
16S rRNA Gene Sequencing Revealed Changes in Gut Microbiota Composition during Pregnancy and Lactation in Mice Model

 , ,
, , 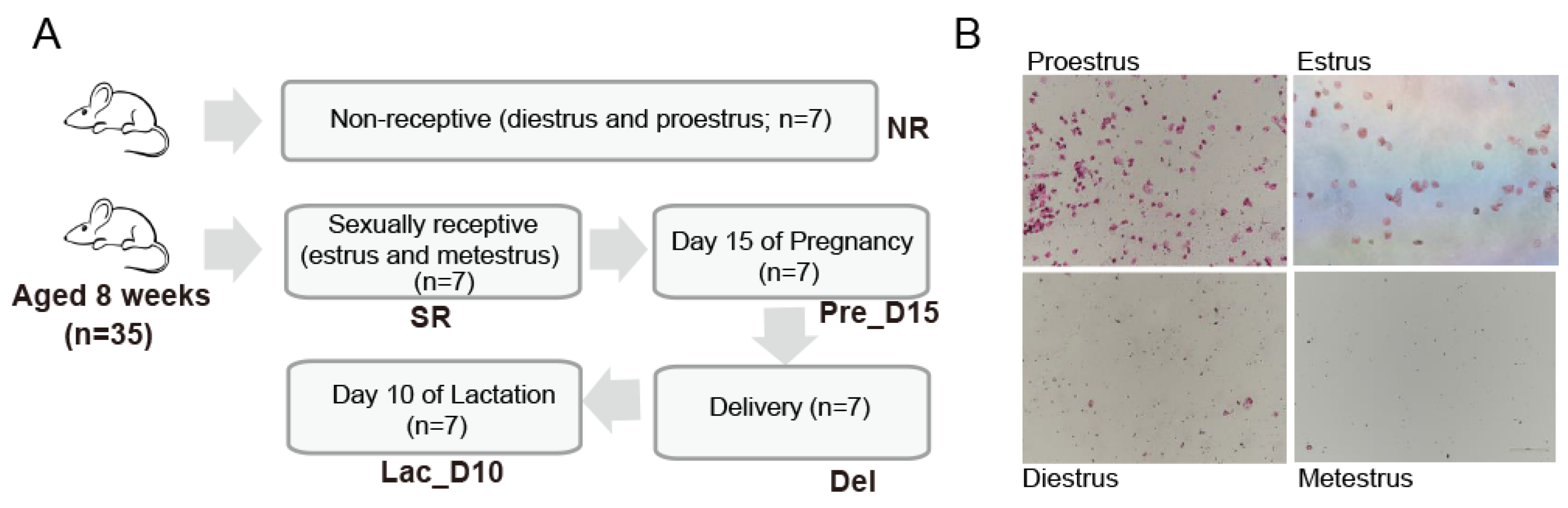{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals, Experimental Design, and Sample Collection
2.2. Identification of Mouse Estrus Cycle Stage
2.3. Sex Hormones Measurements
2.4. Analysis of Cecum Short-Chain Fatty Acids (SCFAs)
2.5. DNA Extraction, PCR Amplification, and 16S rRNA Gene Sequencing
2.6. 16S rRNA Gene Sequencing Data Analysis
2.7. Statistical Analysis
3. Results
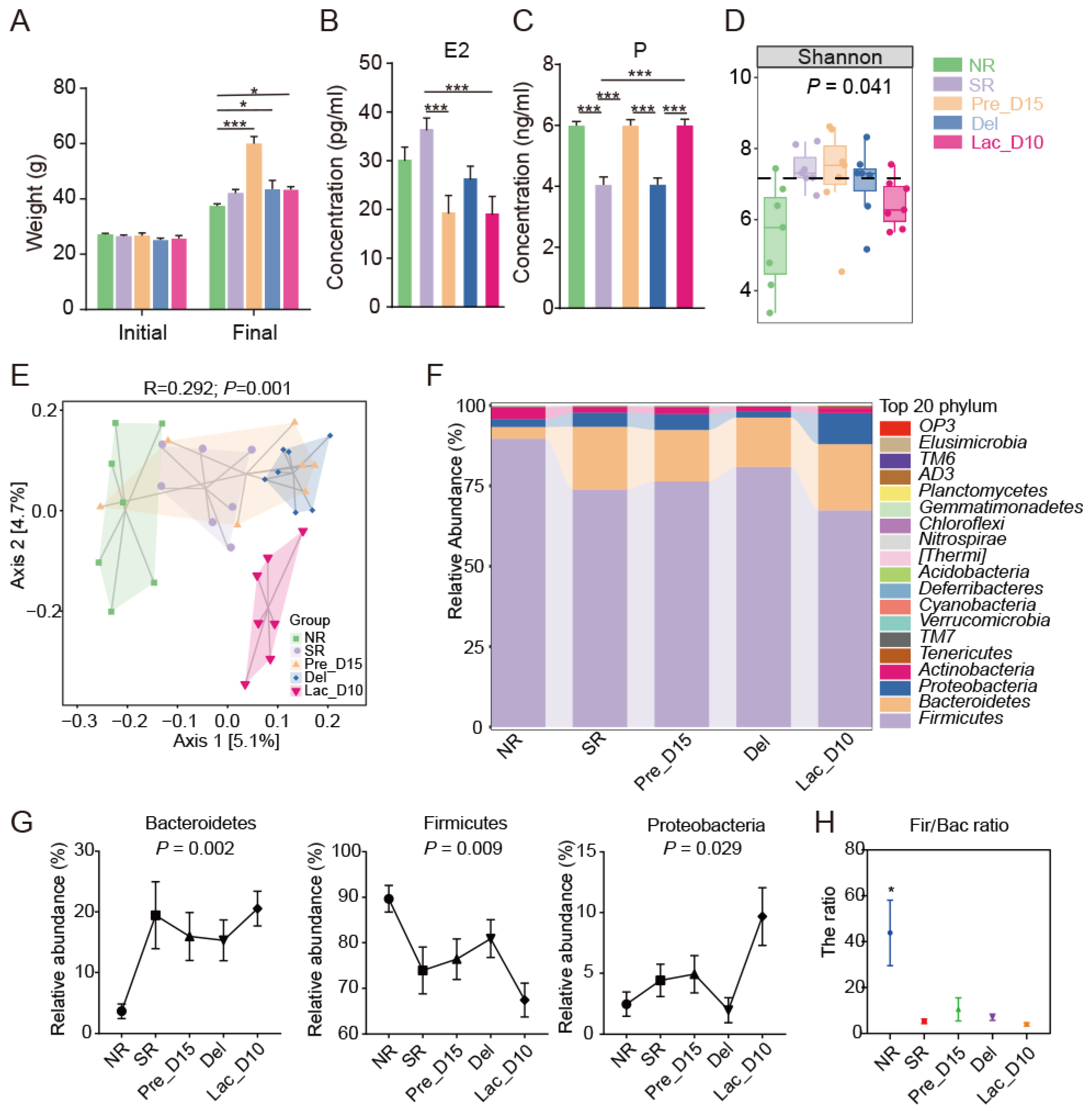
3.1. Weight and SERUM Sex Hormones Changes during Pregnancy and Lactation
3.2. Microbial Community Profiles during Pregnancy and Lactation in Mice
3.3. Comparison of the Gut Microbiota during Pregnancy and Lactation
3.4. Fecal SCFAs Concentrations during Pregnancy and Lactation in Mice
3.5. Correlation Analysis of Caecal Microbiota Composition and SCFAs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lin, L.; Wang, Y.; Xu, L.; Liu, J.; Mao, S. Microbiome–host co-oscillation patterns in remodeling of colonic homeostasis during adaptation to a high-grain diet in a sheep model. Anim. Microbiome 2020, 2, 22. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, P.L. Linking the gut microbiome to metabolism through endocrine hormones. Endocrinology 2018, 159, 2978–2979. [Google Scholar] [CrossRef] [PubMed]
- Mor, G.; Aldo, P.; Alvero, A.B. The unique immunological and microbial aspects of pregnancy. Nat. Rev. Immunol. 2017, 17, 469–482. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Lin, L.; Hu, F.; Zhu, W.; Mao, S. Disruption of ruminal homeostasis by malnutrition involved in systemic ruminal microbiota-host interactions in a pregnant sheep model. Microbiome 2020, 8, 138. [Google Scholar] [CrossRef]
- Edwards, S.M.; Cunningham, S.A.; Dunlop, A.L.; Corwin, E.J. The maternal gut microbiome during pregnancy. MCN. Am. J. Matern. Child Nurs. 2017, 42, 310. [Google Scholar] [CrossRef]
- Trevisanuto, D.; Peruzzetto, C.; Cavallin, F.; Vedovato, S.; Cosmi, E.; Visentin, S.; Chiarelli, S.; Zanardo, V. Fetal placental inflammation is associated with poor neonatal growth of preterm infants: A case-control study. J. Matern.-Fetal Neonatal Med. 2013, 26, 1484–1490. [Google Scholar] [CrossRef]
- Koren, O.; Goodrich, J.K.; Cullender, T.C.; Spor, A.; Laitinen, K.; Bäckhed, H.K.; Gonzalez, A.; Werner, J.J.; Angenent, L.T.; Knight, R. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 2012, 150, 470–480. [Google Scholar] [CrossRef]
- Mulak, A.; Taché, Y.; Larauche, M. Sex hormones in the modulation of irritable bowel syndrome. World J. Gastroenterol. WJG 2014, 20, 2433. [Google Scholar] [CrossRef]
- Brantsæter, A.L.; Myhre, R.; Haugen, M.; Myking, S.; Sengpiel, V.; Magnus, P.; Jacobsson, B.; Meltzer, H.M. Intake of probiotic food and risk of preeclampsia in primiparous women: The Norwegian Mother and Child Cohort Study. Am. J. Epidemiol. 2011, 174, 807–815. [Google Scholar] [CrossRef]
- García-Gómez, E.; González-Pedrajo, B.; Camacho-Arroyo, I. Role of sex steroid hormones in bacterial-host interactions. BioMed Res. Int. 2013, 2013, 928290. [Google Scholar] [CrossRef]
- Mayer, E.A.; Savidge, T.; Shulman, R.J. Brain–gut microbiome interactions and functional bowel disorders. Gastroenterology 2014, 146, 1500–1512. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Xu, Q.; Huang, W.; Yan, Q.; Chen, Y.; Zhang, L.; Tian, Z.; Liu, T.; Yuan, X.; Liu, C. Gestational diabetes mellitus is associated with reduced dynamics of gut microbiota during the first half of pregnancy. MSystems 2020, 5, e00109–e00120. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Hou, C.; Li, N.; Zhang, X.; Zhang, G.; Yang, F.; Zeng, X.; Liu, Z.; Qiao, S. Microbial and metabolic alterations in gut microbiota of sows during pregnancy and lactation. FASEB J. 2019, 33, 4490–4501. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Xu, J.; Lefever, D.E.; Glenn, T.C.; Nagy, T.; Guo, T.L. Genistein prevention of hyperglycemia and improvement of glucose tolerance in adult non-obese diabetic mice are associated with alterations of gut microbiome and immune homeostasis. Toxicol. Appl. Pharmacol. 2017, 332, 138–148. [Google Scholar] [CrossRef]
- Plottel, C.S.; Blaser, M.J. Microbiome and malignancy. Cell Host Microbe 2011, 10, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.M.; Al-Nakkash, L.; Herbst-Kralovetz, M.M. Estrogen–gut microbiome axis: Physiological and clinical implications. Maturitas 2017, 103, 45–53. [Google Scholar] [CrossRef]
- Nuriel-Ohayon, M.; Neuman, H.; Ziv, O.; Belogolovski, A.; Barsheshet, Y.; Bloch, N.; Uzan, A.; Lahav, R.; Peretz, A.; Frishman, S. Progesterone increases Bifidobacterium relative abundance during late pregnancy. Cell Rep. 2019, 27, 730–736.e3. [Google Scholar] [CrossRef]
- Zhu, N.; Jia, H.-X.; Liu, X.-K.; Zhao, X.-E.; Wei, Q.; Ma, B.-H. Measuring the estrus cycle and its effect on superovulation in mice. Zool. Res. 2012, 33, 276–282. [Google Scholar] [CrossRef]
- Zhang, K.; He, C.; Xu, Y.; Zhang, C.; Li, C.; Jing, X.; Wang, M.; Yang, Y.; Suo, L.; Kalds, P. Taxonomic and functional adaption of the gastrointestinal microbiome of goats kept at high altitude (4800 m) under intensive or extensive rearing conditions. FEMS Microbiol. Ecol. 2021, 97, fiab009. [Google Scholar] [CrossRef]
- LI, B.; Zhang, K.; Li, C.; Wang, X.; Chen, Y.; Yang, Y. Characterization and comparison of microbiota in the gastrointestinaltracts of the goat (capra hircus) during preweaning development. Front. Microbiol. 2019, 10, 2125. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed]
- Osman, M.-A.; Neoh, H.-M.; Ab Mutalib, N.-S.; Chin, S.-F.; Jamal, R. 16S rRNA gene sequencing for deciphering the colorectal cancer gut microbiome: Current protocols and workflows. Front. Microbiol. 2018, 9, 767. [Google Scholar] [CrossRef] [PubMed]
- Amato, K.R.; Sanders, J.G.; Song, S.J.; Nute, M.; Metcalf, J.L.; Thompson, L.R.; Morton, J.T.; Amir, A.; McKenzie, V.J.; Humphrey, G. Evolutionary trends in host physiology outweigh dietary niche in structuring primate gut microbiomes. ISME J. 2019, 13, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Lin, W.; Chen, S.; Xiang, T.; Yang, Y.; Yin, Y.; Xu, G.; Liu, Z.; Liu, L.; Pan, J. Gut microbiota as an objective measurement for auxiliary diagnosis of insomnia disorder. Front. Microbiol. 2019, 10, 1770. [Google Scholar] [CrossRef]
- Luan, H.; Meng, N.; Liu, P.; Feng, Q.; Lin, S.; Fu, J.; Davidson, R.; Chen, X.; Rao, W.; Chen, F. Pregnancy-induced metabolic phenotype variations in maternal plasma. J. Proteome Res. 2014, 13, 1527–1536. [Google Scholar] [CrossRef]
- Fernandes, L.; Calegare, B.F.; Cavalcante-Silva, V.; D’Almeida, V. Detraining in pregnancy and/or lactation modulates neuropeptidergic hypothalamic systems in offspring mice. Endocrine 2015, 50, 715–724. [Google Scholar] [CrossRef]
- Teixeira, P.D.; Ramos-Lobo, A.M.; Furigo, I.C.; Donato, J., Jr. Brain STAT5 modulates long-term metabolic and epigenetic changes induced by pregnancy and lactation in female mice. Endocrinology 2019, 160, 2903–2917. [Google Scholar] [CrossRef]
- Chen, L.; Xu, Y.; Chen, X.; Fang, C.; Zhao, L.; Chen, F. The maturing development of gut microbiota in commercial piglets during the weaning transition. Front. Microbiol. 2017, 8, 1688. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Collado, M.C.; Isolauri, E.; Laitinen, K.; Salminen, S. Distinct composition of gut microbiota during pregnancy in overweight and normal-weight women. Am. J. Clin. Nutr. 2008, 88, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Kaliannan, K.; Robertson, R.C.; Murphy, K.; Stanton, C.; Kang, C.; Wang, B.; Hao, L.; Bhan, A.K.; Kang, J.X. Estrogen-mediated gut microbiome alterations influence sexual dimorphism in metabolic syndrome in mice. Microbiome 2018, 6, 205. [Google Scholar] [CrossRef] [PubMed]
- King, J.C. Physiology of pregnancy and nutrient metabolism. Am. J. Clin. Nutr. 2000, 71, 1218S–1225S. [Google Scholar] [CrossRef]
- Khan, I.; Azhar, E.I.; Abbas, A.T.; Kumosani, T.; Barbour, E.K.; Raoult, D.; Yasir, M. Metagenomic analysis of antibiotic-induced changes in gut microbiota in a pregnant rat model. Front. Pharmacol. 2016, 7, 104. [Google Scholar] [CrossRef]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Girard, J.; Ferre, P.; Pegorier, J.; Duee, P. Adaptations of glucose and fatty acid metabolism during perinatal period and suckling-weaning transition. Physiol. Rev. 1992, 72, 507–562. [Google Scholar] [CrossRef]
- Kawase, M.; He, F. Anti-obesity effects of probiotics in fermented milk. J. Intest. Microbiol. 2010, 24, 265–271. [Google Scholar]
- Watanabe, K.; Takahashi, I.; Sawada, K.; Okubo, N.; Matsuzaka, M.; Akimoto, N.; Umeda, T.; Nakaji, S.; Hisada, T.; Benno, Y. Possible relationship between percentage of body fat and Lactobacillales in gut microbiota: Results from a community-based study. Hirosaki Med. J. 2014, 65, 12–20. [Google Scholar]
- Ciampolillo, A. Metabolic syndrome and gut microbiota: There is a gender difference? Ital. J. Gend-Specif. Med. 2019, 5, 21–26. [Google Scholar]
- Azagra-Boronat, I.; Tres, A.; Massot-Cladera, M.; Franch, À.; Castell, M.; Guardiola, F.; Pérez-Cano, F.J.; Rodríguez-Lagunas, M.J. Lactobacillus fermentum CECT5716 supplementation in rats during pregnancy and lactation impacts maternal and offspring lipid profile, immune system and microbiota. Cells 2020, 9, 575. [Google Scholar] [CrossRef] [PubMed]
- Mokkala, K.; Röytiö, H.; Munukka, E.; Pietilä, S.; Ekblad, U.; Rönnemaa, T.; Eerola, E.; Laiho, A.; Laitinen, K. Gut microbiota richness and composition and dietary intake of overweight pregnant women are related to serum zonulin concentration, a marker for intestinal permeability. J. Nutr. 2016, 146, 1694–1700. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Ma, C.; Gao, Q.; Zhang, W.; Azad, M.; Kalam, A.; Kong, X. Alterations in the blood parameters and fecal microbiota and metabolites during pregnant and lactating stages in Bama mini pigs as a model. Mediat. Inflamm. 2020, 2020, 8829072. [Google Scholar] [CrossRef]
- Palacios-González, B.; Ramírez-Salazar, E.G.; Rivera-Paredez, B.; Quiterio, M.; Flores, Y.N.; Macias-Kauffer, L.; Moran-Ramos, S.; Denova-Gutiérrez, E.; Ibarra-González, I.; Vela-Amieva, M. A multi-omic analysis for low bone mineral density in postmenopausal women suggests a relationship between diet, metabolites, and microbiota. Microorganisms 2020, 8, 1630. [Google Scholar] [CrossRef]
- Zhang, J.; Luo, D.; Lin, Z.; Zhou, W.; Rao, J.; Li, Y.; Wu, J.; Peng, H.; Lou, T. Dysbiosis of gut microbiota in adult idiopathic membranous nephropathy with nephrotic syndrome. Microb. Pathog. 2020, 147, 104359. [Google Scholar] [CrossRef]
- Zhang, F.; Yue, L.; Fang, X.; Wang, G.; Li, C.; Sun, X.; Jia, X.; Yang, J.; Song, J.; Zhang, Y. Altered gut microbiota in Parkinson’s disease patients/healthy spouses and its association with clinical features. Parkinsonism Relat. Disord. 2020, 81, 84–88. [Google Scholar] [CrossRef]
- Tirosh, A.; Calay, E.S.; Tuncman, G.; Claiborn, K.C.; Inouye, K.E.; Eguchi, K.; Alcala, M.; Rathaus, M.; Hollander, K.S.; Ron, I. The short-chain fatty acid propionate increases glucagon and FABP4 production, impairing insulin action in mice and humans. Sci. Transl. Med. 2019, 11, eaav0120. [Google Scholar] [CrossRef]
- Johns, E.C.; Denison, F.C.; Norman, J.E.; Reynolds, R.M. Gestational diabetes mellitus: Mechanisms, treatment, and complications. Trends Endocrinol. Metab. 2018, 29, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, Y.; Wen, Z.; Liu, W.; Meng, L.; Huang, H. Oscillospira-a candidate for the next-generation probiotics. Gut Microbes 2021, 13, 1987783. [Google Scholar] [CrossRef] [PubMed]
- Gophna, U.; Konikoff, T.; Nielsen, H.B. Oscillospira and related bacteria–From metagenomic species to metabolic features. Environ. Microbiol. 2017, 19, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Santoru, M.L.; Piras, C.; Murgia, A.; Palmas, V.; Camboni, T.; Liggi, S.; Ibba, I.; Lai, M.A.; Orrù, S.; Blois, S. Cross sectional evaluation of the gut-microbiome metabolome axis in an Italian cohort of IBD patients. Sci. Rep. 2017, 7, 9523. [Google Scholar] [CrossRef] [PubMed]
- Al-Lahham, S.A.; Roelofsen, H.; Rezaee, F.; Weening, D.; Hoek, A.; Vonk, R.; Venema, K. Propionic acid affects immune status and metabolism in adipose tissue from overweight subjects. Eur. J. Clin. Investig. 2012, 42, 357–364. [Google Scholar] [CrossRef]
- Al-Lahham, S.A.H.; Roelofsen, H.; Priebe, M.; Weening, D.; Dijkstra, M.; Hoek, A.; Rezaee, F.; Venema, K.; Vonk, R.J. Regulation of adipokine production in human adipose tissue by propionic acid. Eur. J. Clin. Investig. 2010, 40, 401–407. [Google Scholar] [CrossRef]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef]
- Mariño, E.; Richards, J.L.; McLeod, K.H.; Stanley, D.; Yap, Y.A.; Knight, J.; McKenzie, C.; Kranich, J.; Oliveira, A.C.; Rossello, F.J. Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat. Immunol. 2017, 18, 552–562. [Google Scholar] [CrossRef]
- Duncan, S.H.; Louis, P.; Thomson, J.M.; Flint, H.J. The role of pH in determining the species composition of the human colonic microbiota. Environ. Microbiol. 2009, 11, 2112–2122. [Google Scholar] [CrossRef]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, M.; Cao, X.; Zhang, K.; Pan, M.; Wu, Y.; Langda, S.; Yang, Y.; Chen, Y.; Gui, B.; Ma, B. 16S rRNA Gene Sequencing Revealed Changes in Gut Microbiota Composition during Pregnancy and Lactation in Mice Model. Vet. Sci. 2022, 9, 169. https://doi.org/10.3390/vetsci9040169
Guo M, Cao X, Zhang K, Pan M, Wu Y, Langda S, Yang Y, Chen Y, Gui B, Ma B. 16S rRNA Gene Sequencing Revealed Changes in Gut Microbiota Composition during Pregnancy and Lactation in Mice Model. Veterinary Sciences. 2022; 9(4):169. https://doi.org/10.3390/vetsci9040169
Chicago/Turabian StyleGuo, Mengmeng, Xi Cao, Ke Zhang, Menghao Pan, Yujiang Wu, Suo Langda, Yuxin Yang, Yulin Chen, Ba Gui, and Baohua Ma. 2022. "16S rRNA Gene Sequencing Revealed Changes in Gut Microbiota Composition during Pregnancy and Lactation in Mice Model" Veterinary Sciences 9, no. 4: 169. https://doi.org/10.3390/vetsci9040169
APA StyleGuo, M., Cao, X., Zhang, K., Pan, M., Wu, Y., Langda, S., Yang, Y., Chen, Y., Gui, B., & Ma, B. (2022). 16S rRNA Gene Sequencing Revealed Changes in Gut Microbiota Composition during Pregnancy and Lactation in Mice Model. Veterinary Sciences, 9(4), 169. https://doi.org/10.3390/vetsci9040169