Development of a Sensitive Real-Time Fast-qPCR Based on SYBR® Green for Detection and Quantification of Chicken Parvovirus (ChPV)

 , ,
, , 
Abstract
1. Introduction
2. Material and Methods
2.1. Sampling
2.2. Preparation of Samples and DNA Extraction
2.3. PCR
2.4. Primer Design and Standard DNA Construction
2.5. Real-Time PCR—qPCR Assay
2.6. Determination of Standard Curve for qPCR Assay
2.7. Specificity and Sensitivity of qPCR Assay
2.8. Limit of Detection and Quantification
2.9. DNA Sequencing and Phylogenetic Analysis
2.10. GenBank Accession Numbers
3. Results
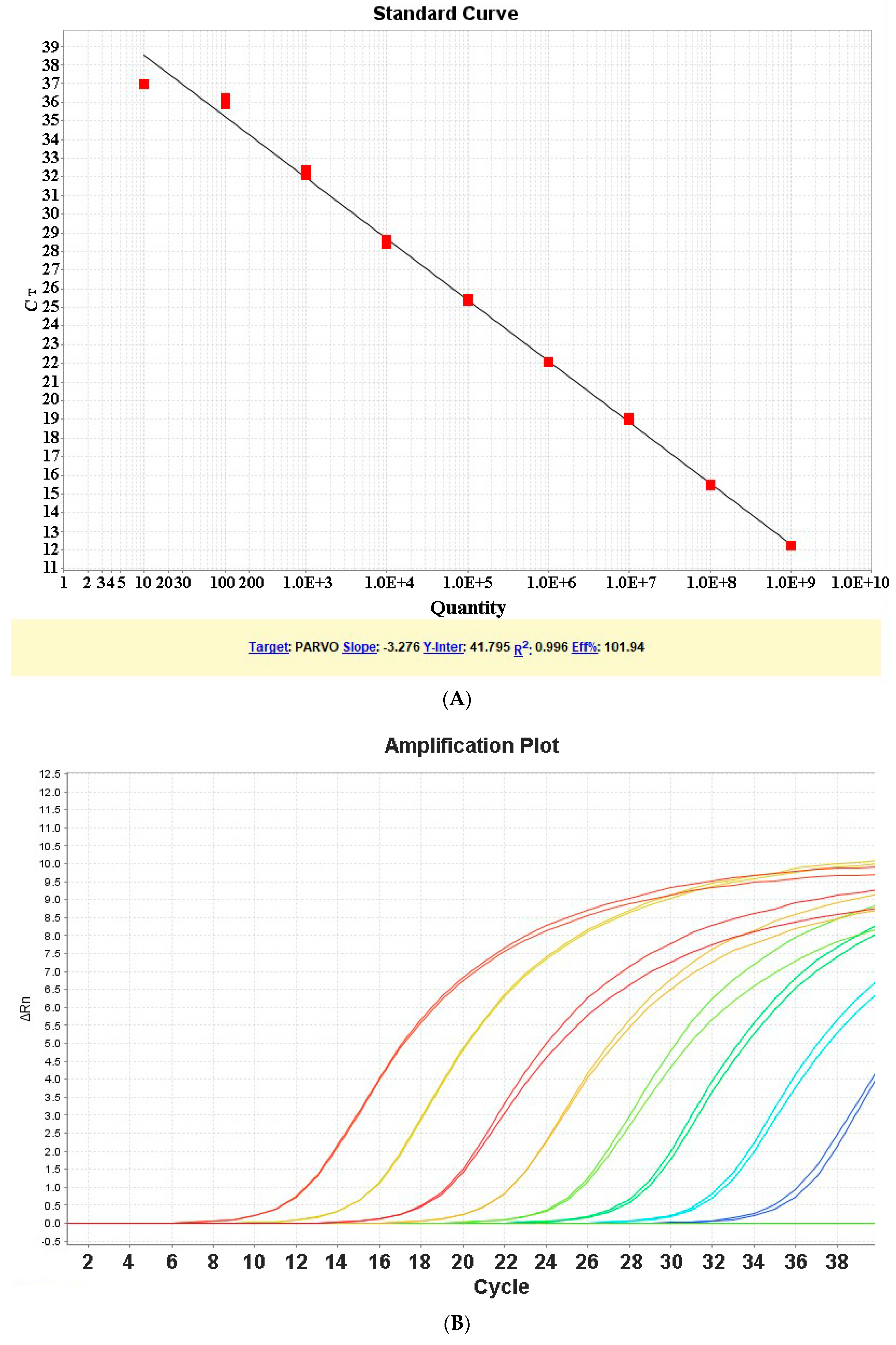
3.1. Determination of Standard Curve
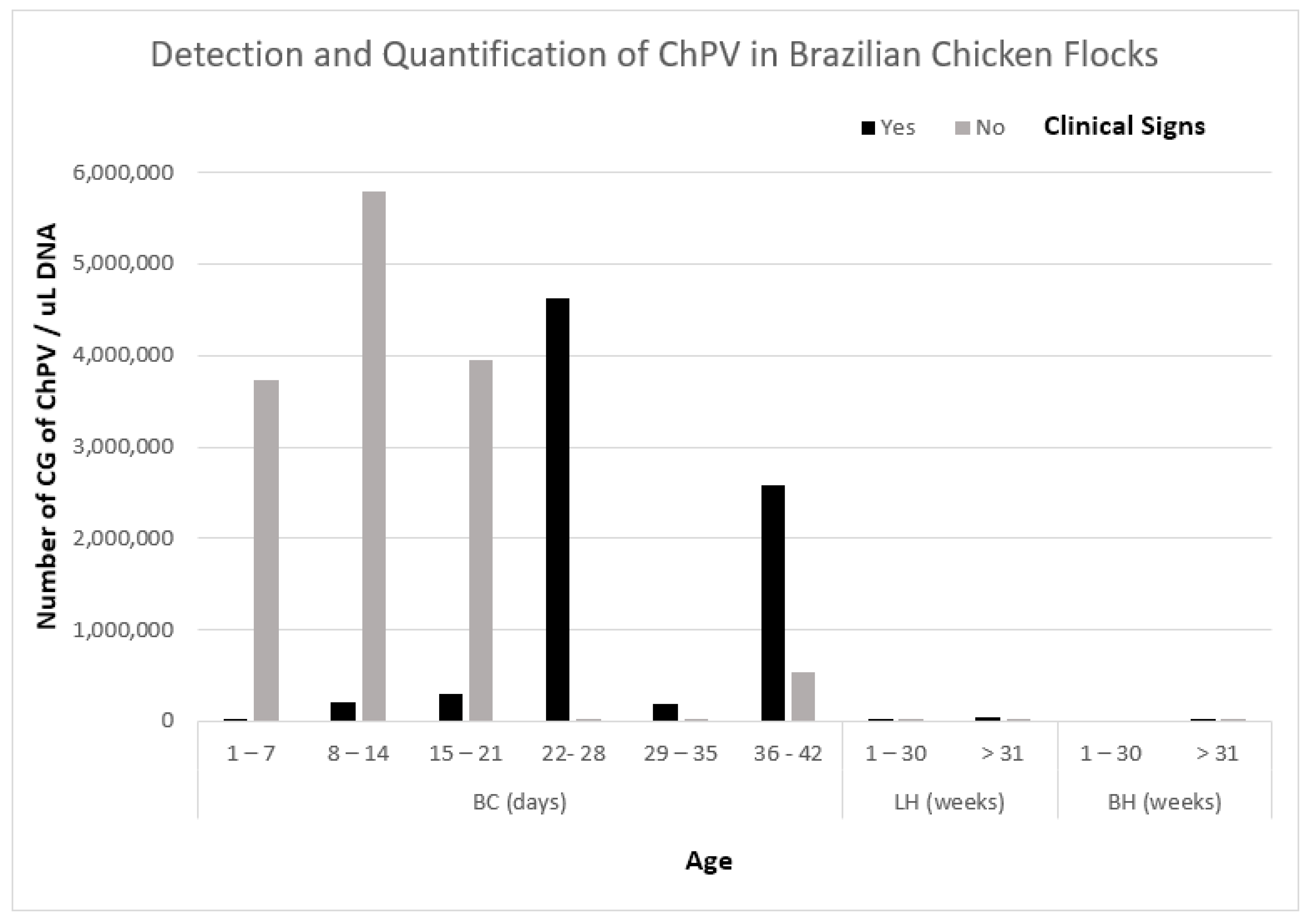
3.2. Limit of Detection and Quantification
3.3. Run Time
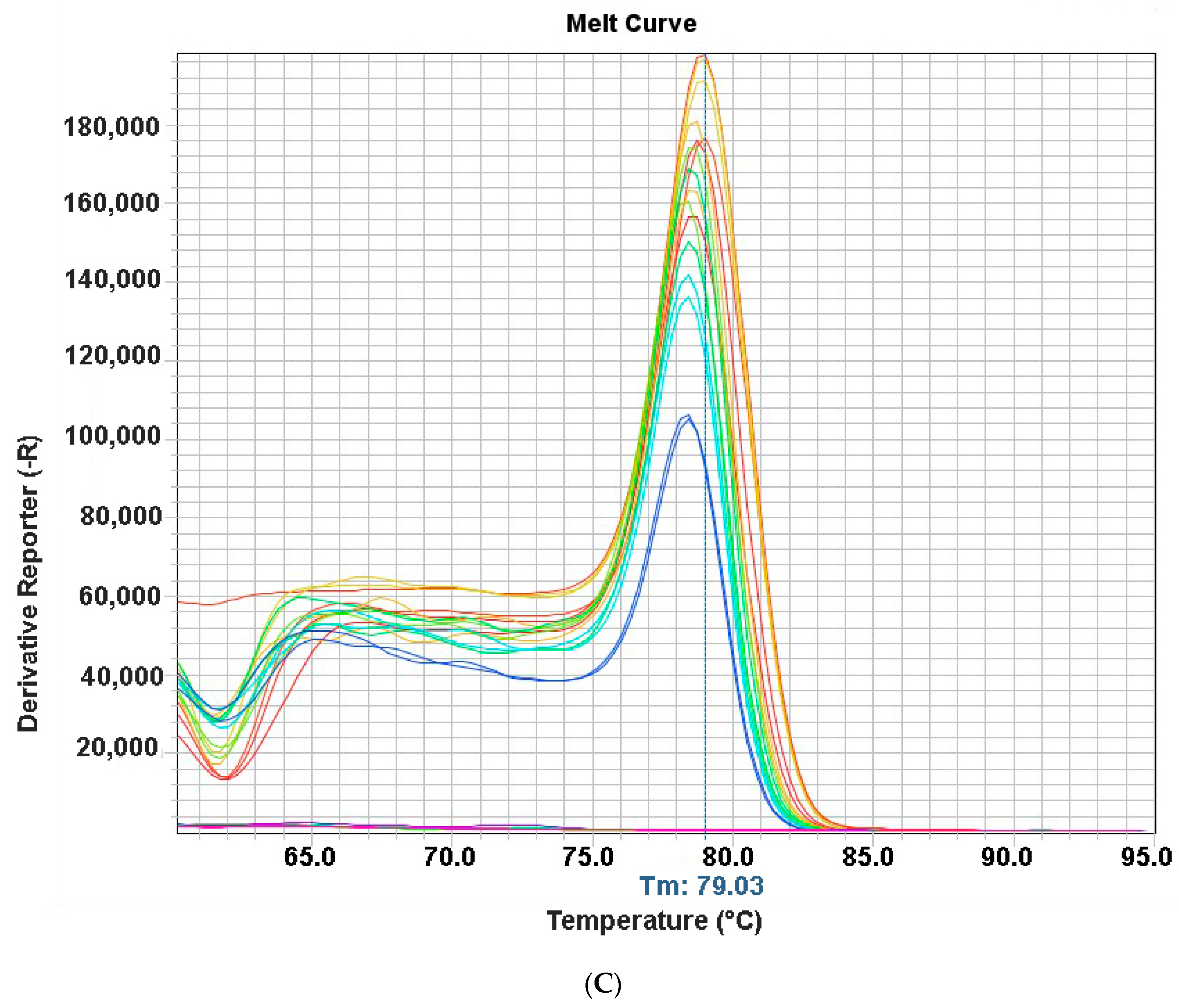
3.4. Specificity and Sensitivity of qPCR Assay
3.5. Evaluation of qPCR Assay for Detection of ChPV
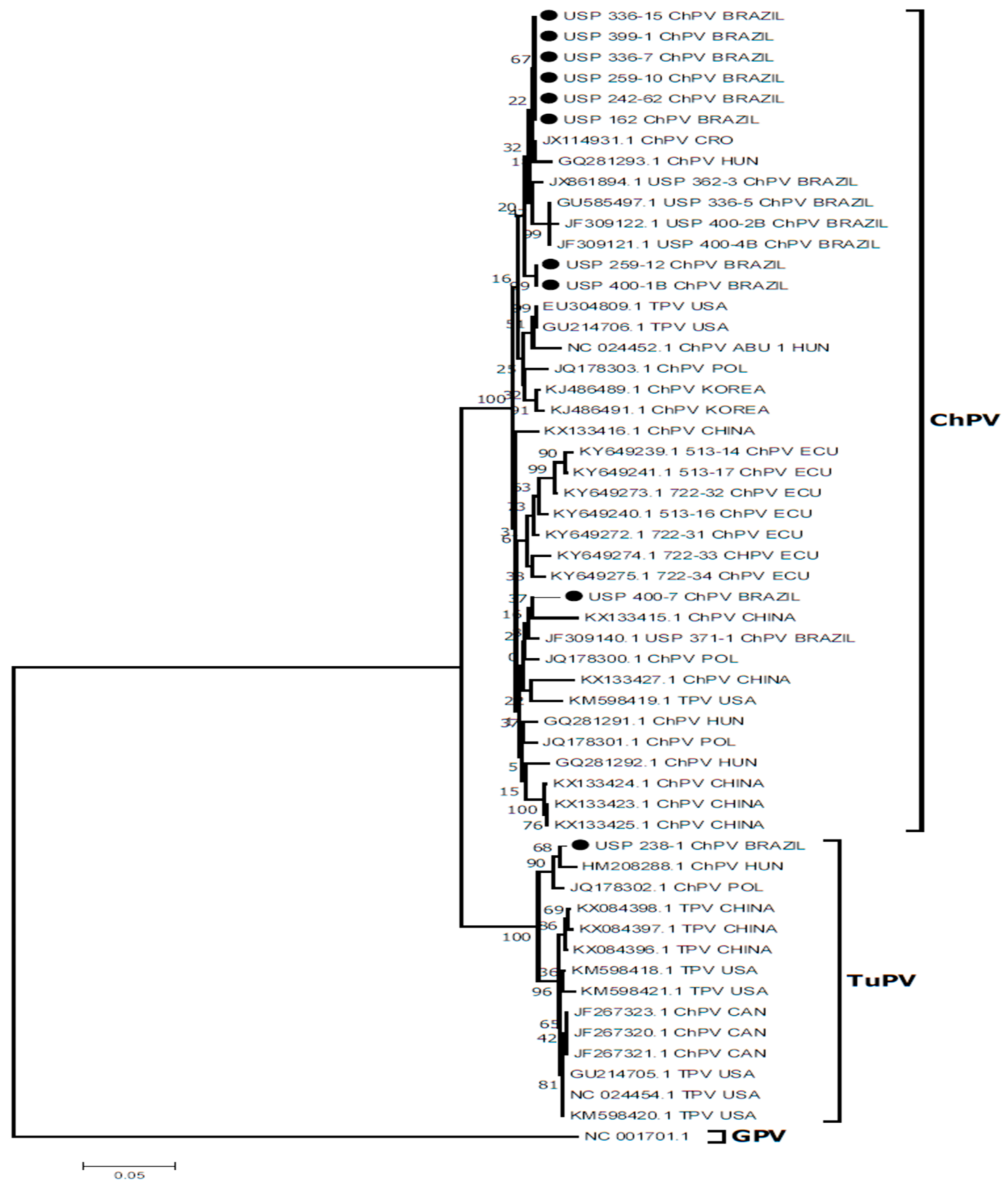
3.6. DNA Sequencing and Molecular Analyses
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sinclair, A.J.; Embury, D.H.; Smart, I.; Barr, D.A.; Reece, R.L.; Hooper, P.T.; Gould, J.A. Pancreatic degeneration in broilers with runting and stunting syndrome. Vet. Rec. 1984, 115, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, M.A.; Davis, J.F.; McNulty, M.S.; Brown, J.; Player, E.C. Enteritis (so-called runting stunting syndrome) in Georgia broiler chicks. Avian Dis. 1993, 37, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Zsak, L.; Cha, R.M.; Day, J.M. Chicken parvovirus-induced runting-stunting syndrome in young broilers. Avian Dis. 2013, 57, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Kisary, J. Experimental infection of chicken embryos and day-old chickens with parvovirus of chicken origin. Avian Pathol. 1985, 14, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.S. Virus infections of the gastrointestinal tract of poultry. Poult. Sci. 1998, 77, 1166–1175. [Google Scholar] [CrossRef] [PubMed]
- Otto, P.; Liebler-Tenorio, E.M.; Elschner, M.; Reetz, J.; Löhren, U.; Diller, R. Detection of rotaviruses and intestinal lesions in broiler chicks from flocks with runting and stunting syndrome (RSS). Avian Dis. 2006, 50, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Pantin-Jackwood, M.J.; Spackman, E.; Woolcock, P.R. Molecular characterization and typing of chicken and turkey astroviruses circulating in the United States: Implications for diagnostics. Avian Dis. 2006, 50, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Zsak, L.; Strother, K.O.; Day, J.M. Development of a polymerase chain reaction procedure for detection of chicken and turkey parvoviruses. Avian Dis. 2009, 53, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Mettifogo, E.; Nuñez, L.F.; Chacón, J.L.; Santander Parra, S.H.; Astolfi-Ferreira, C.S.; Jerez, J.A.; Jones, R.C.; Ferreira, A.J.P. Emergence of enteric viruses in production chickens is a concern for avian health. Sci. World J. 2014, 2014, 450423. [Google Scholar] [CrossRef] [PubMed]
- Devaney, R.; Trudgett, J.; Trudgett, A.; Meharg, C.; Smyth, V. A metagenomic comparison of endemic viruses from broiler chickens with runting-stunting syndrome and from normal birds. Avian Pathol. 2016, 45, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Hueffer, K.; Parrish, C.R. Parvovirus host range, cell tropism and evolution. Curr. Opin. Microbiol. 2003, 6, 392–398. [Google Scholar] [CrossRef]
- Imada, T.; Yamaguchi, S.; Kawamura, H. Pathogenicity for baby chicks of the G-4260 strain of the picornavirus “avian nephritis virus”. Avian Dis. 1979, 23, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.-I.; El-Gazzar, M.; Sellers, H.S.; Dorea, F.; Susan, M.; Kim, T.; Collet, S.; Mundt, E. Investigation into the aetiology of runting and stunting syndrome in chickens. Avian Pathol. 2012, 41, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Kisary, J.; Nagy, B.; Bitay, Z. Presence of parvoviruses in the intestine of chickens showing stunting syndrome. Avian Pathol. 1984, 13, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.; Tseng, C.; Chang, P.; Mei, K.; Wang, S. Genetic variation of viral protein 1 genes of field strains of waterfowl parvoviruses and their attenuated derivatives. Avian Dis. 2004, 48, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Nuñez, L.F.; Sá, L.R.; Santander Parra, S.H.; Astolfi-Ferreira, C.S.; Carranza, C.; Ferreira, A.J.P. Molecular detection of chicken parvovirus in broilers with enteric disorders presenting curving of duodenal loop, pancreatic atrophy, and mesenteritis. Poult. Sci. 2016, 95, 802–810. [Google Scholar] [CrossRef] [PubMed]
- Kisary, J.; Miller-Faures, A.; Rommelaere, J. Presence of fowl parvovirus in fibroblast cell cultures prepared from uninoculated White Leghorn chicken embryos. Avian Pathol. 1987, 16, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Palade, E.A.; Kisary, J.; Benyeda, Z.; Mándoki, M.; Balka, G.; Jakab, C.; Vegh, B.; Demeter, Z.; Rusvai, M. Naturally occurring parvoviral infection in Hungarian broiler flocks. Avian Pathol. 2011, 40, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Strother, K.O.; Zsak, L. Development of an enzyme-linked immunosorbent assay to detect chicken parvovirus-specific antibodies. Avian Dis. 2009, 53, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Day, J.M.; Zsak, L. Determination and analysis of the full-length chicken parvovirus genome. Virology 2010, 399, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Carratalà, A.; Rusinol, M.; Hundesa, A.; Biarnes, M.; Rodriguez-Manzano, J.; Vantarakis, A.; Kern, A.; Suñen, E.; Girones, R.; Bofill-Mas, S. A novel tool for specific detection and quantification of chicken/turkey parvoviruses to trace poultry fecal contamination in the environment. Appl. Environ. Microbiol. 2012, 78, 7496–7499. [Google Scholar] [CrossRef] [PubMed]
- Tarasiuk, K.; Woźniakowski, G.; Samorek-Salamonowicz, E. Occurrence of chicken parvovirus infection in poland. Open Virol. J. 2012, 6, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Finkler, F.; Lima, D.A.; Cerva, C.; Cibulski, S.P.; Teixeira, T.F.; Santos, H.F.; Almeida, L.L.; Roehe, P.M.; Franco, A.C. Chicken parvovirus viral loads in cloacal swabs from malabsorption syndrome-affected and healthy broilers. Trop. Anim. Health Prod. 2016, 48, 1685–1689. [Google Scholar] [CrossRef] [PubMed]
- Nuñez, L.F.; Santander Parra, S.H.; Mettifogo, E.; Astolfi-Ferreira, C.S.; Piantino Ferreira, A.J. Isolation and molecular characterisation of chicken parvovirus from Brazilian flocks with enteric disorders. Br. Poult. Sci. 2015, 56, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Nuñez, L.F.; Santander Parra, S.H.; Astolfi-Ferreira, C.S.; Carranza, C.; De La Torre, D.I.D.; Pedroso, A.C.; Ferreira, A.J.P. Detection of enteric viruses in pancreas and spleen of broilers with runting-stunting syndrome (RSS). Pesqui. Vet. Bras. 2016, 36, 595–599. [Google Scholar] [CrossRef]
- Torre, D.D.; Nuñez, L.F.; Puga, B.; Parra, S.H.S.; Astolfi-Ferreira, C.S.; Piantino Ferreira, A.J. Molecular diagnostic of chicken parvovirus (ChPV) affecting broiler flocks in Ecuador. Braz. J. Poult. Sci. 2018, 20, 121–125. [Google Scholar]
- Murgia, A.M.V.; Rauf, A.; Tang, Y.; Gingerich, E.; Lee, C.; Saif, Y.M. Prevalence of Parvoviruses in Commercial Turkey Flocks. Avian Dis. 2012, 56, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Zsak, L.; Cha, R.M.; Li, F.; Day, J.M. Host Specificity and Phylogenetic Relationships of Chicken and Turkey Parvoviruses. Avian Dis. 2015, 59, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, E.; Allée, C.; Vabret, A.; Eterradossi, N.; Brown, P.A. Single reaction, real time RT-PCR detection of all known avian and human metapneumoviruses. J. Virol. Methods 2018, 251, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7. Molecular evolutionary genetics analysis Version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Day, J.M.; Oakley, B.B.; Seal, B.S.; Zsak, L. Comparative analysis of the intestinal bacterial and RNA viral communities from sentinel birds placed on selected broiler chicken farms. PLoS ONE 2015, 10, e0117210. [Google Scholar] [CrossRef] [PubMed]
- Woolcock, P.R.; Shivaprasad, H.L. Electron microscopic identification of viruses associated with poult enteritis in turkeys grown in California 1993–2003. Avian Dis. 2008, 52, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Dalla-Costa, L.M.; Morello, L.G.; Conte, D.; Pereira, L.A.; Palmeiro, J.K.; Ambrosio, A.; Cardozo, D.; Krieger, M.A.; Raboni, S.M. Comparison of DNA extraction methods used to detect bacterial and yeast DNA from spiked whole blood by real-time PCR. J. Microbiol. Methods 2017, 140, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Klenner, J.; Kohl, C.; Dabrowski, P.W.; Nitsche, A. Comparing viral metagenomic extraction methods. Curr. Issues Mol. Biol. 2017, 24, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Xie, Z.; Deng, X.; Xie, L.; Xie, Z.; Huang, L.; Fan, Q.; Luo, S.; Huang, J.; Zhang, Y.; et al. Genetic and phylogenetic analysis of a novel parvovirus isolated from chickens in Guangxi, China. Arch. Virol. 2016, 161, 3285–3289. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.S.; Lee, H.R.; Jeon, E.O.; Han, M.S.; Min, K.C.; Lee, S.B.; Bae, Y.J.; Cho, S.H.; Mo, J.S.; Kwon, H.M.; et al. Genetic characterization of three novel chicken parvovirus strains based on analysis of their coding sequences. Avian Pathol. 2015, 44, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Day, J.M.; Zsak, L. Recent progress in the characterization of avian enteric viruses. Avian Dis. 2013, 57, 573–580. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, D.I.; Nuñez, L.F.; Astolfi-Ferreira, C.S.; Piantino Ferreira, A.J. Enteric virus diversity examined by molecular methods in Brazilian poultry flocks. Vet. Sci. 2018, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Zsak, L.; Strother, K.O.; Kisary, J. Partial genome sequence analysis of parvoviruses associated with enteric disease in poultry. Avian Pathol. 2008, 37, 435–441. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Gene Target | Assay | Sequences 5’–3’ | Product | Reference |
|---|---|---|---|---|---|
| PVA-F | NS | qPCR | GCA ACT AAC CTG ACC GTG TG | 96 bp | This Study |
| PVA-R | CCC GGA TTC AGA ACC AGT AT | ||||
| PVF-1 | PCR | TTCTAATAACGATATCACTCAAGTTTC | 561 bp | (Zsak et al., 2009) | |
| PVR-1 | TTTGCGCTTGCGGTGAAGTCTGGCTCG |
| Birds | Age | Quantitative PCR | ||||||
|---|---|---|---|---|---|---|---|---|
| Presence or Absence of Clinical Signs of Enteric Disease | ||||||||
| Presence | Absence | |||||||
| Number of Positives/Total of Samples | Result of Detection | Average of CG/µL DNA | Number of Positives/Total of Samples | Result of Detection | Average of CG/µL DNA | |||
| Broilers | Days | 1–7 | 8/9 | + | 1368 | 5/5 | + | 3,722,751 |
| 8–14 | 8/8 | + | 215,461 | 4/4 | + | 5,787,915 | ||
| 15–21 | 16/18 | + | 300,689 | 3/3 | + | 3,943,261 | ||
| 22–28 | 21/21 | + | 4,631,217 | 2/2 | + | 202 | ||
| 29–35 | 13/13 | + | 190,928 | 2/2 | + | 24,621 | ||
| 36–42 | 25/25 | + | 2,586,163 | 8/8 | + | 543,367 | ||
| Layer hens | Weeks | 1–30 | 6/6 | + | 1148 | 5/5 | + | 1073 |
| >31 | 4/4 | + | 37,321 | 1/1 | + | 10 | ||
| Breeder Hens | 1–30 | 0/0 | Na | 0/0 | Na | |||
| >31 | 4/4 | + | 4526 | 3/3 | + | 3294 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nuñez, L.F.; Santander-Parra, S.H.; Chaible, L.; De la Torre, D.I.; Buim, M.R.; Murakami, A.; Zaidan Dagli, M.L.; Astolfi-Ferreira, C.S.; Piantino Ferreira, A.J. Development of a Sensitive Real-Time Fast-qPCR Based on SYBR® Green for Detection and Quantification of Chicken Parvovirus (ChPV). Vet. Sci. 2018, 5, 69. https://doi.org/10.3390/vetsci5030069
Nuñez LF, Santander-Parra SH, Chaible L, De la Torre DI, Buim MR, Murakami A, Zaidan Dagli ML, Astolfi-Ferreira CS, Piantino Ferreira AJ. Development of a Sensitive Real-Time Fast-qPCR Based on SYBR® Green for Detection and Quantification of Chicken Parvovirus (ChPV). Veterinary Sciences. 2018; 5(3):69. https://doi.org/10.3390/vetsci5030069
Chicago/Turabian StyleNuñez, Luis F., Silvana H. Santander-Parra, Lucas Chaible, David I. De la Torre, Marcos R. Buim, Alexandre Murakami, Maria Lucia Zaidan Dagli, Claudete S. Astolfi-Ferreira, and Antonio J. Piantino Ferreira. 2018. "Development of a Sensitive Real-Time Fast-qPCR Based on SYBR® Green for Detection and Quantification of Chicken Parvovirus (ChPV)" Veterinary Sciences 5, no. 3: 69. https://doi.org/10.3390/vetsci5030069
APA StyleNuñez, L. F., Santander-Parra, S. H., Chaible, L., De la Torre, D. I., Buim, M. R., Murakami, A., Zaidan Dagli, M. L., Astolfi-Ferreira, C. S., & Piantino Ferreira, A. J. (2018). Development of a Sensitive Real-Time Fast-qPCR Based on SYBR® Green for Detection and Quantification of Chicken Parvovirus (ChPV). Veterinary Sciences, 5(3), 69. https://doi.org/10.3390/vetsci5030069