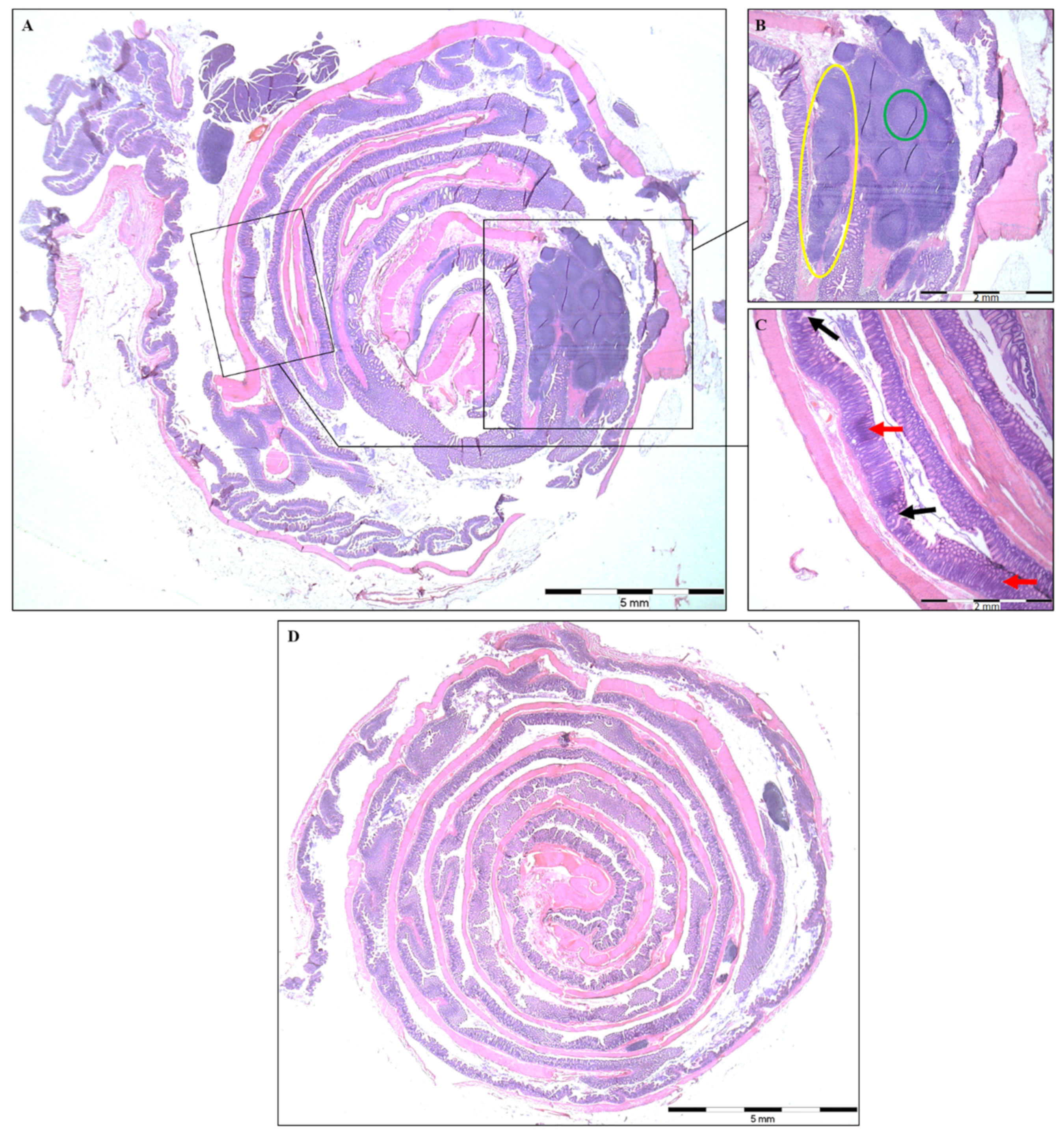
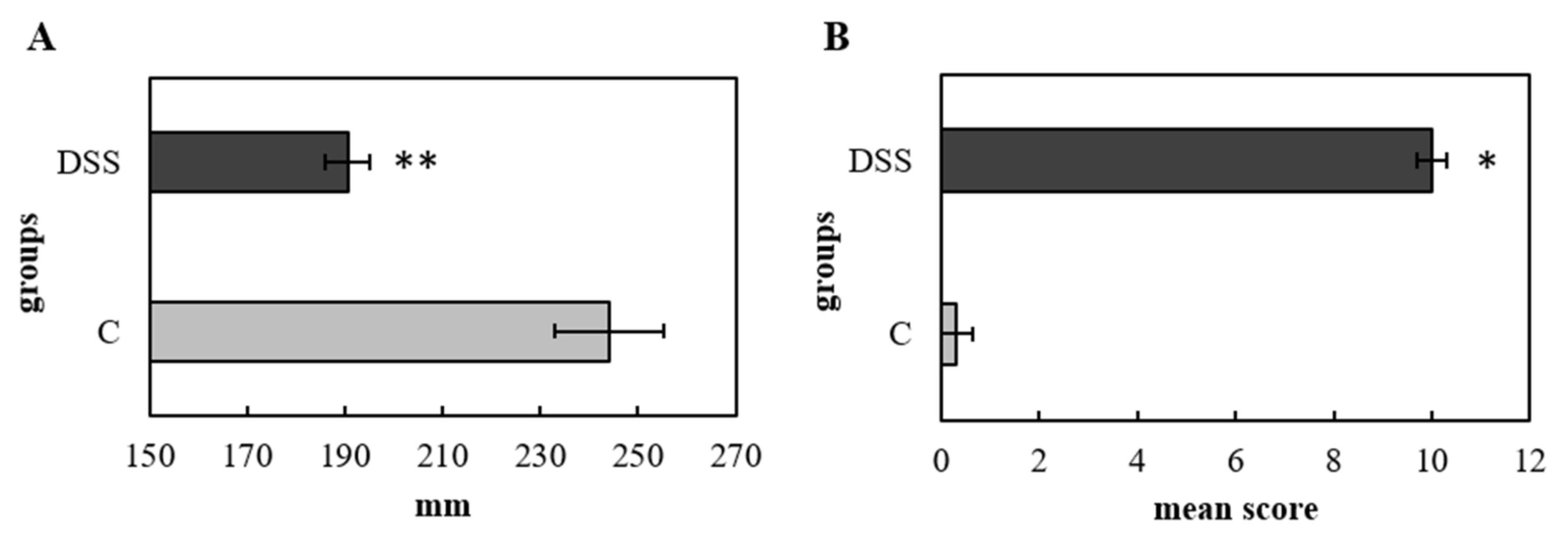
2.2. Histopathological Analysis and Colon Length
2.3. Blood and Serum Analysis
2.4. Multiplex Cytokine/Chemokine Profiling
2.5. Immunophenotyping by Flow Cytometry
2.6. NGS Analysis of Faecal Samples
2.7. Statistical Analysis
3. Results
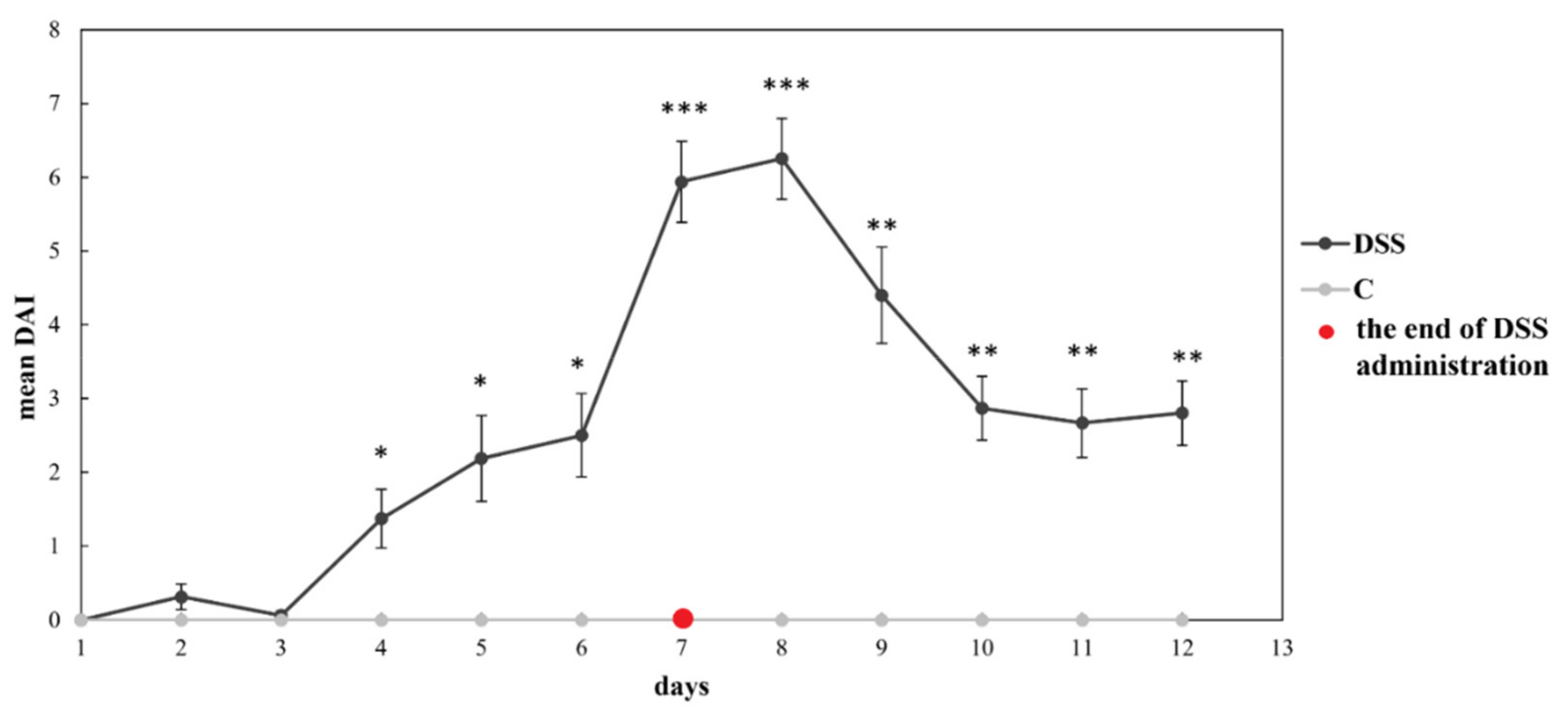
3.1. Disease Severity
3.2. Biochemical, Haematological and Plasma CRP Analyses
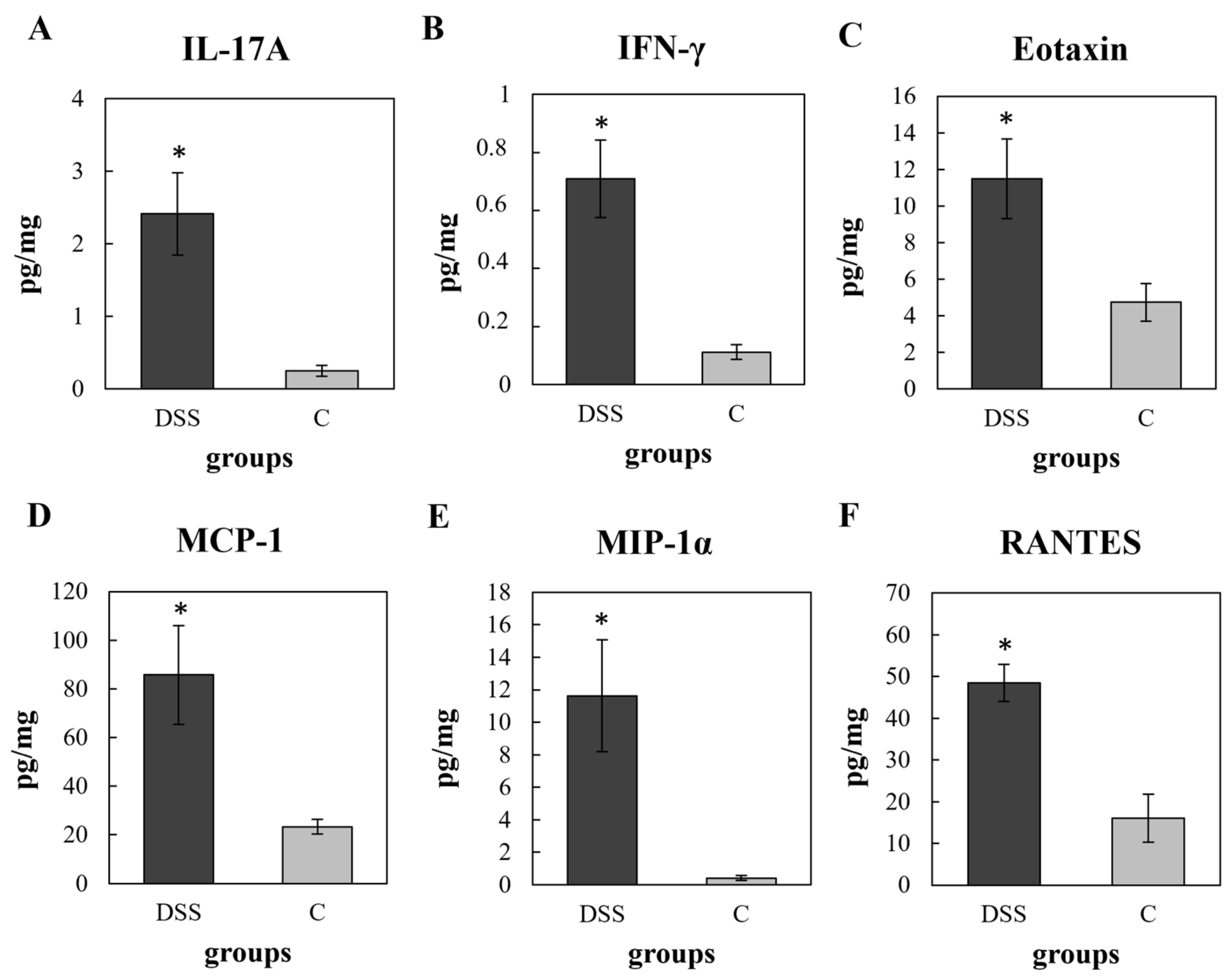
3.3. Multiplex Cytokine/Chemokine Profiling
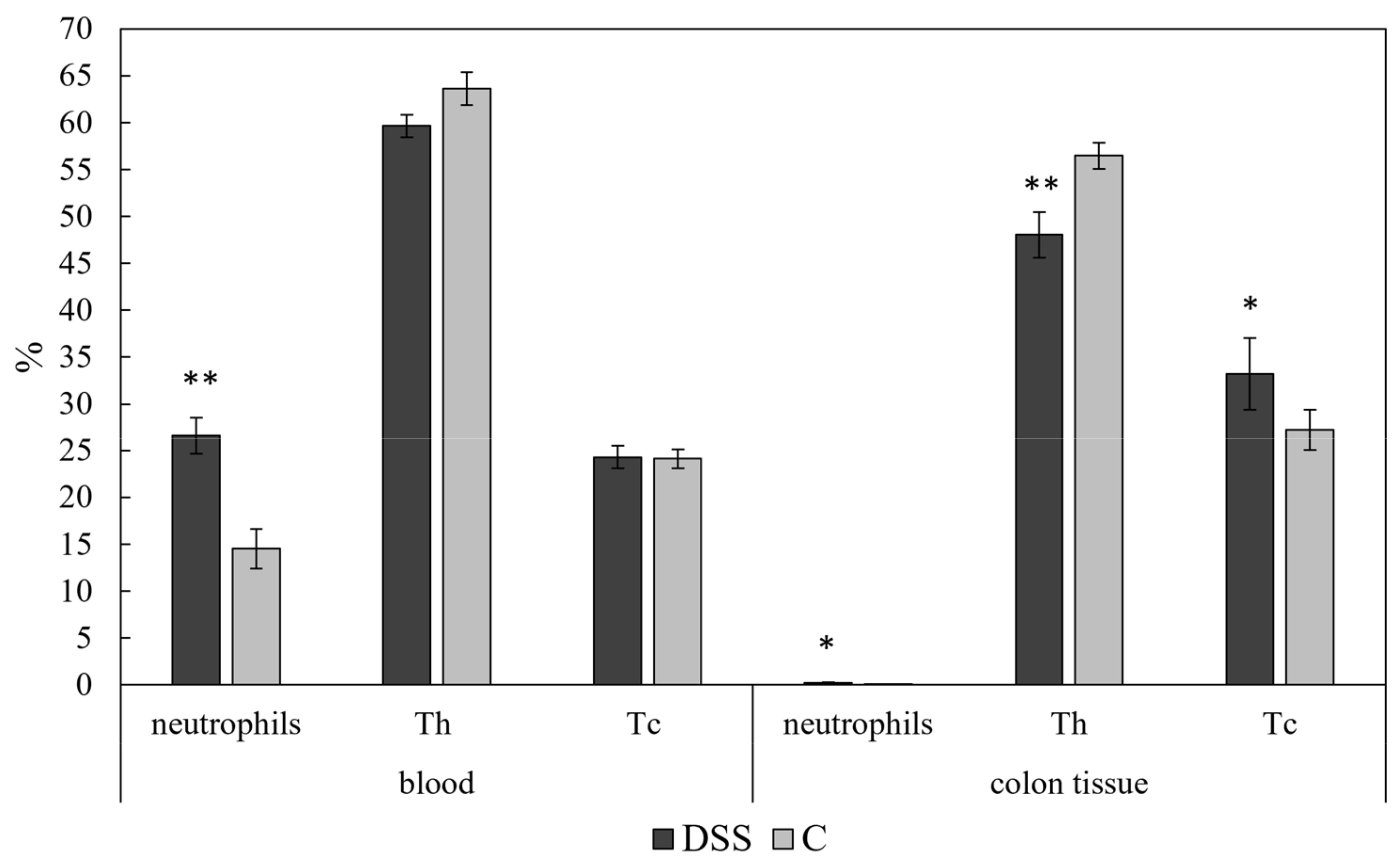
3.4. Flow Cytometric Analysis
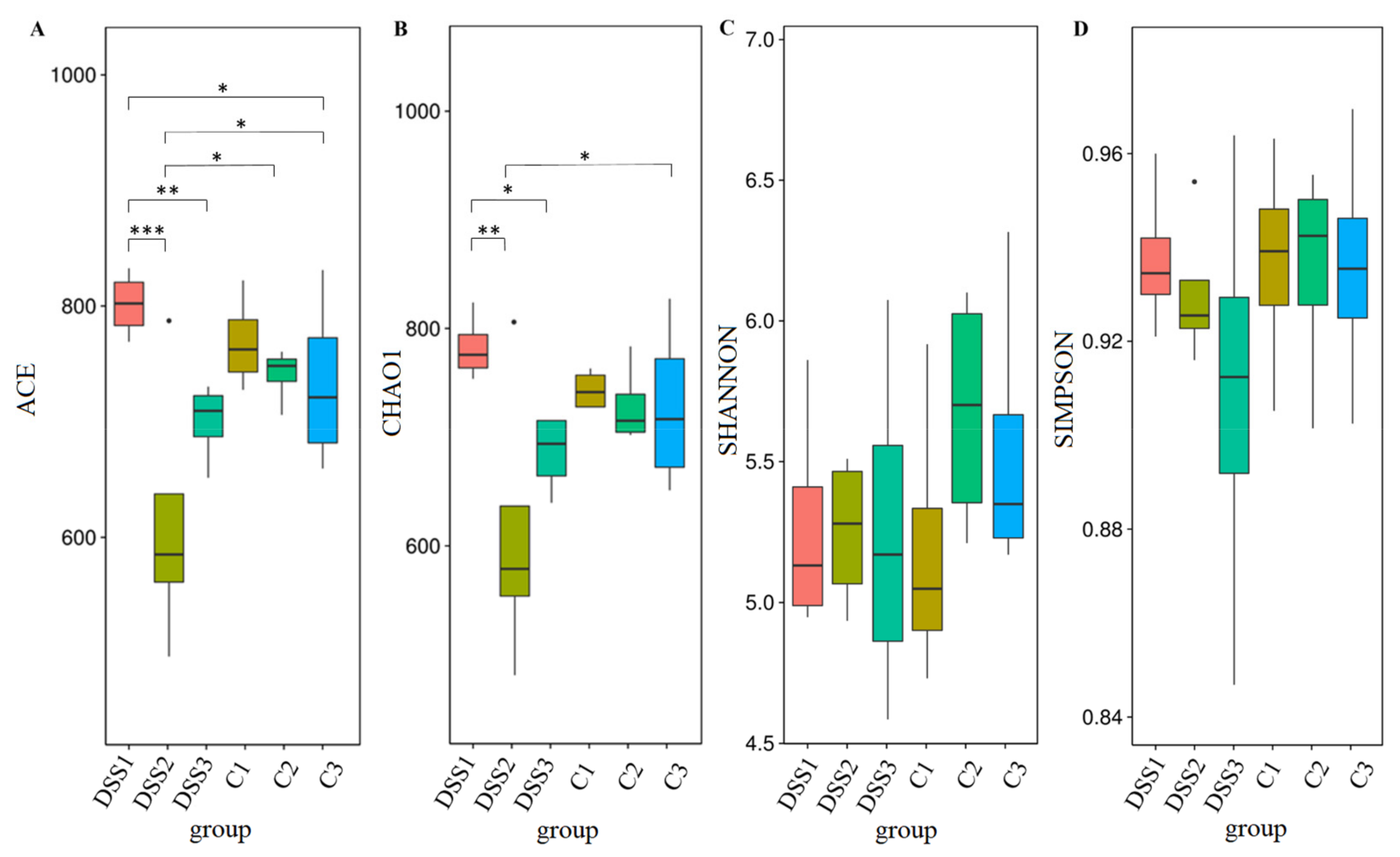
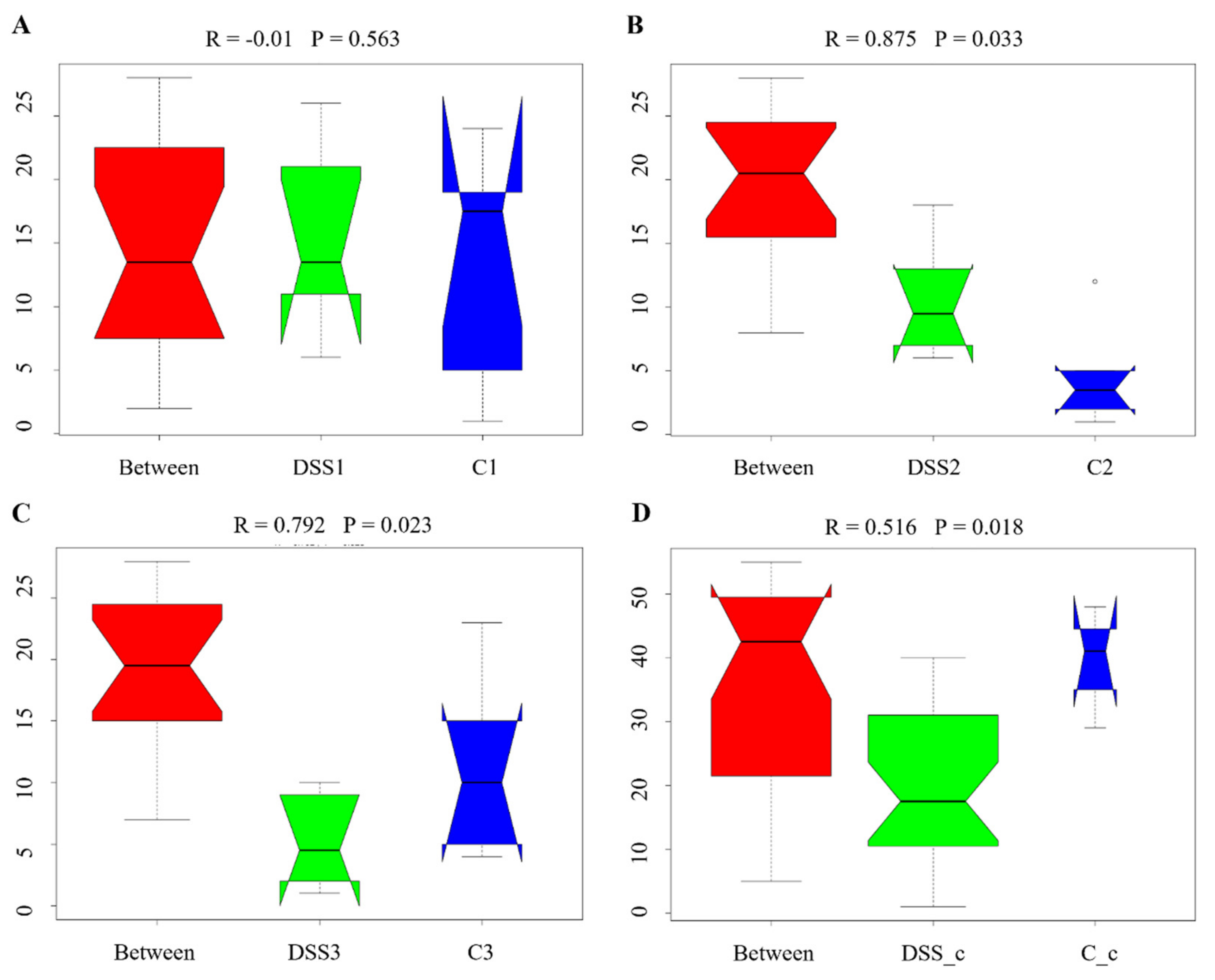
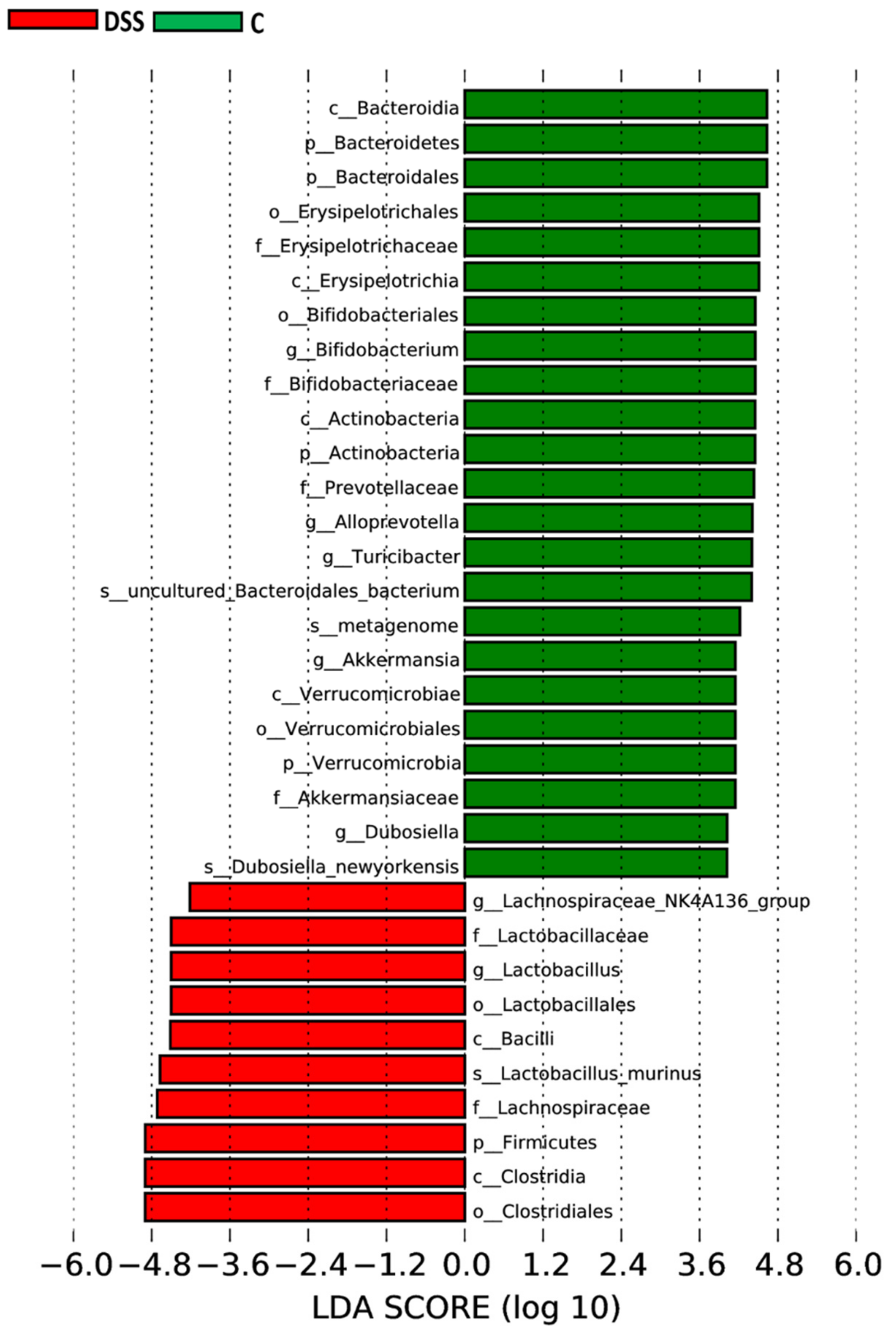
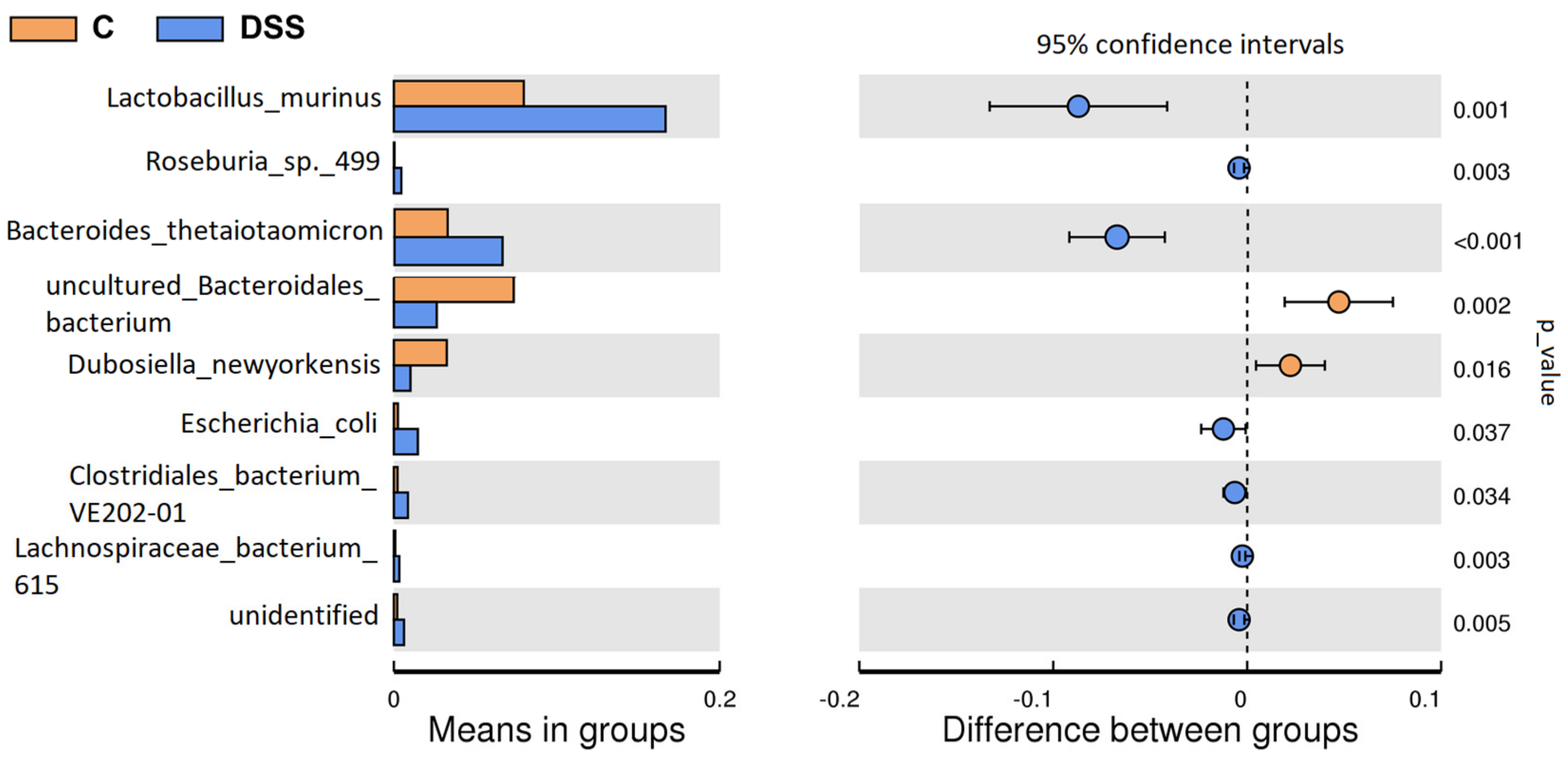
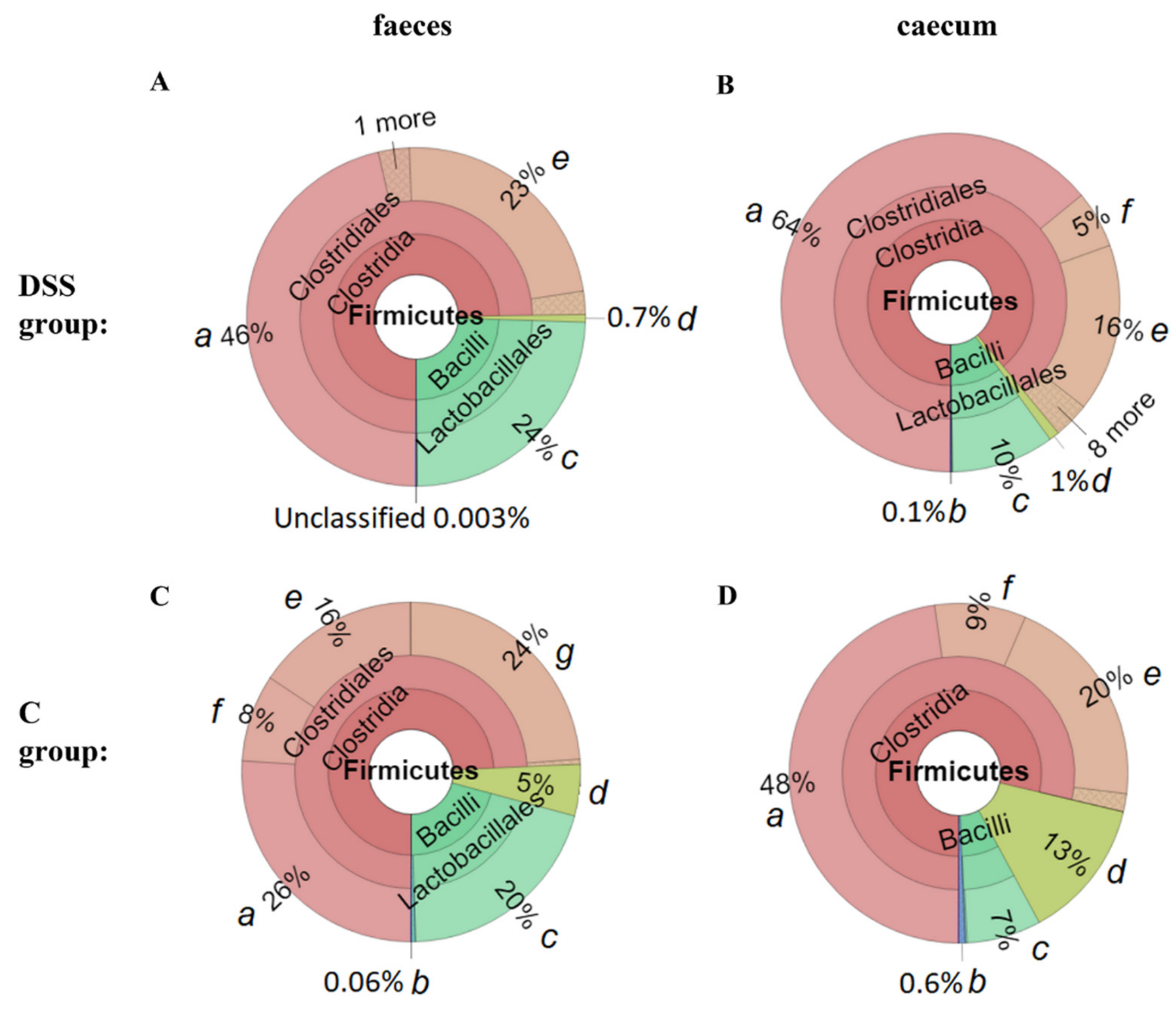
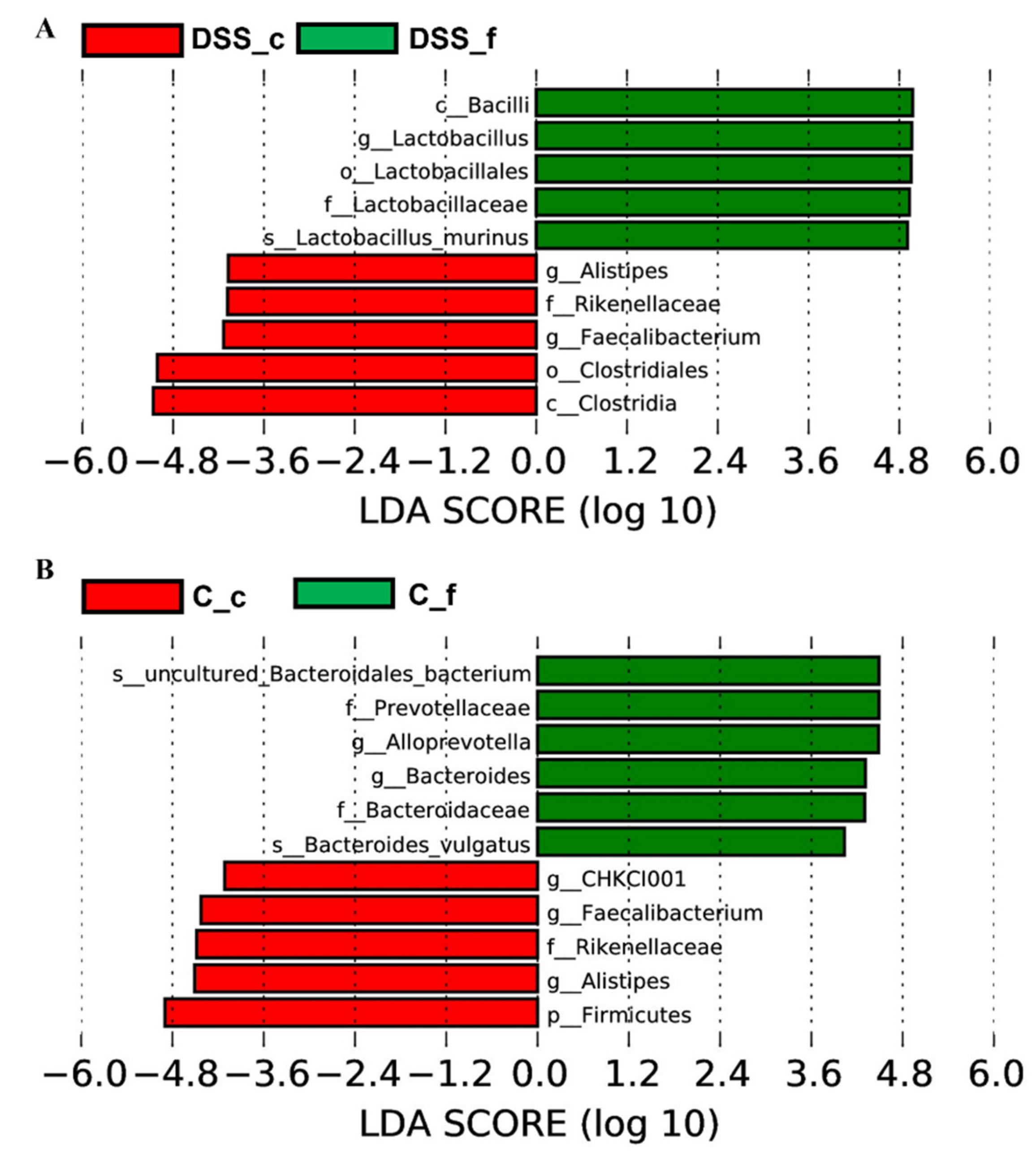
3.5. Microbiome Analysis of Rat Faecal Samples
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GBD 2017 Inflammatory Bowel Disease Collaborators. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990-2017: A systematic analysis of the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 17–30. [Google Scholar] [CrossRef]
- Ogura, Y.; Bonen, D.K.; Inohara, N.; Nicolae, D.L.; Chen, F.F.; Ramos, R.; Britton, H.; Moran, T.; Karaliuskas, R.; Duerr, R.H.; et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 2001, 411, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Harbord, M.; Annese, V.; Vavricka, S.R.; Allez, M.; Barreiro-de Acosta, M.; Boberg, K.M.; Burisch, J.; De Vos, M.; De Vries, A.M.; Dick, A.D.; et al. The First European Evidence-based Consensus on Extra-intestinal Manifestations in Inflammatory Bowel Disease. J. Crohns Colitis 2016, 10, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Elson, C.O.; Sartor, R.B.; Tennyson, G.S.; Riddell, R.H. Experimental models of inflammatory bowel disease. Gastroenterology 1995, 109, 1344–1367. [Google Scholar] [CrossRef]
- Okayasu, I.; Hatakeyama, S.; Yamada, M.; Ohkusa, T.; Inagaki, Y.; Nakaya, R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990, 98, 694–702. [Google Scholar] [CrossRef]
- Melgar, S.; Karlsson, A.; Michaëlsson, E. Acute colitis induced by dextran sulphate, sodium progresses to chronicity in C57BL/6 but not in BALB/c mice: Correlation between symptoms and inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G1328–G1338. [Google Scholar] [CrossRef]
- Mähler, M.; Bristol, I.J.; Leiter, E.H.; Workman, A.E.; Birkenmeier, E.H.; Elson, C.O.; Sundberg, J.P. Differential susceptibility of inbred mouse strains to dextran sulphate sodium-induced colitis. Am. J. Physiol. 1998, 274, G544–G551. [Google Scholar] [CrossRef]
- Gaudio, E.; Taddei, G.; Vetuschi, A.; Sferra, R.; Frieri, G.; Ricciardi, G.; Caprilli, R. Dextran sulphate sodium (DSS) colitis in rats: Clinical, structural, and ultrastructural aspects. Dig. Dis. Sci. 1999, 44, 1458–1475. [Google Scholar] [CrossRef]
- Kiesler, P.; Fuss, I.J.; Strober, W. Experimental Models of Inflammatory Bowel Diseases. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 154–170. [Google Scholar] [CrossRef]
- Axelsson, L.G.; Landström, E.; Goldschmidt, T.J.; Grönberg, A.; Bylund-Fellenius, A.C. Dextran sulphate sodium (DSS) induced experimental colitis in immunodeficient mice: Effects on CD4 (+) -cell depleted, ethnic and NK-cell depleted SCID mice. Inflamm. Res. 1996, 45, 181–191. [Google Scholar] [CrossRef]
- Martin, J.C.; Bériou, G.; Josien, R. Dextran Sulphate Sodium (DSS)-Induced Acute Colitis in the Rat. Methods Mol. Biol. 2016, 1371, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Breider, M.A.; Eppinger, M.; Gough, A. Intercellular adhesion molecule-1 expression in dextran sodium sulphate-induced colitis in rats. Vet. Pathol. 1997, 34, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Iwai, A.; Itoh, K.; Tanaka, M.; Kato, S.; Hokari, R.; Miyahara, T.; Koyama, H.; Miura, S.; Kobayashi, M. Role of inducible nitric oxide synthase in dextran sulphate sodium-induced colitis. Aliment. Pharmacol. Ther. 2000, 14, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Osman, N.; Adawi, D.; Ahrne, S.; Jeppsson, B.; Molin, G. Modulation of the effect of dextran sulphate sodium-induced acute colitis by the administration of different probiotic strains of Lactobacillus and Bifidobacterium. Dig. Dis. Sci. 2004, 49, 320–327. [Google Scholar] [CrossRef]
- Mizuta, Y.; Isomoto, H.; Takahashi, T. Impaired nitrergic innervation in rat colitis induced by dextran sulphate sodium. Gastroenterology 2000, 118, 714–723. [Google Scholar] [CrossRef]
- Wang, F.; Zhao, H.Y.; Zhang, S.T.; Gong, Y.Z.; Zhang, H.F.; Zhang, C. Effect of enteral nutrition on dextran sulphate sodium-induced colitis in rats. J. Dig. Dis. 2011, 12, 453–458. [Google Scholar] [CrossRef]
- Johansson, M.E.; Gustafsson, J.K.; Holmén-Larsson, J.; Jabbar, K.S.; Xia, L.; Xu, H.; Ghishan, F.K.; Carvalho, F.A.; Gewirtz, A.T.; Sjövall, H.; et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut 2014, 63, 281–291. [Google Scholar] [CrossRef]
- Morgan, M.E.; Zheng, B.; Koelink, P.J.; van de Kant, H.J.; Haazen, L.C.; van Roest, M.; Garssen, J.; Folkerts, G.; Kraneveld, A.D. New perspective on dextran sodium sulphate colitis: Antigen-specific T cell development during intestinal inflammation. PLoS ONE 2013, 8, e69936. [Google Scholar] [CrossRef]
- Nishida, A.; Inoue, R.; Inatomi, O.; Bamba, S.; Naito, Y.; And oh, A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin. J. Gastroenterol. 2018, 11, 1–10. [Google Scholar] [CrossRef]
- Andrews, C.N.; Griffiths, T.A.; Kaufman, J.; Vergnolle, N.; Surette, M.G.; Rioux, K.P. Mesalazine (5-aminosalicylic acid) alters faecal bacterial profiles, but not mucosal proteolytic activity in diarrhoea-predominant irritable bowel syndrome. Aliment. Pharmacol. Ther. 2011, 34, 374–383. [Google Scholar] [CrossRef]
- Wills, E.S.; Jonkers, D.M.; Savelkoul, P.H.; Masclee, A.A.; Pierik, M.J.; Penders, J. Fecal microbial composition of ulcerative colitis and Crohn’s disease patients in remission and subsequent exacerbation. PLoS ONE 2014, 9, e90981. [Google Scholar] [CrossRef] [PubMed]
- Adamkova, P.; Hradicka, P.; Gancarcikova, S.; Kassayova, M.; Ambro, L.; Bertkova, I.; Maronek, M.; Farkasova Iannaccone, S.; Demeckova, V. Single Donor FMT Reverses Microbial/Immune Dysbiosis and Induces Clinical Remission in a Rat Model of Acute Colitis. Pathogens 2021, 10, 152. [Google Scholar] [CrossRef] [PubMed]
- Stucchi, A.F.; Shofer, S.; Leeman, S.; Materne, O.; Beer, E.; McClung, J.; Shebani, K.; Moore, F.; O’Brien, M.; Becker, J.M. NK-1 antagonist reduces colonic inflammation and oxidative stress in dextran sulphate-induced colitis in rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G1298–G1306. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.J.; Künzli, B.M.; A-Rahim, Y.I.; Sevigny, J.; Berberat, P.O.; Enjyoji, K.; Csizmadia, E.; Friess, H.; Robson, S.C. From the Cover: CD39 deletion exacerbates experimental murine colitis and human polymorphisms increase susceptibility to inflammatory bowel disease. Proc. Natl. Acad. Sci. USA 2009, 106, 16788–16793. [Google Scholar] [CrossRef]
- Katakura, K.; Lee, J.; Rachmilewitz, D.; Li, G.; Eckmann, L.; Raz, E. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J. Clin. Investig. 2005, 115, 695–702. [Google Scholar] [CrossRef]
- Kupcova Skalnikova, H.; Vodickova Kepkova, K.; Vodicka, P. Luminex xMAP Assays to Quantify Cytokines in Cancer Patient Serum. Methods Mol. Biol. 2020, 2108, 65–88. [Google Scholar] [CrossRef]
- Bayne, L.J.; Vonderheide, R.H. Multicolor flow cytometric analysis of immune cell subsets in tumor-bearing mice. Cold Spring Harb. Protoc. 2013, 2013, 955–960. [Google Scholar] [CrossRef]
- Sproston, N.R.; Ashworth, J.J. Role of C-Reactive Protein at Sites of Inflammation and Infection. Front. Immunol. 2018, 9, 754. [Google Scholar] [CrossRef]
- Low, D.; Nguyen, D.D.; Mizoguchi, E. Animal models of ulcerative colitis and their application in drug research. Drug Des. Dev. Ther. 2013, 7, 1341–1357. [Google Scholar] [CrossRef]
- Munyaka, P.M.; Rabbi, M.F.; Khafipour, E.; Ghia, J.E. Acute dextran sulphate sodium (DSS)-induced colitis promotes gut microbial dysbiosis in mice. J. Basic Microbiol. 2016, 56, 986–998. [Google Scholar] [CrossRef]
- Cooper, H.S.; Murthy, S.N.; Shah, R.S.; Sedergran, D.J. Clinicopathologic study of dextran sulphate, sodium experimental murine colitis. Lab. Investig. 1993, 69, 238–249. [Google Scholar] [PubMed]
- Kim, J.J.; Shajib, M.S.; Manocha, M.M.; Khan, W.I. Investigating intestinal inflammation in DSS-induced model of IBD. J. Vis. Exp. 2012, 60, e3678. [Google Scholar] [CrossRef] [PubMed]
- Wéra, O.; Lancellotti, P.; Oury, C. The Dual Role of Neutrophils in Inflammatory Bowel Diseases. J. Clin. Med. 2016, 5, 118. [Google Scholar] [CrossRef] [PubMed]
- Daniluk, J.; Daniluk, U.; Reszec, J.; Rusak, M.; Dabrowska, M.; Dabrowski, A. The protective effect of cigarette smoke on the course of dextran sulphate sodium-induced colitis are accompanied by lymphocyte subpopulation changes in the blood and colon. Int. J. Colorectal Dis. 2017, 32, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.Y.; Zhao, H.M.; Zhao, N.; Lu, C.; Lu, A.P. Effect of Bawei Xilei powder on CD3, CD4, CD8 T-lymphocytes of rats with ulcerative colitis. Zhongguo Zhong Yao Za Zhi 2008, 33, 1301–1304. [Google Scholar] [PubMed]
- Neil, G.A.; Summers, R.W.; Cheyne, B.A.; Carpenter, C.; Huang, W.L.; Waldschmidt, T.J. Analysis of T-lymphocyte subpopulations in inflammatory bowel diseases by three-color flow cytometry. Dig. Dis. Sci. 1994, 39, 1900–1908. [Google Scholar] [CrossRef]
- Ray, K. Deciphering the role of CD8 + T cells in IBD: From single-cell analysis of biomarkers. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 595. [Google Scholar] [CrossRef]
- Nancey, S.; Holvöet, S.; Graber, I.; Joubert, G.; Philippe, D.; Martin, S.; Nicolas, J.F.; Desreumaux, P.; Flourié, B.; Kaiserlian, D. CD8+ cytotoxic T cells induce relapsing colitis in normal mice. Gastroenterology 2006, 131, 485–496. [Google Scholar] [CrossRef]
- Nikolaus, S.; Schreiber, S. Diagnostics of inflammatory bowel disease. Gastroenterology 2007, 133, 1670–1689. [Google Scholar] [CrossRef]
- Thayer, W.R.; Charland, C.; Field, C.E. The subpopulations of circulating white blood cells in inflammatory bowel disease. Gastroenterology 1976, 71, 379–384. [Google Scholar] [CrossRef]
- Zhou, G.X.; Liu, Z.J. Potential roles of neutrophils in regulating intestinal mucosal inflammation of inflammatory bowel disease. J. Dig. Dis. 2017, 18, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Fournier, B.M.; Parkos, C.A. The role of neutrophils during intestinal inflammation. Mucosal Immunol. 2012, 5, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Kakuta, S.; Shimizu, K.; Kadoki, M.; Kamiya, T.; Shimazu, T.; Kubo, S.; Saijo, S.; Ishigame, H.; Nakae, S.; et al. Suppression of IL-17F, but not of IL-17A, provides protection against colitis by inducing T. Nat. Immunol. 2018, 19, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Uhlig, H.H.; Powrie, F. Dendritic cells and the intestinal bacterial flora: A role for localized mucosal immune responses. J. Clin. Investig. 2003, 112, 648–651. [Google Scholar] [CrossRef]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef]
- Shea-Donohue, T.; Thomas, K.; Cody, M.J.; Zhao, A.; Detolla, L.J.; Kopydlowski, K.M.; Fukata, M.; Lira, S.A.; Vogel, S.N. Mice deficient in the CXCR2 ligand, CXCL1 (KC/GRO-alpha), exhibit increased susceptibility to dextran sodium sulphate (DSS) -induced colitis. Innate Immun. 2008, 14, 117–124. [Google Scholar] [CrossRef]
- Laan, M.; Cui, Z.H.; Hoshino, H.; Lötvall, J.; Sjöstrand, M.; Gruenert, D.C.; Skoogh, B.E.; Lindén, A. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J. Immunol. 1999, 162, 2347–2352. [Google Scholar]
- Ferretti, S.; Bonneau, O.; Dubois, G.R.; Jones, C.E.; Trifilieff, A. IL-17, produced by lymphocytes and neutrophils, is necessary for lipopolysaccharide-induced airway neutrophilia: IL-15 as a possible trigger. J. Immunol. 2003, 170, 2106–2112. [Google Scholar] [CrossRef]
- Soehnlein, O.; Lindbom, L. Phagocyte partnership during the onset and resolution of inflammation. Nat. Rev. Immunol. 2010, 10, 427–439. [Google Scholar] [CrossRef]
- Cherfane, C.E.; Gessel, L.; Cirillo, D.; Zimmerman, M.B.; Polyak, S. Monocytosis and a Low Lymphocyte to Monocyte Ratio Are Effective Biomarkers of Ulcerative Colitis Disease Activity. Inflamm. Bowel. Dis. 2015, 21, 1769–1775. [Google Scholar] [CrossRef]
- Banks, C.; Bateman, A.; Payne, R.; Johnson, P.; Sheron, N. Chemokine expression in IBD. The mucosal chemokine expression is unselectively increased in both ulcerative colitis and Crohn’s disease. J. Pathol. 2003, 199, 28–35. [Google Scholar] [CrossRef] [PubMed]
- McCormack, G.; Moriarty, D.; O’Donoghue, D.P.; McCormick, P.A.; Sheahan, K.; Baird, A.W. Tissue cytokine and chemokine expression in inflammatory bowel disease. Inflamm. Res. 2001, 50, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Puleston, J.; Cooper, M.; Murch, S.; Bid, K.; Makh, S.; Ashwood, P.; Bingham, A.H.; Green, H.; Moss, P.; Dhillon, A.; et al. A distinct subset of chemokines dominates the mucosal chemokine response in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2005, 21, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.F.; Lai, P.S.; Yue, G.; Yin, K.; Nagele, R.G.; Tong, D.M.; Krzesicki, R.F.; Chin, J.E.; Wong, P.Y. Pattern of cytokine and adhesion molecule mRNA in hapten-induced relapsing colon inflammation in the rat. Inflammation 2001, 25, 33–45. [Google Scholar] [CrossRef]
- Luther, S.A.; Cyster, J.G. Chemokines as regulators of T cell differentiation. Nat. Immunol. 2001, 2, 102–107. [Google Scholar] [CrossRef]
- Ka, M.B.; Daumas, A.; Textoris, J.; Mege, J.L. Phenotypic diversity and emerging new tools to study macrophage activation in bacterial infectious diseases. Front Immunol. 2014, 5, 500. [Google Scholar] [CrossRef]
- Ahrens, R.; Waddell, A.; Seidu, L.; Blanchard, C.; Carey, R.; Forbes, E.; Lampinen, M.; Wilson, T.; Cohen, E.; Stringer, K.; et al. Intestinal macrophage/epithelial cell-derived CCL11/eotaxin-1 mediates eosinophil recruitment and function in pediatric ulcerative colitis. J. Immunol. 2008, 181, 7390–7399. [Google Scholar] [CrossRef]
- Kamada, N.; Hisamatsu, T.; Okamoto, S.; Chinen, H.; Kobayashi, T.; Sato, T.; Sakuraba, A.; Kitazume, M.T.; Sugita, A.; Koganei, K.; et al. Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-gamma axis. J. Clin. Investig. 2008, 118, 2269–2280. [Google Scholar] [CrossRef]
- Lissner, D.; Schumann, M.; Batra, A.; Kredel, L.I.; Kühl, A.A.; Erben, U.; May, C.; Schulzke, J.D.; Siegmund, B. Monocyte and M1 Macrophage-induced Barrier Defect Contributes to Chronic Intestinal Inflammation in IBD. Inflamm. Bowel Dis. 2015, 21, 1297–1305. [Google Scholar] [CrossRef]
- Barman, S.; Kayama, H.; Okuzaki, D.; Ogino, T.; Osawa, H.; Matsuno, H.; Mizushima, T.; Mori, M.; Nishimura, J.; Takeda, K. Identification of a human intestinal myeloid cell subset that regulates gut homeostasis. Int. Immunol. 2016, 28, 533–545. [Google Scholar] [CrossRef]
- Friedrich, M.; Pohin, M.; Powrie, F. Cytokine Networks in the Pathophysiology of Inflammatory Bowel Disease. Immunity 2019, 50, 992–1006. [Google Scholar] [CrossRef] [PubMed]
- Ajuebor, M.N.; Hogaboam, C.M.; Kunkel, S.L.; Proudfoot, A.E.; Wallace, J.L. The chemokine RANTES is a crucial mediator of the progression from acute to chronic colitis in the rat. J. Immunol. 2001, 166, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Ansari, N.; Abdulla, J.; Zayyani, N.; Brahmi, U.; Taha, S.; Satir, A.A. Comparison of RANTES expression in Crohn's disease and ulcerative colitis: An aid in the differential diagnosis? J. Clin. Pathol. 2006, 59, 1066–1072. [Google Scholar] [CrossRef] [PubMed]
- Schrödl, W.; Büchler, R.; Wendler, S.; Reinhold, P.; Muckova, P.; Reindl, J.; Rhode, H. Acute phase proteins as promising biomarkers: Perspectives and limitations of human and veterinary medicine. Proteom. Clin. Appl. 2016, 10, 1077–1092. [Google Scholar] [CrossRef]
- Diaz Padilla, N.; Bleeker, W.K.; Lubbers, Y.; Rigter, G.M.; Van Mierlo, G.J.; Daha, M.R.; Hack, C.E. Rat C-reactive protein activates the autologous complement system. Immunology 2003, 109, 564–571. [Google Scholar] [CrossRef]
- Żyła, E.; Dziendzikowska, K.; Kamola, D.; Wilczak, J.; Sapierzyński, R.; Harasym, J.; Gromadzka-Ostrowska, J. Anti-Inflammatory Activity of Oat Beta-Glucans in a Crohn’s Disease Model: Time- and Molar Mass-Dependent Effects. Int. J. Mol. Sci. 2021, 22, 4485. [Google Scholar] [CrossRef]
- Kirdak, T.; Uysal, E.; Sezgin, E.; Cecen, G.S.; Cavun, S. Inflammatory response markers in rats undergoing abdominal surgical procedures. Ann. Gastroenterol. 2020, 33, 528–535. [Google Scholar] [CrossRef]
- Costa, C.A.; Tanimoto, A.; Quaglio, A.E.; Almeida, L.D.; Severi, J.A.; Di Stasi, L.C. Anti-inflammatory effects of Brazilian ginseng (Pfaffia paniculata) on TNBS-induced intestinal inflammation: Experimental evidence. Int. Immunopharmacol. 2015, 28, 459–469. [Google Scholar] [CrossRef]
- Mitaka, C. Clinical laboratory differentiation of infected versus non-infectious systemic inflammatory response syndrome. Clin. Chim. Acta 2005, 351, 17–29. [Google Scholar] [CrossRef]
- Ridker, P.M. Clinical application of C-reactive protein for cardiovascular disease detection and prevention. Circulation 2003, 107, 363–369. [Google Scholar] [CrossRef]
- Mürüvvet, A.; Pınar, A.U. Acute Phase Protein Levels in Rats with Experimentally Induced Infectious and Noninfectious Inflammation. J. Dairy Vet. Sci. 2017, 4, 555642. [Google Scholar] [CrossRef]
- Vavricka, S.R.; Schoepfer, A.; Scharl, M.; Lakatos, P.L.; Navarini, A.; Rogler, G. Extraintestinal Manifestations of Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 1982–1992. [Google Scholar] [CrossRef]
- Kim, S.H.; Lee, W.; Kwon, D.; Lee, S.; Son, S.W.; Seo, M.S.; Kim, K.S.; Lee, Y.H.; Kim, S.; Jung, Y.S. Metabolomic Analysis of the Liver of a Dextran Sodium Sulphate-Induced Acute Colitis Mouse Model: Implications of the Gut-Liver Connection. Cells 2020, 9, 341. [Google Scholar] [CrossRef]
- Mehta, R.L.; Kellum, J.A.; Shah, S.V.; Molitoris, B.A.; Ronco, C.; Warnock, D.G.; Levin, A.; Network, A.K.I. Acute Kidney Injury Network: Report of an initiative to improve outcomes in acute kidney injury. Crit. Care 2007, 11, R31. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, N.J.; Wolfe, G.I. Asymptomatic/pause-symptomatic creatine kinase elevations (hyperckemia). Muscle Nerve 2013, 47, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Herrlinger, K.R.; Noftz, M.K.; Fellermann, K.; Schmidt, K.; Steinhoff, J.; Stange, E.F. Minimal renal dysfunction in inflammatory bowel disease is related to disease activity but not to 5-ASA use. Aliment. Pharmacol. Ther. 2001, 15, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, P.; Jayakumar, C.; Santhakumar, M.; Ramesh, G. Netrin-1 regulates colon-kidney crosstalk through suppression of IL-6 function in a mouse model of DSS-colitis. Am. J. Physiol. Ren. Physiol. 2013, 304, F1187–F1197. [Google Scholar] [CrossRef]
- Mahmoodpoor, F.; Rahbar Saadat, Y.; Barzegari, A.; Ardalan, M.; Zununi Vahed, S. The impact of gut microbiota on kidney function and pathogenesis. Biomed. Pharmacother. 2017, 93, 412–419. [Google Scholar] [CrossRef]
- Collins, C.M.; D’Orazio, S.E. Bacterial ureas: Structure, regulation of expression and role in pathogenesis. Mol. Microbiol. 1993, 9, 907–913. [Google Scholar] [CrossRef]
- Suzuki, K.; Benno, Y.; Mitsuoka, T.; Takebe, S.; Kobashi, K.; Hase, J. Areas-producing species of intestinal anaerobes and their activities. Appl. Environ. Microbiol. 1979, 37, 379–382. [Google Scholar] [CrossRef]
- Wozny, M.A.; Bryant, M.P.; Holdeman, L.V.; Moore, W.E. Urease assay and ureas-producing species of anaerobes in the bovine rumen and human feces. Appl. Environ. Microbiol. 1977, 33, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Song, I.H. The clinical impact of gut microbiota in chronic kidney disease. Korean J. Intern. Med. 2020, 35, 1305–1316. [Google Scholar] [CrossRef] [PubMed]
- Heikius, B.; Niemelä, S.; Lehtola, J.; Karttunen, T.J. Elevated pancreatic enzymes in inflammatory bowel disease are associated with extensive disease. Am. J. Gastroenterol. 1999, 94, 1062–1069. [Google Scholar] [CrossRef]
- Ramos, L.R.; Sachar, D.B.; DiMaio, C.J.; Colombel, J.F.; Torres, J. Inflammatory Bowel Disease and Pancreatitis: A Review. J. Crohns Colitis 2016, 10, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Fousekis, F.S.; Theopistos, V.I.; Katsanos, K.H.; Christodoulou, D.K. Pancreatic Involvement in Inflammatory Bowel Disease: A Review. J. Clin. Med. Res. 2018, 10, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Bokemeyer, B. Asymptomatic elevation of serum lipase and amylase in conjunction with Crohn’s disease and ulcerative colitis. Z. Gastroenterol. 2002, 40, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Barthet, M.; Lesavre, N.; Desplats, S.; Panuel, M.; Gasmi, M.; Bernard, J.P.; Dagorn, J.C.; Grimaud, J.C. Frequency and characteristics of pancreatitis in patients with inflammatory bowel disease. Pancreatology 2006, 6, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, M.; Strisciuglio, C.; Illiceto, M.T.; Cardile, S.; Guariso, G.; Vignola, S.; Aloi, M.; D’Altilia, M.R.; Alvisi, P.; Salvatore, S.; et al. Natural history of pancreatic involvement in paediatric inflammatory bowel disease. Dig. Liver Dis. 2015, 47, 384–389. [Google Scholar] [CrossRef]
- Ray, P.; Van Arsdall, M.R. Elevated Lipase during Initial Presentation of Ulcerative Colitis in a Pediatric Patient: Do We Check for It. Case Rep. Gastroenterol. 2016, 10, 568–573. [Google Scholar] [CrossRef]
- Bilski, J.; Mazur-Bialy, A.; Wojcik, D.; Zahradnik-Bilska, J.; Brzozowski, B.; Magierowski, M.; Mach, T.; Magierowska, K.; Brzozowski, T. The Role of Intestinal Alkaline Phosphatase in Inflammatory Disorders of Gastrointestinal Tract. Mediat. Inflamm. 2017, 2017, 9074601. [Google Scholar] [CrossRef]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Malo, M.S.; Alam, S.N.; Mostafa, G.; Zeller, S.J.; Johnson, P.V.; Mohammad, N.; Chen, K.T.; Moss, A.K.; Ramasamy, S.; Faruqui, A.; et al. Intestinal alkaline phosphatase preserves the normal homeostasis of gut microbiota. Gut 2010, 59, 1476–1484. [Google Scholar] [CrossRef] [PubMed]
- Molnár, K.; Vannay, A.; Szebeni, B.; Bánki, N.F.; Sziksz, E.; Cseh, A.; Győrffy, H.; Lakatos, P.L.; Papp, M.; Arató, A.; et al. Intestinal alkaline phosphatase in the colonic mucosa of children with inflammatory bowel disease. World J. Gastroenterol. 2012, 18, 3254–3259. [Google Scholar] [CrossRef] [PubMed]
- Niepel, D.; Klag, T.; Malek, N.P.; Wehkamp, J. Practical guidance for the management of iron deficiency in patients with inflammatory bowel disease. Therap. Adv. Gastroenterol. 2018, 11, 1756284818769074. [Google Scholar] [CrossRef]
- González Alayón, C.; Pedrajas Crespo, C.; Marín Pedrosa, S.; Benítez, J.M.; Iglesias Flores, E.; Salgueiro Rodríguez, I.; Medina Medina, R.; García-Sánchez, V. Prevalence of iron deficiency without anaemia in inflammatory bowel disease and impact on health-related quality of life. Gastroenterol. Hepatol. 2018, 41, 22–29. [Google Scholar] [CrossRef]
- Seyoum, Y.; Baye, K.; Humblot, C. Iron homeostasis in host and gut bacteri—A complex interrelationship. Gut Microbes 2021, 13, 1–19. [Google Scholar] [CrossRef]
- Dostal, A.; Fehlbaum, S.; Chassard, C.; Zimmermann, M.B.; Lacroix, C. Low iron availability in continuous in vitro colonic fermentations induces strong dispose of the child gut microbial consortium and a decrease in main metabolites. FEMS Microbiol. Ecol. 2013, 83, 161–175. [Google Scholar] [CrossRef]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut microbiota and IBD: Causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef]
- Knox, N.C.; Forbes, J.D.; Van Domselaar, G.; Bernstein, C.N. The Gut Microbiome as a Target for IBD Treatment: Are We There Yet? Curr. Treat Opt. Gastroenterol. 2019, 17, 115–126. [Google Scholar] [CrossRef]
- Buttó, L.F.; Haller, D. Dysbiosis in intestinal inflammation: Cause or consequence. Int. J. Med. Microbiol. 2016, 306, 302–309. [Google Scholar] [CrossRef]
- Gong, D.; Gong, X.; Wang, L.; Yu, X.; Dong, Q. Involvement of Reduced Microbial Diversity in Inflammatory Bowel Disease. Gastroenterol. Res. Pract. 2016, 2016, 6951091. [Google Scholar] [CrossRef] [PubMed]
- Zakerska-Banaszak, O.; Tomczak, H.; Gabryel, M.; Baturo, A.; Wolko, L.; Michalak, M.; Malinska, N.; Mankowska-Wierzbicka, D.; Eder, P.; Dobrowolska, A.; et al. Dysbiosis of gut microbiota in Polish patients with ulcerative colitis: A pilot study. Sci. Rep. 2021, 11, 2166. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, D.; Moran, C.; Shanahan, F. The microbiota in inflammatory bowel disease. J. Gastroenterol. 2015, 50, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Sha, S.; Xu, B.; Wang, X.; Zhang, Y.; Wang, H.; Kong, X.; Zhu, H.; Wu, K. The biodiversity and composition of the dominant fecal microbiota in patients with inflammatory bowel disease. Diagn. Microbiol. Infect. Dis. 2013, 75, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Papa, E.; Docktor, M.; Smillie, C.; Weber, S.; Preheim, S.P.; Gevers, D.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Ingram, J.; et al. Non-invasive mapping of the gastrointestinal microbiota identifies children with inflammatory bowel disease. PLoS ONE 2012, 7, e39242. [Google Scholar] [CrossRef] [PubMed]
- Ling, Z.; Jin, C.; Xie, T.; Cheng, Y.; Li, L.; Wu, N. Alterations in the Fecal Microbiota of Patients with HIV-1 Infection: An Observational Study in A Chinese Population. Sci. Rep. 2016, 6, 30673. [Google Scholar] [CrossRef]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef]
- Schierová, D.; Březina, J.; Mrázek, J.; Fliegerová, K.O.; Kvasnová, S.; Bajer, L.; Drastich, P. Gut Microbiome Changes in Patients with Active Left-Sided Ulcerative Colitis after Fecal Microbiome Transplantation and Topical 5-aminosalicylic Acid Therapy. Cells 2020, 9, 2283. [Google Scholar] [CrossRef]
- Noor, S.O.; Ridgway, K.; Scovell, L.; Kemsley, E.K.; Lund, E.K.; Jamieson, C.; Johnson, I.T.; Narbad, A. Ulcerative colitis and irritable bowel patients exhibit distinct abnormalities of the gut microbiota. BMC Gastroenterol. 2010, 10, 134. [Google Scholar] [CrossRef]
- Conte, M.P.; Schippa, S.; Zamboni, I.; Penta, M.; Chiarini, F.; Seganti, L.; Osborn, J.; Falconieri, P.; Borrelli, O.; Cucchiara, S. Gut-associated bacterial microbiota in paediatric patients with inflammatory bowel disease. Gut 2006, 55, 1760–1767. [Google Scholar] [CrossRef]
- Round, J.L.; Mazmanian, S.K. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc. Natl. Acad. Sci USA 2010, 107, 12204–12209. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.J.; Huang, Y.; Peterson, D.A.; Goeser, L.; Fan, T.J.; Chang, E.B.; Sartor, R.B. The colitis-associated transcriptional profile of commercial Bacteroides thetaiotaomicron enhances adaptive immune responses to a bacterial antigen. PLoS ONE 2012, 7, e42645. [Google Scholar] [CrossRef] [PubMed]
- Bloom, S.M.; Bijanki, V.N.; Nava, G.M.; Sun, L.; Malvin, N.P.; Donermeyer, D.L.; Dunne, W.M.; Allen, P.M.; Stappenbeck, T.S. Commensal Bacteroides species induce colitis in host-genotype-specific fashion in a mouse model of inflammatory bowel disease. Cell Host Microbe 2011, 9, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.R.; Lee, J.C.; Lee, H.Y.; Kim, M.S.; Whon, T.W.; Lee, M.S.; Bae, J.W. An increase in the Akkermansia spp. Population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 2014, 63, 727–735. [Google Scholar] [CrossRef]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef]
- Lepage, P.; Häsler, R.; Spehlmann, M.E.; Rehman, A.; Zvirbliene, A.; Begun, A.; Ott, S.; Kupcinskas, L.; Doré, J.; Raedler, A.; et al. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology 2011, 141, 227–236. [Google Scholar] [CrossRef]
- Vacca, M.; Celano, G.; Calabrese, F.M.; Portincasa, P.; Gobbetti, M.; De Angelis, M. The Controversial Role of Human Gut Lachnospiraceae. Microorganisms 2020, 8, 573. [Google Scholar] [CrossRef]
- Schwiertz, A.; Jacobi, M.; Frick, J.S.; Richter, M.; Rusch, K.; Köhler, H. Microbiota in pediatric inflammatory bowel disease. J. Pediatr. 2010, 157, 240–244, e241. [Google Scholar] [CrossRef]
- Nagao-Kitamoto, H.; Shreiner, A.B.; Gillilland, M.G.; Kitamoto, S.; Ishii, C.; Hirayama, A.; Kuffa, P.; El-Zaatari, M.; Grasberger, H.; Seekatz, A.M.; et al. Functional Characterization of Inflammatory Bowel Disease-Associated Gut Dysbiosis in Gnotobiotic Mice. Cell Mol. Gastroenterol. Hepatol. 2016, 2, 468–481. [Google Scholar] [CrossRef]
- Yang, H.; Mirsepasi-Lauridsen, H.C.; Struve, C.; Allaire, J.M.; Sivignon, A.; Vogl, W.; Bosman, E.S.; Ma, C.; Fotovati, A.; Reid, G.S.; et al. Ulcerative Colitis-associated. Gut Microbes 2020, 12, 1847976. [Google Scholar] [CrossRef]
- Keighley, M.R.; Arabi, Y.; Dimock, F.; Burdon, D.W.; Allan, R.N.; Alexander-Williams, J. Influence of inflammatory bowel disease on intestinal microflora. Gut 1978, 19, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Chen, D.; Zhang, J.N.; Lv, X.; Wang, K.; Duan, L.P.; Nie, Y.; Wu, X.L. Bacterial community mapping of the mouse gastrointestinal tract. PLoS ONE 2013, 8, e74957. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Tannock, G.W. The bowel microbiota and inflammatory bowel diseases. Int. J. Inflam. 2010, 2010, 954051. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 2009, 294, 1–8. [Google Scholar] [CrossRef]
- Song, C.S.; Park, D.I.; Yoon, M.Y.; Seok, H.S.; Park, J.H.; Kim, H.J.; Cho, Y.K.; Sohn, C.I.; Jeon, W.K.; Kim, B.I. Association between red cell distribution width and disease activity in patients with inflammatory bowel disease. Dig. Dis. Sci. 2012, 57, 1033–1038. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Markers | Conjugate | Isotype | Reactivity | Clone |
|---|---|---|---|---|
| CD3 | PE | Mouse BALB/c IgG1, κ | Rat | G4.18 |
| CD4 | BV421 | Mouse BALB/c IgG1, κ | Rat | OX-35 |
| CD8α | BB700 | Mouse BALB/c IgG1, κ | Rat | OX-8 |
| CD11b | APC | Mouse BALB/c IgG1, κ | Rat | WT.5 |
| CD45 | APC-Cy7 | Mouse BALB/c IgG1, κ | Rat | OX-1 |
| Granulocytes | FITC | Mouse IgM, κ | Rat | HIS48 |
| DSS | C | p-Value | |
|---|---|---|---|
| Body Weight Gain (g) | 76.34 ± 7.54 | 96.33 ± 4.50 | 0.035 |
| FER (%) | 25.06 ± 2.51 | 32.86 ± 0.62 | 0.008 |
| Food Intake (g/rat/day) | 24.98 ± 0.45 | 24.80 ± 0.38 | 0.760 |
| DSS | C | p-Value | |
|---|---|---|---|
| Lymphoid Aggregates (LA) per Rat | 1.57 ± 0.61 | 0.33 ± 0.33 | 0.119 |
| Lymphoid Follicles per Rat | 5.00 ± 2.27 | 0.33 ± 0.33 | 0.088 |
| Mean Length of LA (μm) | 2495.00 ± 382.00 | 150.00 ± 150.00 | <0.001 |
| Mean Width of LA (μm) | 481.80 ± 67.80 | 66.70 ± 66.70 | 0.005 |
| 1st Collection | 2nd Collection | 3rd Collection | ||||
|---|---|---|---|---|---|---|
| DSS | C | DSS | C | DSS | C | |
| WBC (109/L) | 14.52 ± 0.53 α | 15.93 ± 1.03 | 22.37 ± 0.52 *β | 18.52 ± 0.93 | 20.08 ± 1.36 β | 18.88 ± 0.90 |
| LYM (109/L) | 10.97 ± 0.37 α | 12.66 ± 0.86 | 15.69 ± 0.44 *β | 13.13 ± 0.97 | 15.37 ± 1.19 β | 14.74 ± 0.94 |
| MON (109/L) | 0.50 ± 0.05 A | 0.45 ± 0.04 | 1.07 ± 0.07 *B | 0.63 ± 0.13 | 0.83 ± 0.12 B | 0.78 ± 0.15 |
| GRA (109/L) | 3.10 ± 0.16 α | 3.53 ± 0.34 | 5.03 ± 0.21 *β | 3.62 ± 0.53 | 4.64 ± 0.45 β | 4.58 ± 0.42 |
| HGB (g/L) | 131.32 ± 2.25 α | 132.38 ± 3.19 γ | 145.57 ± 1.17 β | 150.00 ± 1.86 δ | 142.20 ± 1.08 β | 150.6 ± 3.39 δ |
| RBC (1012/L) | 5.61 ± 0.10 α | 5.65 ± 0.12 γ | 6.43 ± 0.09 β | 6.59 ± 0.09 δ | 6.29 ± 0.12 *β | 6.77 ± 0.14 δ |
| PLT (109/L) | 488.60 ± 24.90 | 431.50 ± 45.00 c | 513.70 ± 29.20 * | 633.80 ± 41.10 d | 521.40 ± 85.70 | 488.6 ± 61.9 cd |
| PLR | 41.98 ± 1.95 α | 34.52 ± 3.52 | 34.69 ± 2.89 β | 44.64 ± 5.04 | 36.44 ± 4.36 αβ | 34.25 ± 5.76 |
| LMR | 27.32 ± 1.72 α | 29.10 ± 2.15 | 17.98 ± 1.14 *β | 30.13 ± 2.46 | 18.48 ± 1.85 β | 21.04 ± 3.26 |
| GLR | 0.28 ± 0.01 α | 0.28 ± 0.02 | 0.32 ± 0.01 β | 0.28 ± 0.03 | 0.32 ± 0.03 β | 0.31 ± 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adamkova, P.; Hradicka, P.; Kupcova Skalnikova, H.; Cizkova, V.; Vodicka, P.; Farkasova Iannaccone, S.; Kassayova, M.; Gancarcikova, S.; Demeckova, V. Dextran Sulphate Sodium Acute Colitis Rat Model: A Suitable Tool for Advancing Our Understanding of Immune and Microbial Mechanisms in the Pathogenesis of Inflammatory Bowel Disease. Vet. Sci. 2022, 9, 238. https://doi.org/10.3390/vetsci9050238
Adamkova P, Hradicka P, Kupcova Skalnikova H, Cizkova V, Vodicka P, Farkasova Iannaccone S, Kassayova M, Gancarcikova S, Demeckova V. Dextran Sulphate Sodium Acute Colitis Rat Model: A Suitable Tool for Advancing Our Understanding of Immune and Microbial Mechanisms in the Pathogenesis of Inflammatory Bowel Disease. Veterinary Sciences. 2022; 9(5):238. https://doi.org/10.3390/vetsci9050238
Chicago/Turabian StyleAdamkova, Petra, Petra Hradicka, Helena Kupcova Skalnikova, Veronika Cizkova, Petr Vodicka, Silvia Farkasova Iannaccone, Monika Kassayova, Sona Gancarcikova, and Vlasta Demeckova. 2022. "Dextran Sulphate Sodium Acute Colitis Rat Model: A Suitable Tool for Advancing Our Understanding of Immune and Microbial Mechanisms in the Pathogenesis of Inflammatory Bowel Disease" Veterinary Sciences 9, no. 5: 238. https://doi.org/10.3390/vetsci9050238
APA StyleAdamkova, P., Hradicka, P., Kupcova Skalnikova, H., Cizkova, V., Vodicka, P., Farkasova Iannaccone, S., Kassayova, M., Gancarcikova, S., & Demeckova, V. (2022). Dextran Sulphate Sodium Acute Colitis Rat Model: A Suitable Tool for Advancing Our Understanding of Immune and Microbial Mechanisms in the Pathogenesis of Inflammatory Bowel Disease. Veterinary Sciences, 9(5), 238. https://doi.org/10.3390/vetsci9050238