
Anaplasma phagocytophilum Manipulates Host Cell Apoptosis by Different Mechanisms to Establish Infection




Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
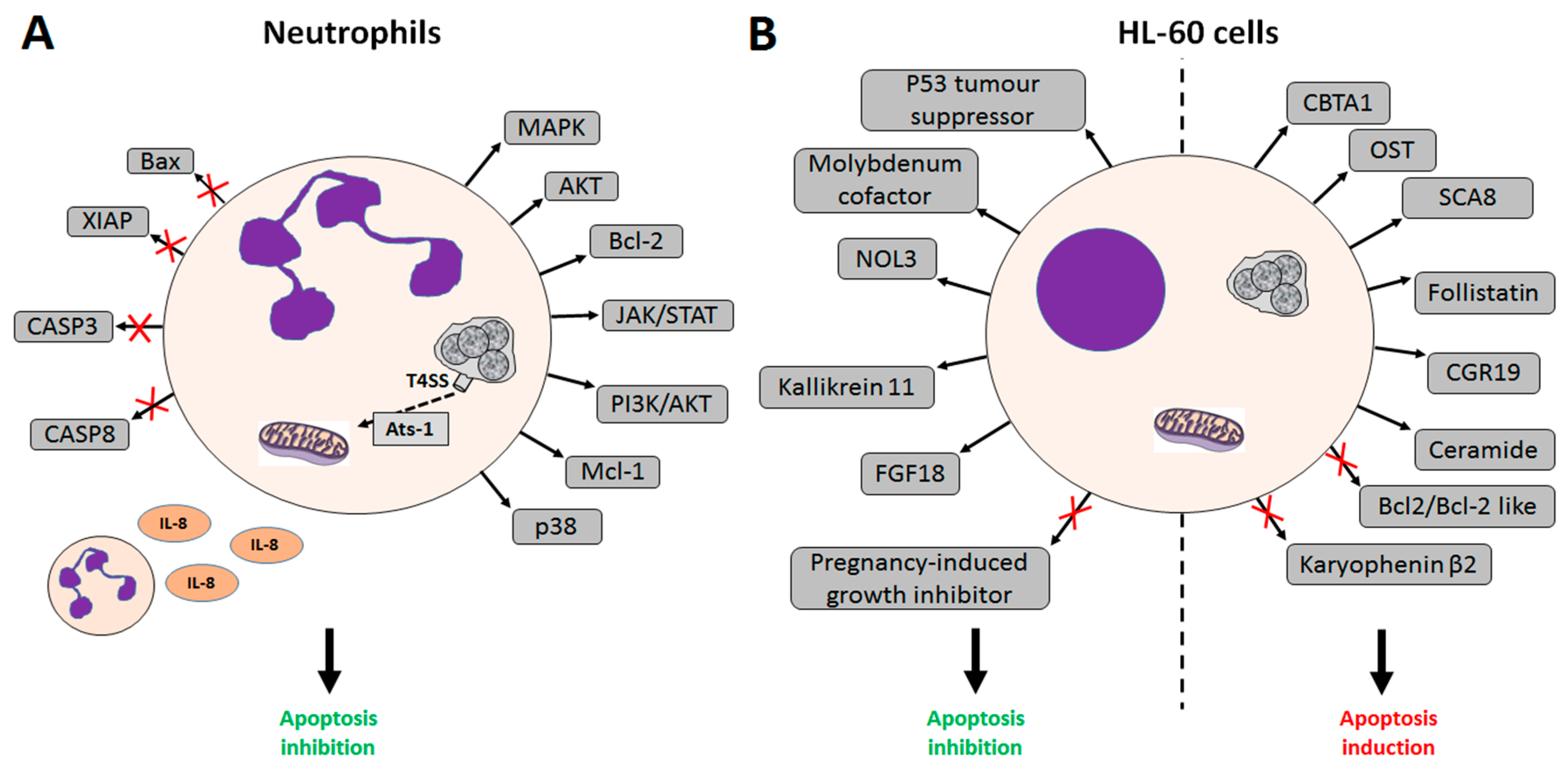
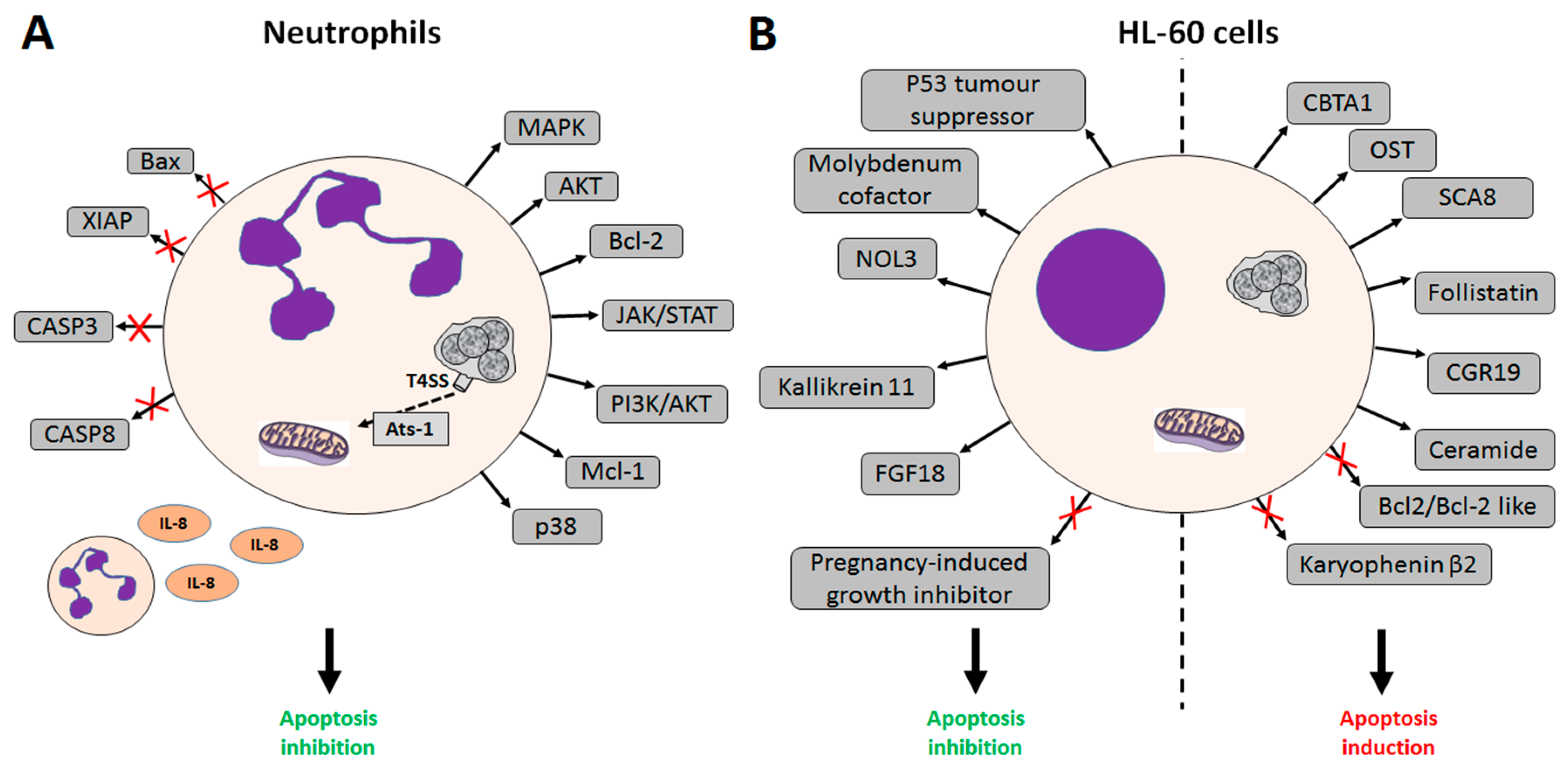
2. A. phagocytophilum Inhibits Apoptosis in Human Cells
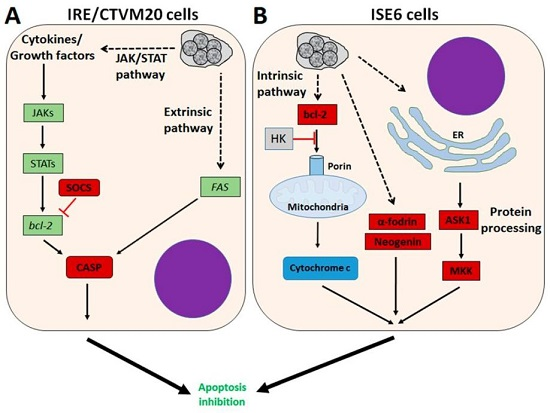
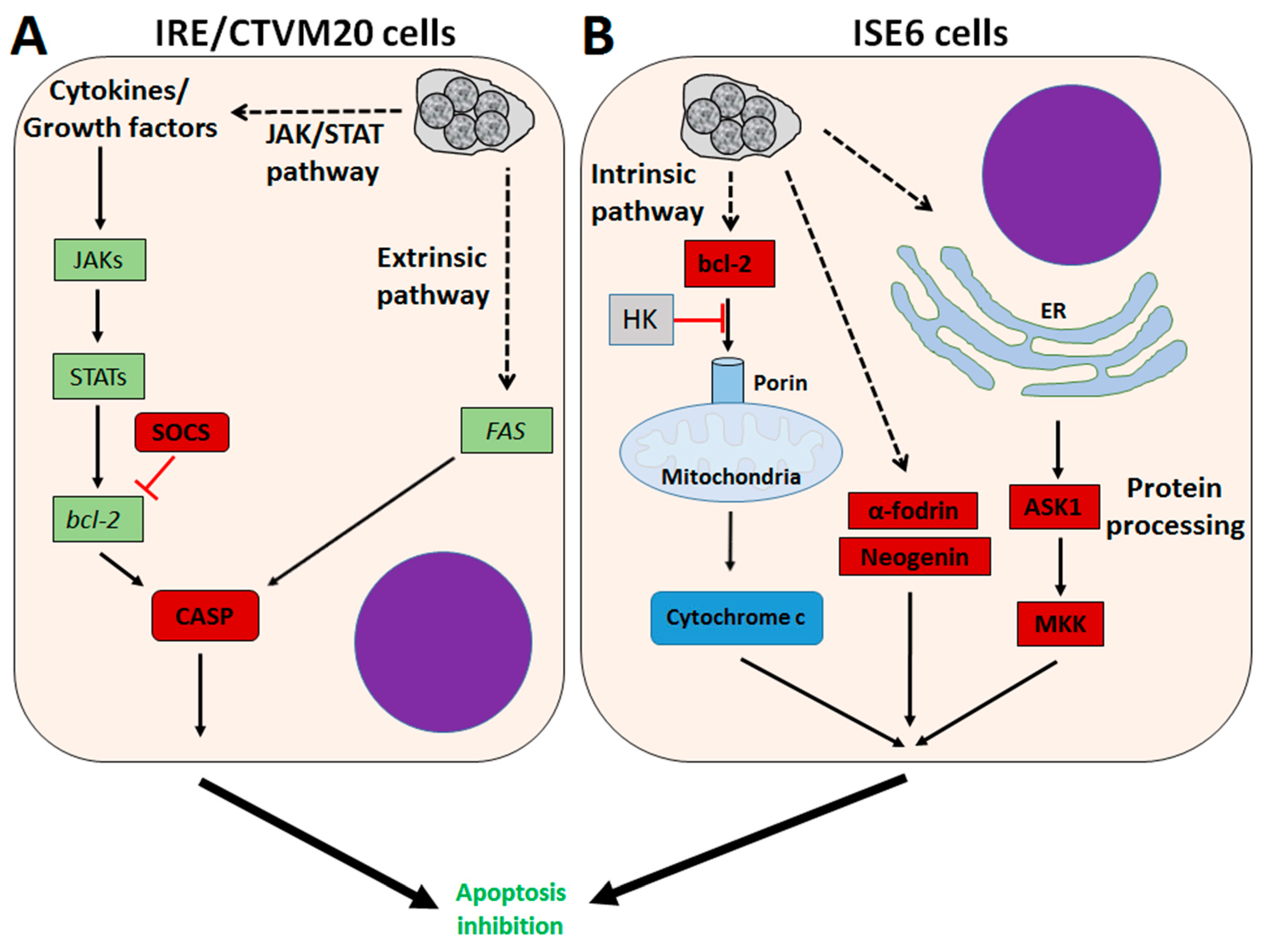
3. A. phagocytophilum Inhibits Apoptosis in Vertebrate Host Cells
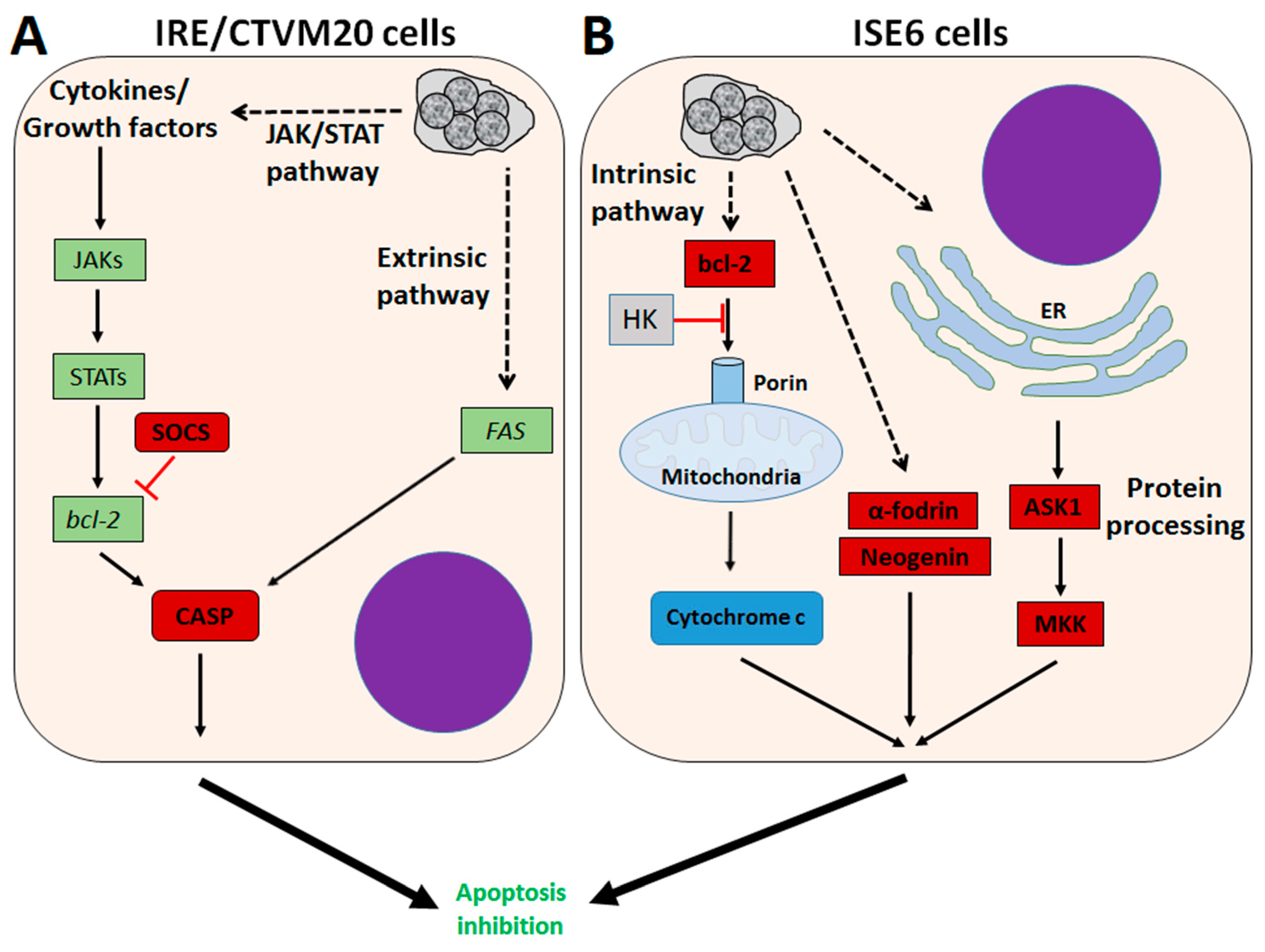
4. A. phagocytophilum Inhibits Apoptosis in Ticks and Tick Cells
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| HGA | Human Granulocytic Anaplasmosis |
| TBF | Tick-borne Fever |
| Ats-1 | Anaplasma translocated substrate 1 protein |
| T4SS | Type IV secretion system |
| PARP | Poly-ADP-ribose polymerase |
| bcl-2 | B cell lymphoma 2 |
| XIAP | X-linked inhibitor of apoptosis protein |
| JAK/STAT | Janus kinase/signal transducers and activators of transcription |
| PI3K | phosphoinositide kinase-3 |
| ROS | Reactive oxygen species |
| AKT | protein kinase B |
| MAP | mitogen-activated protein kinase |
| Mcl-1 | myeloid leukemia cell |
| NOL3 | nucleolar protein 3 |
| edg-2 | G-protein linked receptor |
| SOCS | suppressor of cytokine signalling proteins |
| HK | hexokinase |
| FAS | Fatty Acid Synthase |
| CASP | caspase |
| PEPCK | phosphoenolpyruvate carboxykinase |
| MKK | Mitogen-activated protein kinase kinase |
| ASK1 | apoptosis signal-regulating kinase 1 |
| ER | endoplasmic reticulum |
| HME | histone modifying enzymes |
| GM-CSF | Granulocyte macrophage colony-stimulating factor |
| FGF18 | fibroblast growth factor 18 |
| CBTA1 | Calmodulin- binding transcription activator 1 |
| OST | Heparan sulphate (glucosamine) OST |
| SCA8 | human spinocerebellar ataxia type 8 |
| CGR19 | cell growth regulator 19 |
References
- Severo, M.S.; Pedra, J.H.F.; Ayllón, N.; Kocan, K.M.; de la Fuente, J. Anaplasma. In Molecular Medical Microbiology, 2nd ed.; Tang, Y.W., Sussman, M., Liu, D., Poxton, I., Schwartzman, J., Eds.; Academic Press, Elsevier: New York, NY, USA, 2015; pp. 2033–2042. [Google Scholar]
- Bakken, J.S.; Dumler, J.S. Human granulocytic anaplasmosis. Infect. Dis. Clin. N. Am. 2015, 29, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Woldehiwet, Z. The natural history of Anaplasma phagocytophilum. Vet. Parasitol. 2010, 167, 108–122. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente, J.; Massung, R.F.; Wong, S.J.; Chu, F.K.; Lutz, H.; Meli, M.; von Loewenich, F.D.; Grzeszczuk, A.; Torina, A.; Caracappa, S.; et al. Sequence analysis of the msp4 gene of Anaplasma phagocytophilum strains. J. Clin. Microbiol. 2005, 43, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente, J.; Estrada-Peña, A.; Cabezas-Cruz, A.; Brey, R. Flying ticks: Anciently evolved associations that constitute a risk of infectious disease spread. Parasit. Vectors 2015, 8. [Google Scholar] [CrossRef] [PubMed]
- Estrada-Peña, A.; de la Fuente, J.; Ostfeld, R.S.; Cabezas-Cruz, A. Interactions between tick and transmitted pathogens evolved to minimise competition through nested and coherent networks. Sci. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Torina, A.; Alongi, A.; Naranjo, V.; Scimeca, S.; Nicosia, S.; Di Marco, V.; Caracappa, S.; Kocan, K.M.; de la Fuente, J. Characterization of Anaplasma infections in Sicily, Italy. Ann. N. Y. Acad. Sci. 2008, 1149, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Rar, V.; Golovljova, I. Anaplasma, Ehrlichia, and Candidatus Neoehrlichia bacteria: Pathogenicity, biodiversity, and molecular genetic characteristics, a review. Infect. Genet. Evol. 2011, 11, 1842–1861. [Google Scholar] [CrossRef] [PubMed]
- Kocan, K.M.; de la Fuente, J.; Cabezas-Cruz, A. The genus Anaplasma: New challenges after reclassification. Rev. Sci. Tech. Off. Int. Epiz. 2015, 34, 577–586. [Google Scholar] [CrossRef]
- Stuen, S. Anaplasma phagocytophilum—The most widespread tick-borne infection in animals in Europe. Vet. Res. Commun. 2007, 31, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.H.; Meinken, C.; Bastian, M.; Bruns, H.; Legaspi, A.; Ochoa, M.T.; Krutzik, S.R.; Bloom, B.R.; Ganz, T.; Modlin, R.L.; Stenger, S. Macrophages acquire neutrophil granules for antimicrobial activity against intracellular pathogens. J. Immunol. 2006, 177, 1864–1871. [Google Scholar] [CrossRef] [PubMed]
- Helaine, S.; Thompson, J.A.; Watson, K.G.; Liu, M.; Boyle, C.; Holden, D.W. Dynamics of intracellular bacterial replication at the single cell level. Proc. Natl. Acad. Sci. USA 2010, 107, 3746–3751. [Google Scholar] [CrossRef] [PubMed]
- Ashida, H.; Mimuro, H.; Ogawa, M.; Kobayashi, T.; Sanada, T.; Kim, M.; Sasakawa, C. Host-pathogen interactions cell death and infection: A double-edged sword for host and pathogen survival. J. Cell Biol. 2011, 195, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Rudel, T.; Kepp, O.; Kozjak-Pavlovic, V. Interactions between bacterial pathogens and mitochondrial cell death pathways. Nat. Rev. Microbiol. 2010, 8, 693–705. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente, J.; Estrada-Peña, A.; Cabezas-Cruz, A.; Kocan, K.M. Anaplasma phagocytophilum uses common strategies for infection of ticks and vertebrate hosts. Trends Microbiol. 2016, 24, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Carlyon, J.A.; Fikrig, E. Invasion and survival strategies of Anaplasma phagocytophilum. Cell. Microbiol. 2003, 5, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Galindo, R.C.; de la Fuente, J. Transcriptomics data integration reveals Jak-STAT as a common pathway affected by pathogenic intracellular bacteria in natural reservoir hosts. J. Proteom. Bioinform. 2012, 5, 108–115. [Google Scholar] [CrossRef]
- Lee, H.C.; Goodman, J.L. Anaplasma phagocytophilum causes global induction of antiapoptosis in human neutrophils. Genomics 2006, 88, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Kioi, M.; Han, J.; Puri, R.K.; Goodman, J.L. Anaplasma phagocytophilum-induced gene expression in both human neutrophils and HL-60 cells. Genomics 2008, 92, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Rikihisa, Y. Molecular pathogenesis of Anaplasma phagocytophilum. Clin. Microbiol. Rev. 2011, 24, 469–489. [Google Scholar] [CrossRef] [PubMed]
- Woldehiwet, Z.; Yavari, C. Anaplasma phagocytophilum up-regulates some anti-apoptotic genes in neutrophils and pro-inflammatory genes in mononuclear cells of sheep. J. Comp. Pathol. 2014, 150, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Sprick, M.R.; Walczak, H. The interplay between the Bcl-2 family and death receptor-mediated apoptosis. Biochim. Biophys. Acta 2004, 1644, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Ayllón, N.; Villar, M.; Galindo, R.C.; Kocan, K.M.; Šíma, R.; López, J.A.; Vázquez, J.; Alberdi, P.; Cabezas-Cruz, A.; Kopáček, P.; et al. Systems biology of tissue-specific response to Anaplasma phagocytophilum reveals differentiated apoptosis in the tick vector Ixodes scapularis. PLoS Genet. 2015. [Google Scholar] [CrossRef] [PubMed]
- Cabezas-Cruz, A.; Alberdi, P.; Ayllón, N.; Valdés, J.J.; Pierce, R.; Villar, M.; de la Fuente, J. Anaplasma phagocytophilum increases the levels of histone modifying enzymes to inhibit cell apoptosis and facilitate pathogen infection in the tick vector Ixodes scapularis. Epigenetics 2016. [Google Scholar] [CrossRef] [PubMed]
- Niu, H.; Kozjak-Pavlovic, V.; Rudel, T.; Rikihisa, Y. Anaplasma phagocytophilum Ats-1 is imported into host cell mitochondria and interferes with apoptosis induction. PLoS Pathog. 2010. [Google Scholar] [CrossRef] [PubMed]
- Niu, H.; Rikihisa, Y. Ats-1: A novel bacterial molecule that links autophagy to bacterial nutrition. Autophagy 2013, 9, 787–788. [Google Scholar] [CrossRef] [PubMed]
- Goodman, J.L. Human granulocytic anaplasmosis (Ehrlichiosis). In Tick-Borne Diseases of Humans; Goodman, J.L., Dennis, D.T., Sonenshine, D.E., Eds.; ASM Press: Washington, DC, USA, 2005; pp. 218–238. [Google Scholar]
- Yoshiie, K.; Kim, H.Y.; Mott, J.; Rikihisa, Y. Intracellular infection by the human granulocytic ehrlichiosis agent inhibits human neutrophil apoptosis. Infect. Immun. 2000, 68, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Borjesson, D.L.; Kobayashi, S.D.; Whitney, A.R.; Voyich, J.M.; Argue, C.M.; DeLeo, F.R. Insights into pathogen immune evasion mechanisms: Anaplasma phagocytophilum fails to induce an apoptosis differentiation program in human neutrophils. J. Immunol. 2005, 174, 6364–6372. [Google Scholar] [CrossRef] [PubMed]
- Scaife, H.; Woldehiwet, Z.; Hart, C.A.; Edwards, S.W. Anaplasma phagocytophilum reduces neutrophil apoptosis in vivo. Infect. Immun. 2003, 71, 1995–2001. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Yoshiie, K.; Kuribayashi, F.; Lin, M.; Rikihisa, Y. Anaplasma phagocytophilum inhibits human neutrophil apoptosis via upregulation of bfl-1 maintenance of mitochondrial membrane potential and prevention of caspase 3 activation. Cell. Microbiol. 2005, 7, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Rikihisa, Y. Anaplasma phagocytophilum delays spontaneous human neutrophil apoptosis by modulation of multiple apoptotic pathways. Cell. Microbiol. 2006, 8, 1406–1416. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.S.; Park, J.T.; Dumler, J.S. Anaplasma phagocytophilum delay of neutrophil apoptosis through the p38 mitogen-activated protein kinase signal pathway. Infect. Immun. 2005, 73, 8209–8218. [Google Scholar] [CrossRef] [PubMed]
- Severo, M.S.; Stephens, K.D.; Kotsyfakis, M.; Pedra, J.H. Anaplasma phagocytophilum: Deceptively simple or simply deceptive? Future Microbiol. 2012, 7, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Chow, V.T.; Quek, H.H.; Tock, E.P. Alternative splicing of the p53 tumor suppressor gene in the Molt-4 T-lymphoblastic leukemia cell line. Cancer Lett. 1993, 73, 141–148. [Google Scholar] [CrossRef]
- Goodman, J.L.; Nelson, C.; Vitale, B.; Madigan, J.E.; Dumler, J.S.; Kurtti, T.J.; Munderloh, U.G. Direct cultivation of the causative agent of human granulocytic ehrlichiosis. N. Engl. J. Med. 1996, 334, 209–215. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, J.; Ayoubi, P.; Blouin, E.F.; Almazán, C.; Naranjo, V.; Kocan, K.M. Gene expression profiling of human promyelocytic cells in response to infection with Anaplasma phagocytophilum. Cell. Microbiol. 2005, 7, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Dumler, J.S.; Barbet, A.C.; Bekker, C.P.J.; Dasch, G.A.; Palmer, G.H.; Ray, S.C.; Rikihisa, Y.; Rurangirwa, F.R. Reorganization of the genera in the families Rickettsiaceae and Anaplasmataceae in the order Rickettsiales: Unification of some species of Ehrlichia with Anaplasma, Cowdria with Ehrlichia and Ehrlichia with Neorickettsia, descriptions of six new species combinations and designation of Ehrlichia equi and ‘HGE agent’ as subjective synonyms of Ehrlichia phagocytophila. Int. J. Syst. Evol. Microbiol. 2001, 51, 2145–2165. [Google Scholar] [PubMed]
- Kocan, K.M.; Busby, A.T.; Allison, R.W.; Breshears, M.A.; Coburn, L.; Galindo, R.C.; Ayllón, N.; Blouin, E.F.; de la Fuente, J. Sheep experimentally infected with a human isolate of Anaplasma phagocytophilum serve as a host for infection of Ixodes scapularis ticks. Ticks Tick Borne Dis. 2012, 3, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Reppert, E.; Galindo, R.C.; Ayllón, N.; Breshears, M.A.; Kocan, K.M.; Blouin, E.F.; de la Fuente, J. Studies of Anaplasma phagocytophilum in sheep experimentally infected with the human NY-18 isolate: Characterization of tick feeding sites. Ticks Tick Borne Dis. 2014, 5, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Galindo, R.C.; Ayoubi, P.; García-Pérez, A.L.; Naranjo, V.; Kocan, K.M.; Gortazar, C.; de la Fuente, J. Differential expression of inflammatory and immune response genes in sheep infected with Anaplasma phagocytophilum. Vet. Immunol. Immunopathol. 2008, 126, 27–34. [Google Scholar] [CrossRef] [PubMed]
- López, V.; Alberdi, P.; Fernández de Mera, I.G.; Barasona, J.A.; Vicente, V.; Garrido, J.M.; Torina, A.; Caracappa, S.; Lelli, R.C.; Gortázar, C.; et al. Evidence of co-infection with Mycobacterium bovis and tick-borne pathogens in a naturally infected sheep flock. Ticks Tick Borne Dis. 2015, 7, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.C.; Graeler, M.; Shankar, G.; Spencer, J.; Goetzl, E.J. Lysophospholipid mediators of immunity and neoplasia. Biochim. Biophys. Acta 2002, 1582, 161–167. [Google Scholar] [CrossRef]
- Galindo, R.C.; Ayllón, N.; Smrdel, K.S.; Boadella, M.; Beltrán-Beck, B.; Mazariegos, M.; García, N.; Pérez de la Lastra, J.M.; Avsic-Zupanc, T.; Kocan, K.M.; et al. Gene expression profile suggests that pigs (Sus scrofa) are susceptible to Anaplasma phagocytophilum but control infection. Parasit. Vectors 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Fuente, J.; Gortazar, C. Wild boars as hosts of human-pathogenic Anaplasma phagocytophilum variants. Emerg. Infect. Dis. 2012, 18, 2094–2095. [Google Scholar] [CrossRef] [PubMed]
- Mercado-Curiel, R.F.; Palmer, G.H.; Guerrero, F.D.; Brayton, K.A. Temporal characterisation of the organ-specific Rhipicephalus microplus transcriptional response to Anaplasma marginale infection. Int. J. Parasitol. 2011, 41, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Hajdušek, O.; Šíma, R.; Ayllón, N.; Jalovecká, M.; Perner, J.; de la Fuente, J.; Kopáček, P. Interaction of the tick immune system with transmitted pathogens. Front. Cell. Infect. Microbiol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Villar, M.; Ayllón, N.; Alberdi, P.; Moreno, A.; Moreno, M.; Tobes, R.; Mateos-Hernández, L.; Weisheit, S.; Bell-Sakyi, L.; de la Fuente, J. Integrated metabolomics, transcriptomics and proteomics identifies metabolic pathways affected by Anaplasma phagocytophilum infection in tick cells. Mol. Cell. Proteom. 2015, 14, 3154–3172. [Google Scholar] [CrossRef] [PubMed]
- Ayllón, N.; Villar, M.; Busby, A.T.; Kocan, K.M.; Blouin, E.F.; Bonzón-Kulichenko, E.; Galindo, R.C.; Mangold, A.J.; Alberdi, P.; Pérez de la Lastra, J.M.; et al. Anaplasma phagocytophilum inhibits apoptosis and promotes cytoskeleton rearrangement for infection of tick cells. Infect. Immun. 2013, 81, 2415–2425. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, S.; Fikrig, E. Tick microbiome: The force within. Trends Parasitol. 2015, 31, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Severo, M.S.; Choy, A.; Stephens, K.D.; Sakhon, O.S.; Chen, G.; Chung, D.W.; Le Roch, K.G.; Blaha, G.; Pedra, J.H. The E3 ubiquitin ligase XIAP restricts Anaplasma phagocytophilum colonization of Ixodes scapularis ticks. J. Infect. Dis. 2013, 208, 1830–1840. [Google Scholar] [CrossRef] [PubMed]
- Severo, M.S.; Sakhon, O.S.; Choy, A.; Stephens, K.D.; Pedra, J.H. The “ubiquitous” reality of vector immunology. Cell. Microbiol. 2013, 15, 1070–1078. [Google Scholar] [CrossRef] [PubMed]
- Munderloh, U.G.; Liu, Y.; Wang, M.; Chen, C.; Kurtti, T.J. Establishment, maintenance and description of cell lines from the tick Ixodes scapularis. J. Parasitol. 1994, 80, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Munderloh, U.G.; Madigan, J.E.; Dumler, J.S.; Goodman, J.L.; Hayes, S.F.; Barlough, J.E.; Nelson, C.M.; Kurtti, T.J. Isolation of the equine granulocytic ehrlichiosis agent, Ehrlichia equi, in tick cell culture. J. Clin. Microbiol. 1996, 34, 664–670. [Google Scholar] [PubMed]
- Munderloh, U.G.; Tate, C.M.; Lynch, M.J.; Howerth, E.W.; Kurtti, T.J.; Davidson, W.R. Isolation of an Anaplasma sp. organism from white-tailed deer by tick cell culture. J. Clin. Microbiol. 2003, 41, 4328–4335. [Google Scholar] [CrossRef] [PubMed]
- Bell-Sakyi, L.; Zweygarth, E.; Blouin, E.F.; Gould, E.A.; Jongejan, F. Tick cell lines: Tools for tick and tick-borne disease research. Trends Parasitol. 2007, 23, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Alberdi, P.; Ayllón, N.; Cabezas-Cruz, A.; Bell-Sakyi, L.; Zweygarth, E.; Stuen, S.; de la Fuente, J. Infection of Ixodes spp. tick cells with different Anaplasma phagocytophilum isolates induces the inhibition of apoptotic cell death. Ticks Tick Borne Dis. 2015, 6, 758–767. [Google Scholar] [PubMed]
- Alberdi, P.; Mansfield, K.L.; Manzano-Román, R.; Cook, C.; Ayllón, N.; Villar, M.; Johnson, N.; Fooks, A.R.; de la Fuente, J. Tissue-specific signatures in the transcriptional response to Anaplasma phagocytophilum infection of Ixodes scapularis and Ixodes ricinus tick cell lines. Front. Cell. Infect. Microbiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, E.; Tauszig-Delamasure, S.; Monnier, P.P.; Mueller, B.K.; Strittmatter, S.M.; Mehlen, P.; Chédotal, A. RGM and its receptor neogenin regulate neuronal survival. Nat. Cell Biol. 2004, 6, 749–755. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente, J.; Villar, M.; Cabezas-Cruz, A.; Estrada-Peña, A.; Ayllón, N.; Alberdi, P. Tick-host-pathogen interactions: Conflict and cooperation. PLoS Pathog. 2016. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alberdi, P.; Espinosa, P.J.; Cabezas-Cruz, A.; De la Fuente, J. Anaplasma phagocytophilum Manipulates Host Cell Apoptosis by Different Mechanisms to Establish Infection. Vet. Sci. 2016, 3, 15. https://doi.org/10.3390/vetsci3030015
Alberdi P, Espinosa PJ, Cabezas-Cruz A, De la Fuente J. Anaplasma phagocytophilum Manipulates Host Cell Apoptosis by Different Mechanisms to Establish Infection. Veterinary Sciences. 2016; 3(3):15. https://doi.org/10.3390/vetsci3030015
Chicago/Turabian StyleAlberdi, Pilar, Pedro J. Espinosa, Alejandro Cabezas-Cruz, and José De la Fuente. 2016. "Anaplasma phagocytophilum Manipulates Host Cell Apoptosis by Different Mechanisms to Establish Infection" Veterinary Sciences 3, no. 3: 15. https://doi.org/10.3390/vetsci3030015
APA StyleAlberdi, P., Espinosa, P. J., Cabezas-Cruz, A., & De la Fuente, J. (2016). Anaplasma phagocytophilum Manipulates Host Cell Apoptosis by Different Mechanisms to Establish Infection. Veterinary Sciences, 3(3), 15. https://doi.org/10.3390/vetsci3030015