
Mechanisms of Drug Resistance in Veterinary Oncology— A Review with an Emphasis on Canine Lymphoma


Abstract
:1. Introduction. Treatment Failure versus Drug Resistance
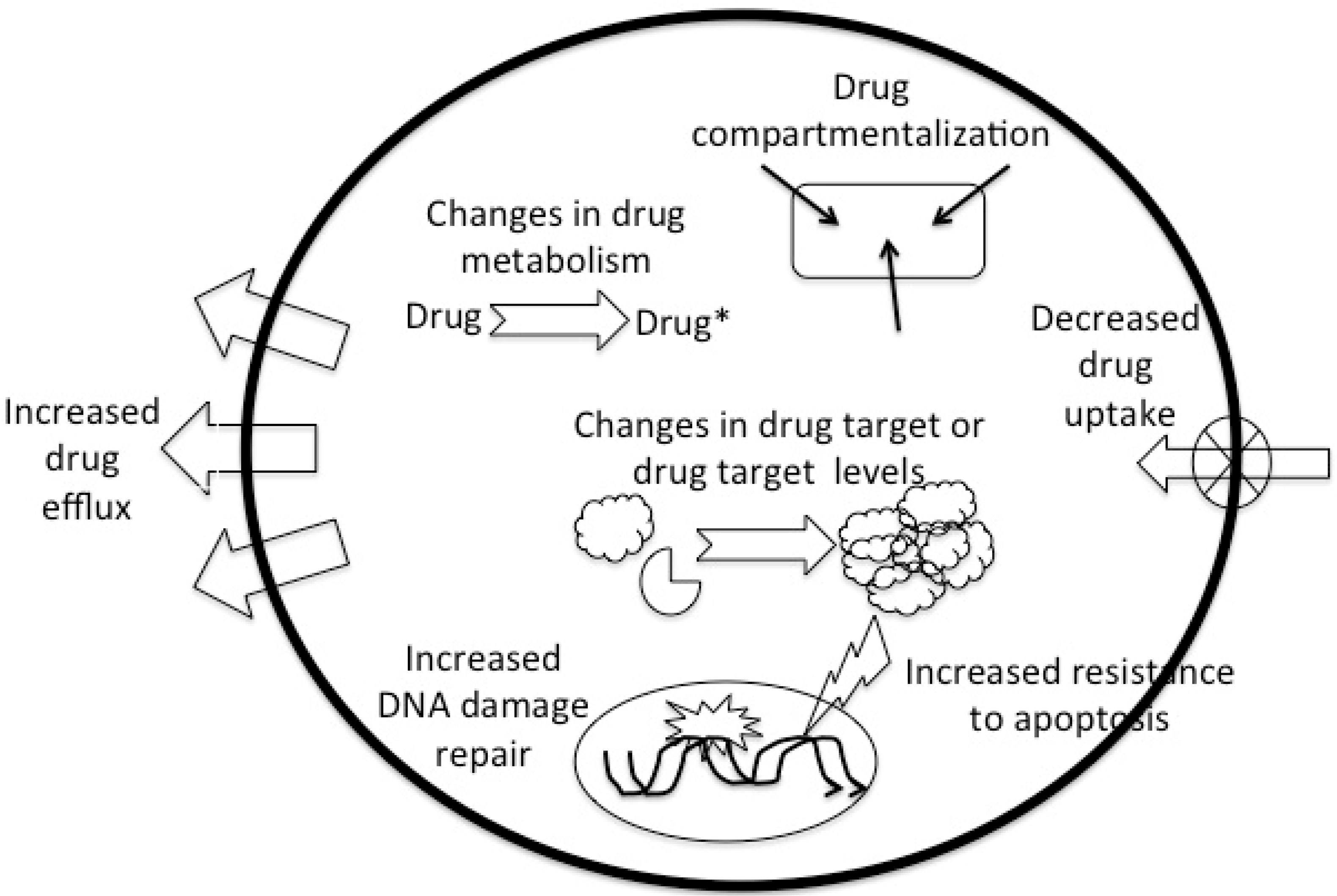
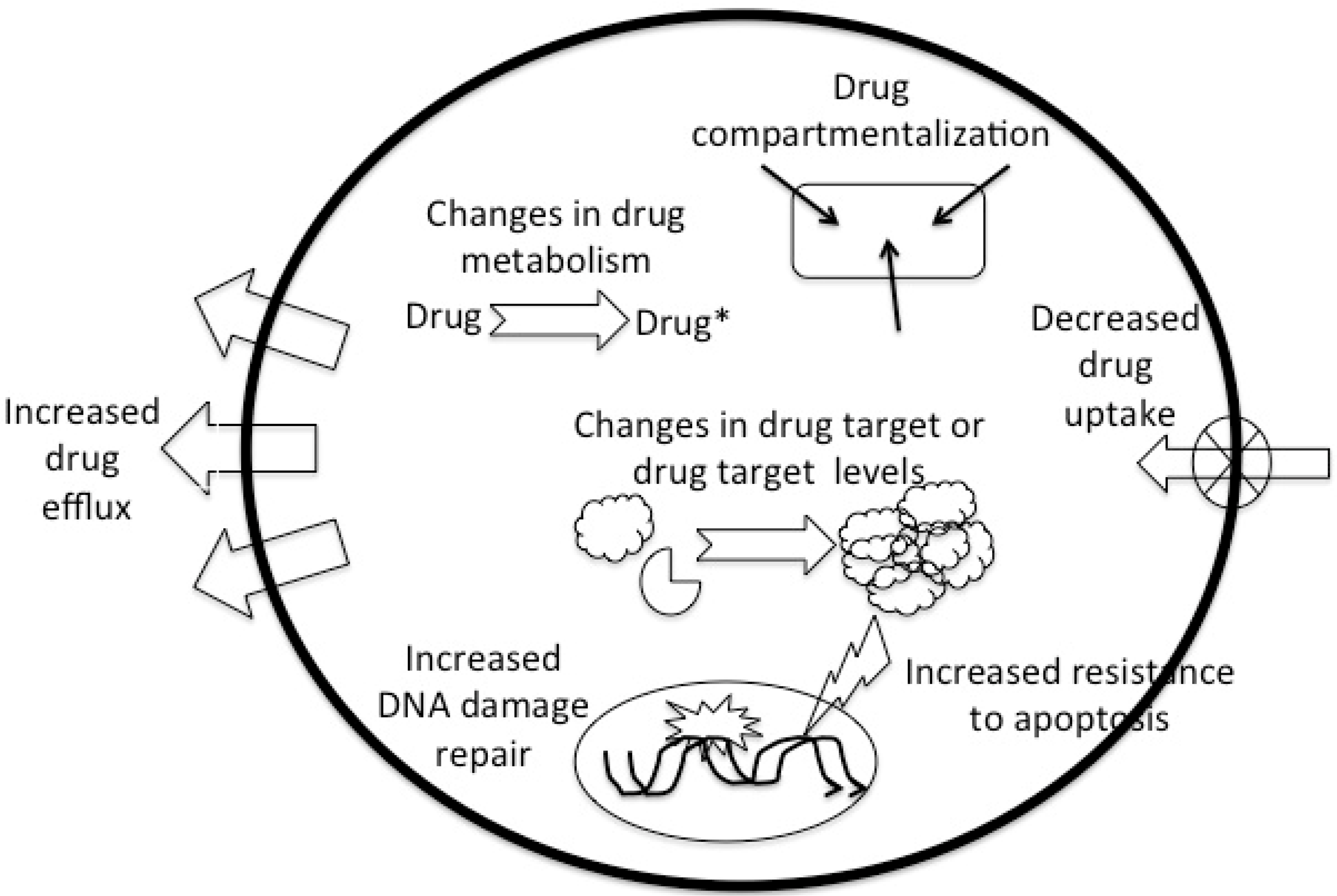
2. Drug Resistance-Mechanisms

{kind=link}
{kind=link}
| Mechanism | Drugs |
|---|---|
| Decreased uptake | methotrexate, other antimetabolites, nitrogen mustard, melphalan, cisplatin |
| Changes in drug metabolism (changes in activation or inactivation) | many antimetabolites (e.g., 5-fluorouracil, cytosine arabinoside), alkylating agents, cisplatin |
| Increased efflux (including compartmentalization) | methotrexate, melphalan, vinca-alkaloids, taxanes, etoposide, anthracyclines |
| Modifications in target enzyme | methotrexate, other antimetabolites, topoisomerase inhibitors |
| Increased DNA repair | alkylating agents, cisplatin, anthracyclines, etoposide |
| Resistance to apoptosis | alkylating agents, cisplatin, anthracyclines, etoposide |
3. Development of Drug Resistance
4. Pharmacodynamic Mechanisms Associated with Drug Resistance
4.1. Decreased Drug Uptake
4.2. Changes in Drug Metabolism
4.3. Increased Drug Efflux
4.4. Changes in Drug Target
4.5. Repair of Drug-Induced DNA Damage
4.6. Resistance to Apoptosis
5. ATP-Binding Cassette Transporters
5.1. History
5.2. ABC-Transporters
5.3. Assessing ABC-Transporter Expression and Function
5.4. ABC-Transporters in Pharmacology
| Tissue | P-gp | MRP1 | MRP2 | BCRP |
|---|---|---|---|---|
| Lung | apical | basolateral | not detected | apical |
| Intestine | ||||
| Duodenum | apical | basolateral | apical | apical |
| Jejunum | apical | basolateral | apical | apical |
| Ileum | apical | basolateral | apical | apical |
| Colon | apical | basolateral | apical | apical |
| Liver | apical | basolateral | not detected | apical |
| Kidney | apical | basolateral | apical | not detected |
| Brain | ||||
| BBB | apical | apical | apical | apical |
| BCSFB | apical | basolateral | - | - |
| Testis | apical | basolateral | not detected | not detected |
| Placenta | apical | basolateral | apical | apical |
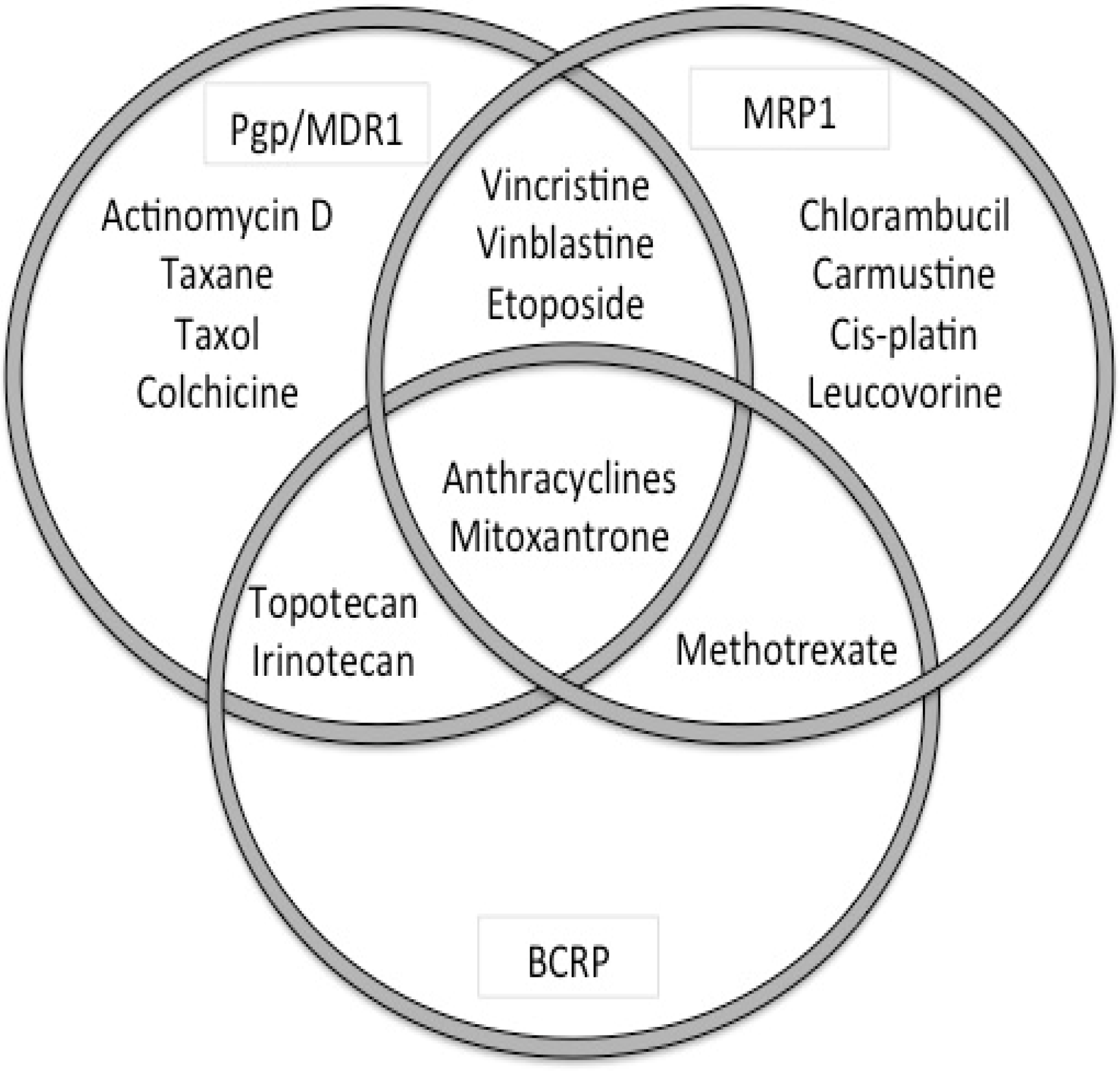
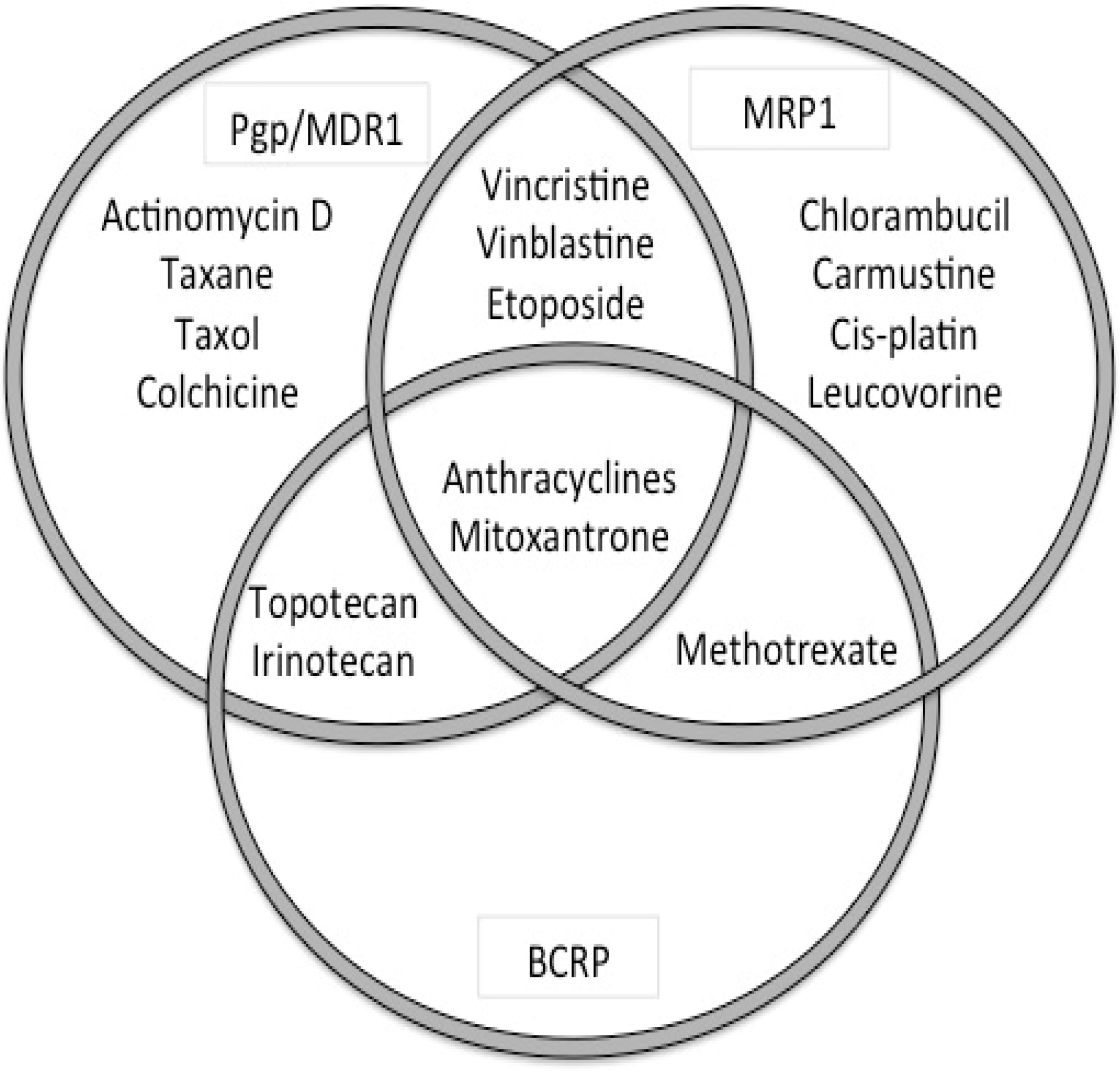
5.5. ABC-Transporters in Drug Resistance
| Gene | Substrates | Inhibitors |
|---|---|---|
| ABCB1 (MDR1, P-gp) | colchicine, doxorubicin, vincristine, vinblastine, paclitaxel, etoposide, digoxin, saquinivir | verapamil, PSC833, GF120918 (GG918), V-104, Pluronic L61, LY335979, XR9576, OC144-093 |
| ABCC1 (MRP1) | doxorubicin, daunorubicin, vincristine, vinblastine, etoposide, colchicine | Cyclosporin A, V-104, MK571 |
| ABCC2 (MRP2, cMOAT) | vinblastine, sulfinpyrazone | PSC833, MK571 |
| ABCC3 | methotrexate, etoposide | |
| ABCC4 | nucleoside monophosphates (thiopurines) | MK571 |
| ABCC5 | nucleoside monophosphates | |
| ABCG2 (BCRP) | mitoxantrone, topotecan, doxorubicin*, daunorubicin*, CPT-11 | fumitremorgin C, Ko143, GF120918 |
5.5.1. ABCB1 or MDR1/P-gp
| Drugs Used in Oncology | Non-Oncological Drugs | |||
|---|---|---|---|---|
| Cytotoxic agents | Anthracyclines | doxorubicin, daunorubicin, epirubicin | Antibiotics | doxycycline, erythromycin, rifampin, tetracycline |
| Anthracenes | mitoxantrone, bisantrene | Antifungals | itraconazole, ketoconazole | |
| Antitumor antibiotics | actinomycin-D, mitomycin-C, plicamycin mithramycin) | Antiparasiticides | ivermectin, moxidectin, selamectin | |
| Taxanes | paclitaxel, docetaxel | Antiemetics | domperideone, ondansetron | |
| Topoisomerase I inhibitors (campothecins) | irinotecan, topotecan | Antidiarrheal agents | loperamide | |
| Topoisomerase II inhibitors (epipodophyllotoxins) | etoposide, teniposide | Anticonvulsant drugs | phenobarbital, phenytoin, levetiracetam | |
| Vinca-alkaloids | vincristine, vinblastine, vinorelbine, vindesine | Cardiac drugs | digoxin, diltiazem, quinidine, verapamil losartan | |
| Antihistamines (H2-antagonsits) | cimetidine, ranitidine, terfenadine | |||
| Steroids | Glucocorticoids | cortisol, dexamethasone, hydrocortisone, methylprednisolone, prednisolone, triamcinolone | Immunosupppressants | cyclosporine A, tacrolimus |
| Mineralocorticoids | aldosterone | Opioids | butorphanol, morphine | |
| Sex steroids | estradiol, progesterone | Miscellaneous | amitryptiline, colchicine, phenothiazines | |
| Tyrosine kinase inhibitors | imatinib, gefitinib | |||
| Collie | Shetland sheepdog |
| Old English sheepdog | Australian shepherd |
| White German shepherd dog | Miniature Australian shepherd |
| English shepherd | Silken windhound |
| Longhaired whippet | McNab |
5.5.2. ABCC1 or MRP1
| Drugs Used in Oncology | Non-Oncological Drugs | |||
|---|---|---|---|---|
| Cytotoxic agents | Anthracyclines | doxorubicin, daunorubicin, epirubicin, idarubicin | Antibiotics | difloxacin grepafloxicin |
| Topoisomerase I inhibitors (campothecins) | irinotecan, topotecan | Metalloids | arsenite trivalent antimonite | |
| Topoisomerase II inhibitors (epipodophyllotoxins) | etoposide, teniposide | Peptides | glutathione | |
| Vinca-alkaloids | vincristine, vinblastine, vinorelbine, vindesine | Glutathione conjugates | cyclophosphamide-SG, doxorubicin-SG melphalan-SG, | |
| Antifolates | methotrexate | Sulfate conjugates | dehydroepiandrosterone-3-sulfate estrone-3-sulfate, | |
| Tyrosine kinase inhibitors | imatinib, gefitinib | Glucuronide conjugates | estradiol-17-β-D-glucuronide, etoposide-glucuronide, irinotecan-glucuronide, | |
| Folates | folic acid, L-leucovorin | |||
5.5.3. ABCG2 or BCRP
| Drugs Used in Oncology | Non-Oncological Drugs | |||
|---|---|---|---|---|
| Cytotoxic agents | Anthracyclines | doxorubicin (mutant form), daunorubicin | Antibiotics | ciprofloxacin, norfloxacin, nitrofurantoin |
| Anthracenedione | mitoxantrone, bisantrene (mutant form) | Anthelmintics | albendazole, oxfendazole | |
| Topoisomerase I inhibitors (campothecins) | irinotecan, topotecan | Diuretics | furosemide, hydrochlorothiazide | |
| Topoisomerase II inhibitors (epipodophyllotoxins) | etoposide, teniposide | Porphyrins | pheophorbide A, protoporphyrin IX, hematoporphyrin | |
| Antifolates | methotrexate | Flavonoids | genestein, quercetin | |
| Tyrosine kinase inhibitors | imatinib, gefitinib, lapatinib | Fungal toxins | alfatoxin B, fumitremorgin C, Ko143 | |
| Drug & metabolite compounds | acetaminophen-sulfate, estrone-3-sulfate, dehydroepiandrosterone-3-sulfate, estradiol-17-β-D-glucuronide, dinitrophenyl-S-glutathione | |||
5.6. Induction and Regulation of ABC-Transporter Expression
6. Drug Resistance due to ABC-Transporters in Canine Lymphoma
7. Glucocorticoids and Drug Resistance
8. Targeting Drug Resistance
8.1. Prevention of Drug Resistance-Use of Cytotoxic Agents
8.2. Modulation of Drug Resistance-ABC-Transporters Modulators
| Generation | P-gp | MRP1 | BCRP |
|---|---|---|---|
| First | verapamil quinidine cyclosporine A | Ko143 Pantoprazole Gefitinib? Imatinib? Quercetin? | |
| Second | PSC833 (valspodar) VX-710 (biricodar) | VX-710 (biricodar) | VX-710 (biricodar) |
| Third | GF120918 (elacridar) XR9576 (tariquidar) LY335979 (zosuquidar) ONT–093 (ontogen) | GF120918 (elacridar) XR9576 (tariquidar) |
8.3. Alternative Drugs and Therapies
9. Summary, Concluding Remarks and Future Studies
Conflicts of Interest
References
- Tredan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Lage, H. An overview of cancer multidrug resistance: A still unsolved problem. Cell Mol. Life Sci. 2008, 65, 3145–3167. [Google Scholar] [CrossRef] [PubMed]
- Goldie, J.H.; Coldman, A.J. The genetic origin of drug resistance in neoplasms: Implications for systemic therapy. Cancer Res. 1984, 44, 3643–3653. [Google Scholar] [PubMed]
- Skipper, H.E.; Schabel, F.M., Jr.; Wilcox, W.S. Experimental evaluation of potential anticancer agents. XIII. On the criteria and kinetics associated with “curability” of experimental leukemia. Cancer Chemother. Rep. 1964, 35, 1–111. [Google Scholar] [PubMed]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer. 2005, 5, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Giovannetti, E.; Erozenci, A.; Smit, J.; Danesi, R.; Peters, G.J. Molecular mechanisms underlying the role of microRNAs (miRNAs) in anticancer drug resistance and implications for clinical practice. Crit. Rev. Oncol. Hematol. 2012, 81, 103–122. [Google Scholar] [CrossRef] [PubMed]
- Wilting, R.H.; Dannenberg, J.H. Epigenetic mechanisms in tumorigenesis, tumor cell heterogeneity and drug resistance. Drug Resist. Updat 2012, 15, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Hediger, M.A.; Romero, M.F.; Peng, J.B.; Rolfs, A.; Takanaga, H.; Bruford, E.A. The ABCs of solute carriers: Physiological, pathological and therapeutic implications of human membrane transport proteinsIntroduction. Pflugers Arch. 2004, 447, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Kuhne, A.; Tzvetkov, M.V.; Hagos, Y.; Lage, H.; Burckhardt, G.; Brockmoller, J. Influx and efflux transport as determinants of melphalan cytotoxicity: Resistance to melphalan in MDR1 overexpressing tumor cell lines. Biochem. Pharmacol. 2009, 78, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Drori, S.; Jansen, G.; Mauritz, R.; Peters, G.J.; Assaraf, Y.G. Clustering of mutations in the first transmembrane domain of the human reduced folate carrier in GW1843U89-resistant leukemia cells with impaired antifolate transport and augmented folate uptake. J. Biol. Chem. 2000, 275, 30855–30863. [Google Scholar] [CrossRef] [PubMed]
- Galmarini, C.M.; Mackey, J.R.; Dumontet, C. Nucleoside analogues: Mechanisms of drug resistance and reversal strategies. Leukemia 2001, 15, 875–890. [Google Scholar] [CrossRef] [PubMed]
- Michael, M.; Doherty, M.M. Drug metabolism by tumours: Its nature, relevance and therapeutic implications. Expert Opin. Drug Metab. Toxicol. 2007, 3, 783–803. [Google Scholar] [CrossRef] [PubMed]
- Ban, N.; Takahashi, Y.; Takayama, T.; Kura, T.; Katahira, T.; Sakamaki, S.; Niitsu, Y. Transfection of glutathione S-transferase (GST)-pi antisense complementary DNA increases the sensitivity of a colon cancer cell line to adriamycin, cisplatin, melphalan, and etoposide. Cancer Res. 1996, 56, 3577–3582. [Google Scholar] [PubMed]
- Gorlick, R.; Goker, E.; Trippett, T.; Waltham, M.; Banerjee, D.; Bertino, J.R. Intrinsic and acquired resistance to methotrexate in acute leukemia. N. Engl. J. Med. 1996, 335, 1041–1048. [Google Scholar] [PubMed]
- Surowiak, P.; Materna, V.; Maciejczyk, A.; Pudelko, M.; Markwitz, E.; Spaczynski, M.; Dietel, M.; Zabel, M.; Lage, H. Nuclear metallothionein expression correlates with cisplatin resistance of ovarian cancer cells and poor clinical outcome. Virchows Arch. 2007, 450, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Hifumi, T.; Miyoshi, N.; Kawaguchi, H.; Nomura, K.; Yasuda, N. Immunohistochemical detection of proteins associated with multidrug resistance to anti-cancer drugs in canine and feline primary pulmonary carcinoma. J. Vet. Med. Sci. 2010, 72, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Arancia, G.; Calcabrini, A.; Meschini, S.; Molinari, A. Intracellular distribution of anthracyclines in drug resistant cells. Cytotechnology 1998, 27, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, M.A.; Neefjes, J.J.; Mathari, A.E.; Flens, M.J.; Scheffer, G.L.; Scheper, R.J. Overexpression of the ABC transporter TAP in multidrug-resistant human cancer cell lines. Br. J. Cancer 1996, 74, 1961–1967. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, M.A.; Scheffer, G.L.; Flens, M.J.; Schroeijers, A.B.; van der Valk, P.; Scheper, R.J. Major vault protein LRP-related multidrug resistance. Eur. J. Cancer 1996, 32A, 979–984. [Google Scholar] [CrossRef]
- Tomiyasu, H.; Goto-Koshino, Y.; Takahashi, M.; Fujino, Y.; Ohno, K.; Tsujimoto, H. Quantitative analysis of mRNA for 10 different drug resistance factors in dogs with lymphoma. J. Vet. Med. Sci. 2010, 72, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, Y.C.; Sharma, R.; Yadav, S.; Dwivedi, S.; Sharma, A.; Awasthi, S. The non-ABC drug transporter RLIP76 (RALBP-1) plays a major role in the mechanisms of drug resistance. Curr. Drug Metab. 2007, 8, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D.; Mayer-Kuckuk, P.; Capiaux, G.; Budak-Alpdogan, T.; Gorlick, R.; Bertino, J.R. Novel aspects of resistance to drugs targeted to dihydrofolate reductase and thymidylate synthase. Biochim. Biophys. Acta 2002, 1587, 164–173. [Google Scholar] [CrossRef]
- Kavallaris, M. Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer. 2010, 10, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y. DNA topoisomerase I and II in cancer chemotherapy: Update and perspectives. Cancer Chemother. Pharmacol. 1993, 32, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Slupphaug, G.; Kavli, B.; Krokan, H.E. The interacting pathways for prevention and repair of oxidative DNA damage. Mutat. Res. 2003, 531, 231–251. [Google Scholar] [CrossRef] [PubMed]
- Scartozzi, M.; Franciosi, V.; Campanini, N.; Benedetti, G.; Barbieri, F.; Rossi, G.; Berardi, R.; Camisa, R.; Silva, R.R.; Santinelli, A.; Ardizzoni, A.; Crino, L.; Rindi, G.; Cascinu, S. Mismatch repair system (MMR) status correlates with response and survival in non-small cell lung cancer (NSCLC) patients. Lung Cancer 2006, 53, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Drablos, F.; Feyzi, E.; Aas, P.A.; Vaagbo, C.B.; Kavli, B.; Bratlie, M.S.; Pena-Diaz, J.; Otterlei, M.; Slupphaug, G.; Krokan, H.E. Alkylation damage in DNA and RNA--repair mechanisms and medical significance. DNA Repair (Amst) 2004, 3, 1389–1407. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Kaina, B. Regulation of repair of alkylation damage in mammalian genomes. Prog. Nucleic Acid Res. Mol. Biol. 1993, 44, 109–142. [Google Scholar] [PubMed]
- Esteller, M.; Garcia-Foncillas, J.; Andion, E.; Goodman, S.N.; Hidalgo, O.F.; Vanaclocha, V.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med. 2000, 343, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Gaidano, G.; Goodman, S.N.; Zagonel, V.; Capello, D.; Botto, B.; Rossi, D.; Gloghini, A.; Vitolo, U.; Carbone, A.; Baylin, S.B.; Herman, J.G. Hypermethylation of the DNA repair gene O(6)-methylguanine DNA methyltransferase and survival of patients with diffuse large B-cell lymphoma. J. Natl. Cancer Inst. 2002, 94, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.M.; Chen, Z.P.; Xu, Z.Y.; Christodoulopoulos, G.; Bello, V.; Mohr, G.; Aloyz, R.; Panasci, L.C. In vitro evidence for homologous recombinational repair in resistance to melphalan. J. Natl. Cancer Inst. 2001, 93, 1473–1478. [Google Scholar] [CrossRef] [PubMed]
- Klopfleisch, R.; Gruber, A.D. Increased expression of BRCA2 and RAD51 in lymph node metastases of canine mammary adenocarcinomas. Vet. Pathol. 2009, 46, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Klopfleisch, R.; Schutze, M.; Gruber, A.D. RAD51 protein expression is increased in canine mammary carcinomas. Vet. Pathol. 2010, 47, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Munday, J.S.; French, A.F.; Gibson, I.R.; Gwynne, K. Widespread mismatch repair protein expression in canine cutaneous mast cell tumors. Vet. Pathol. 2009, 46, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Nagano, H.; Sakon, M.; Yamamoto, T.; Ota, H.; Wada, H.; Damdinsuren, B.; Noda, T.; Marubashi, S.; Miyamoto, A.; Takeda, Y.; Umeshita, K.; Nakamori, S.; Dono, K.; Monden, M. Role of the Fas/FasL pathway in combination therapy with interferon-alpha and fluorouracil against hepatocellular carcinoma in vitro. J. Hepatol. 2007, 46, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Morad, S.A.; Cabot, M.C. Ceramide-orchestrated signalling in cancer cells. Nat. Rev. Cancer. 2013, 13, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Ocker, M.; Hopfner, M. Apoptosis-modulating drugs for improved cancer therapy. Eur. Surg. Res. 2012, 48, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Lee, S.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 2002, 109, 335–346. [Google Scholar] [CrossRef]
- Sokolowska, J.; Cywinska, A.; Malicka, E. P53 Expression in Canine Lymphoma. J. Vet. Med. A Physiol. Pathol. Clin. Med. 2005, 52, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Sueiro, F.A.; Alessi, A.C.; Vassallo, J. Canine lymphomas: A morphological and immunohistochemical study of 55 cases, with observations on p53 immunoexpression. J. Comp. Pathol. 2004, 131, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, N.; Stewart, J.; Brown, R.; Milner, J. Mutations of the p53 gene in canine lymphoma and evidence for germ line p53 mutations in the dog. Oncogene 1998, 16, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Setoguchi, A.; Sakai, T.; Okuda, M.; Minehata, K.; Yazawa, M.; Ishizaka, T.; Watari, T.; Nishimura, R.; Sasaki, N.; Hasegawa, A.; Tsujimoto, H. Aberrations of the p53 tumor suppressor gene in various tumors in dogs. Am. J. Vet. Res. 2001, 62, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Kornblau, S.M.; Ruvolo, P.P.; May, W.S., Jr. Regulation of Bcl2 phosphorylation and potential significance for leukemic cell chemoresistance. J. Natl. Cancer. Inst. Monogr. 2001, 28, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Sjostrom, J.; Blomqvist, C.; von Boguslawski, K.; Bengtsson, N.O.; Mjaaland, I.; Malmstrom, P.; Ostenstadt, B.; Wist, E.; Valvere, V.; Takayama, S.; Reed, J.C.; Saksela, E. The predictive value of bcl-2, bax, bcl-xL, bag-1, fas, and fasL for chemotherapy response in advanced breast cancer. Clin. Cancer Res. 2002, 8, 811–816. [Google Scholar] [PubMed]
- van Oosterwijk, J.G.; Herpers, B.; Meijer, D.; Briaire-de Bruijn, I.H.; Cleton-Jansen, A.M.; Gelderblom, H.; van de Water, B.; Bovee, J.V. Restoration of chemosensitivity for doxorubicin and cisplatin in chondrosarcoma in vitro: BCL-2 family members cause chemoresistance. Ann. Oncol. 2012, 23, 1617–1626. [Google Scholar] [CrossRef] [PubMed]
- Rebhun, R.B.; Lana, S.E.; Ehrhart, E.J.; Charles, J.B.; Thamm, D.H. Comparative analysis of survivin expression in untreated and relapsed canine lymphoma. J. Vet. Intern. Med. 2008, 22, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.B.; Minden, M.D.; McCulloch, E.A. Direct evidence for the participation of bcl-2 in the regulation by retinoic acid of the Ara-C sensitivity of leukemic stem cells. Leukemia 1995, 9, 1667–1673. [Google Scholar] [PubMed]
- Tannock, I.F.; Lee, C. Evidence against apoptosis as a major mechanism for reproductive cell death following treatment of cell lines with anti-cancer drugs. Br. J. Cancer 2001, 84, 100–105. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Kempf, C.R.; Chappell, W.H.; Abrams, S.L.; Stivala, F.; Malaponte, G.; Nicoletti, F.; Libra, M.; Basecke, J.; Maksimovic-Ivanic, D.; Mijatovic, S.; Montalto, G.; Cervello, M.; Cocco, L.; Martelli, A.M. Therapeutic resistance resulting from mutations in Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR signaling pathways. J. Cell. Physiol. 2011, 226, 2762–2781. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Cole, S.P.; Bhardwaj, G.; Gerlach, J.H.; Mackie, J.E.; Grant, C.E.; Almquist, K.C.; Stewart, A.J.; Kurz, E.U.; Duncan, A.M.; Deeley, R.G. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science 1992, 258, 1650–1654. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.A.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H.; Smit, J.J.; van Tellingen, O.; Beijnen, J.H.; Wagenaar, E.; van Deemter, L.; Mol, C.A.; van der Valk, M.A.; Robanus-Maandag, E.C.; te Riele, H.P. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell 1994, 77, 491–502. [Google Scholar] [CrossRef]
- Mealey, K.L.; Bentjen, S.A.; Gay, J.M.; Cantor, G.H. Ivermectin sensitivity in collies is associated with a deletion mutation of the mdr1 gene. Pharmacogenetics 2001, 11, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H.; Jonker, J.W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: An overview. Adv. Drug Deliv. Rev. 2003, 55, 3–29. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Ambudkar, S.V. Overview: ABC transporters and human disease. J. Bioenerg. Biomembr. 2001, 33, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Shirasaka, Y.; Konishi, R.; Funami, N.; Kadowaki, Y.; Nagai, Y.; Sakaeda, T.; Yamashita, S. Expression levels of human P-glycoprotein in in vitro cell lines: Correlation between mRNA and protein levels for P-glycoprotein expressed in cells. Biopharm. Drug Dispos. 2009, 30, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.D.; Brinkhuis, R.F.; van Deemter, L.; Wijnholds, J.; Schinkel, A.H. Extensive contribution of the multidrug transporters P-glycoprotein and Mrp1 to basal drug resistance. Cancer Res. 2000, 60, 5761–5766. [Google Scholar] [PubMed]
- Kourti, M.; Vavatsi, N.; Gombakis, N.; Sidi, V.; Tzimagiorgis, G.; Papageorgiou, T.; Koliouskas, D.; Athanassiadou, F. Expression of multidrug resistance 1 (MDR1), multidrug resistance-related protein 1 (MRP1), lung resistance protein (LRP), and breast cancer resistance protein (BCRP) genes and clinical outcome in childhood acute lymphoblastic leukemia. Int. J. Hematol. 2007, 86, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Amiri-Kordestani, L.; Basseville, A.; Kurdziel, K.; Fojo, A.T.; Bates, S.E. Targeting MDR in breast and lung cancer: Discriminating its potential importance from the failure of drug resistance reversal studies. Drug Resist. Update 2012, 15, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Page, R.L.; Hughes, C.S.; Huyan, S.; Sagris, J.; Trogdon, M. Modulation of P-glycoprotein-mediated doxorubicin resistance in canine cell lines. Anticancer Res. 2000, 20, 3533–3538. [Google Scholar] [PubMed]
- Zandvliet, M.; Teske, E.; Schrickx, J.A. Multi-drug resistance in a canine lymphoid cell line due to increased P-glycoprotein expression, a potential model for drug-resistant canine lymphoma. Toxicol. In Vitro 2014, 28, 1498–1506. [Google Scholar] [CrossRef] [PubMed]
- Leslie, E.M.; Deeley, R.G.; Cole, S.P. Multidrug resistance proteins: Role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol. Appl. Pharmacol. 2005, 204, 216–237. [Google Scholar] [CrossRef] [PubMed]
- Schrickx, J.A. Spinosad is a potent inhibitor of canine P-glycoprotein. Vet. J. 2014, 200, 195–196. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.K.; He, S.M.; Liu, L.; Liu, J.P.; Wei, M.Q.; Zhou, S.F. Herbal interactions with anticancer drugs: Mechanistic and clinical considerations. Curr. Med. Chem. 2010, 17, 1635–1678. [Google Scholar] [CrossRef] [PubMed]
- Lal, S.; Mahajan, A.; Chen, W.N.; Chowbay, B. Pharmacogenetics of target genes across doxorubicin disposition pathway: A review. Curr. Drug Metab. 2010, 11, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer. 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Sharom, F.J. ABC multidrug transporters: Structure, function and role in chemoresistance. Pharmacogenomics 2008, 9, 105–127. [Google Scholar] [CrossRef] [PubMed]
- Nieth, C.; Lage, H. Induction of the ABC-transporters Mdr1/P-gp (Abcb1), mrpl (Abcc1), and bcrp (Abcg2) during establishment of multidrug resistance following exposure to mitoxantrone. J. Chemother. 2005, 17, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Cordon-Cardo, C.; O'Brien, J.P.; Boccia, J.; Casals, D.; Bertino, J.R.; Melamed, M.R. Expression of the multidrug resistance gene product (P-glycoprotein) in human normal and tumor tissues. J. Histochem. Cytochem. 1990, 38, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Fojo, A.T.; Ueda, K.; Slamon, D.J.; Poplack, D.G.; Gottesman, M.M.; Pastan, I. Expression of a multidrug-resistance gene in human tumors and tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Borst, P.; Elferink, R.O. Mammalian ABC transporters in health and disease. Annu. Rev. Biochem. 2002, 71, 537–592. [Google Scholar] [CrossRef] [PubMed]
- Leschziner, G.D.; Andrew, T.; Pirmohamed, M.; Johnson, M.R. ABCB1 genotype and PGP expression, function and therapeutic drug response: A critical review and recommendations for future research. Pharmacogenomics J. 2007, 7, 154–179. [Google Scholar] [CrossRef] [PubMed]
- Steingold, S.F.; Sharp, N.J.; McGahan, M.C.; Hughes, C.S.; Dunn, S.E.; Page, R.L. Characterization of canine MDR1 mRNA: Its abundance in drug resistant cell lines and in vivo. Anticancer Res. 1998, 18, 393–400. [Google Scholar] [PubMed]
- Ginn, P.E. Immunohistochemical detection of P-glycoprotein in formalin-fixed and paraffin-embedded normal and neoplastic canine tissues. Vet. Pathol. 1996, 33, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Conrad, S.; Viertelhaus, A.; Orzechowski, A.; Hoogstraate, J.; Gjellan, K.; Schrenk, D.; Kauffmann, H.M. Sequencing and tissue distribution of the canine MRP2 gene compared with MRP1 and MDR1. Toxicology 2001, 156, 81–91. [Google Scholar] [CrossRef]
- Schleis, S.E.; LeBlanc, A.K.; Neilsen, N.R.; LeBlanc, C.J. Flow cytometric evaluation of multidrug resistance proteins on grossly normal canine nodal lymphocyte membranes. Am. J. Vet. Res. 2008, 69, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Petterino, C.; Rossetti, E.; Bertoncello, D.; Martini, M.; Zappulli, V.; Bargelloni, L.; Castagnaro, M. Immunohistochemical detection of P-glycoprotein (clone C494) in canine mammary gland tumours. J. Vet. Med. A Physiol. Pathol. Clin. Med. 2006, 53, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Badowska-Kozakiewicz, A.M.; Malicka, E. Evaluation of immunohistochemical expression of P-glycoprotein in neoplasms of the mammary gland in bitches. Pol. J. Vet. Sci. 2010, 13, 343–347. [Google Scholar] [PubMed]
- Neff, M.W.; Robertson, K.R.; Wong, A.K.; Safra, N.; Broman, K.W.; Slatkin, M.; Mealey, K.L.; Pedersen, N.C. Breed distribution and history of canine mdr1–1Delta, a pharmacogenetic mutation that marks the emergence of breeds from the collie lineage. Proc. Natl. Acad. Sci. USA 2004, 101, 11725–11730. [Google Scholar] [CrossRef] [PubMed]
- Henik, R.A.; Kellum, H.B.; Bentjen, S.A.; Mealey, K.L. Digoxin and mexiletine sensitivity in a Collie with the MDR1 mutation. J. Vet. Intern. Med. 2006, 20, 415–417. [Google Scholar] [CrossRef] [PubMed]
- Mealey, K.L.; Northrup, N.C.; Bentjen, S.A. Increased toxicity of P-glycoprotein-substrate chemotherapeutic agents in a dog with the MDR1 deletion mutation associated with ivermectin sensitivity. J. Am. Vet. Med. Assoc. 2003, 223, 1434, 1453–1455. [Google Scholar] [CrossRef]
- Barbet, J.L.; Snook, T.; Gay, J.M.; Mealey, K.L. ABCB1-1 Delta (MDR1-1 Delta) genotype is associated with adverse reactions in dogs treated with milbemycin oxime for generalized demodicosis. Vet. Dermatol. 2009, 20, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Sartor, L.L.; Bentjen, S.A.; Trepanier, L.; Mealey, K.L. Loperamide toxicity in a collie with the MDR1 mutation associated with ivermectin sensitivity. J. Vet. Intern. Med. 2004, 18, 117–118. [Google Scholar] [CrossRef] [PubMed]
- Lind, D.L.; Fidel, J.L.; Gay, J.M.; Mealey, K.L. Evaluation of vincristine-associated myelosuppression in Border Collies. Am. J. Vet. Res. 2013, 74, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Uozurmi, K.; Nakaichi, M.; Yamamoto, Y.; Une, S.; Taura, Y. Development of multidrug resistance in a canine lymphoma cell line. Res. Vet. Sci. 2005, 78, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Nakaichi, M.; Takeshita, Y.; Okuda, M.; Nakamoto, Y.; Itamoto, K.; Une, S.; Sasaki, N.; Kadosawa, T.; Takahashi, T.; Taura, Y. Expression of the MDR1 gene and P-glycoprotein in canine mast cell tumor cell lines. J. Vet. Med. Sci. 2007, 69, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Bergman, P.J.; Ogilvie, G.K.; Powers, B.E. Monoclonal antibody C219 immunohistochemistry against P-glycoprotein: Sequential analysis and predictive ability in dogs with lymphoma. J. Vet. Intern. Med. 1996, 10, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, N.; Tojo, E.; Oishi, A.; Fujiki, M.; Misumi, K.; Sakamoto, H.; Kameyama, K.; Shimizu, T.; Yasuda, N. Immunohistochemical detection of P-glycoprotein (PGP) and multidrug resistance-associated protein (MRP) in canine cutaneous mast cell tumors. J. Vet. Med. Sci. 2002, 64, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Tanabe, S.; Shimohira, H.; Kobayashi, Y.; Oomachi, T.; Azuma, S.; Ogihara, K.; Inokuma, H. Expression of cyclooxygenase-2, P-glycoprotein and multi-drug resistance-associated protein in canine transitional cell carcinoma. Res. Vet. Sci. 2007, 83, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Teng, S.P.; Hsu, W.L.; Chiu, C.Y.; Wong, M.L.; Chang, S.C. Overexpression of P-glycoprotein, STAT3, phospho-STAT3 and KIT in spontaneous canine cutaneous mast cell tumours before and after prednisolone treatment. Vet. J. 2012, 193, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Nooter, K.; Westerman, A.M.; Flens, M.J.; Zaman, G.J.; Scheper, R.J.; van Wingerden, K.E.; Burger, H.; Oostrum, R.; Boersma, T.; Sonneveld, P. Expression of the multidrug resistance-associated protein (MRP) gene in human cancers. Clin. Cancer Res. 1995, 1, 1301–1310. [Google Scholar] [PubMed]
- Conseil, G.; Deeley, R.G.; Cole, S.P. Polymorphisms of MRP1 (ABCC1) and related ATP-dependent drug transporters. Pharmacogenet. Genomics 2005, 15, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yuan, H.; Li, N.; Song, G.; Zheng, Y.; Baratta, M.; Hua, F.; Thurston, A.; Wang, J.; Lai, Y. Identification of interspecies difference in efflux transporters of hepatocytes from dog, rat, monkey and human. Eur. J. Pharm. Sci. 2008, 35, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Tashbaeva, R.E.; Hwang, D.N.; Song, G.S.; Choi, N.H.; Lee, J.H.; Lyoo, Y.S.; Lee, S.J.; Jung, D.I.; Kim, H.Y.; Sur, J.H. Cellular characterization of multidrug resistance P-glycoprotein, alpha fetoprotein, and neovascular endothelium-associated antigens in canine hepatocellular carcinoma and cirrhotic liver. Vet. Pathol. 2007, 44, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Honscha, K.U.; Schirmer, A.; Reischauer, A.; Schoon, H.A.; Einspanier, A.; Gabel, G. Expression of ABC-transport proteins in canine mammary cancer: Consequences for chemotherapy. Reprod. Domest. Anim. 2009, 44 (Suppl. 2), 218–223. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Pratt, S.E.; Cao, J.; Dantzig, A.H.; Moore, R.E.; Slapak, C.A. Identification and characterization of the canine multidrug resistance-associated protein. Mol. Cancer. Ther. 2002, 1, 1335–1342. [Google Scholar] [PubMed]
- Maliepaard, M.; Scheffer, G.L.; Faneyte, I.F.; van Gastelen, M.A.; Pijnenborg, A.C.; Schinkel, A.H.; van De Vijver, M.J.; Scheper, R.J.; Schellens, J.H. Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues. Cancer Res. 2001, 61, 3458–3464. [Google Scholar] [PubMed]
- Scharenberg, C.W.; Harkey, M.A.; Torok-Storb, B. The ABCG2 transporter is an efficient Hoechst 33342 efflux pump and is preferentially expressed by immature human hematopoietic progenitors. Blood 2002, 99, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Goldman, I.D. Resistance to antifolates. Oncogene 2003, 22, 7431–7457. [Google Scholar] [CrossRef] [PubMed]
- Ozvegy-Laczka, C.; Cserepes, J.; Elkind, N.B.; Sarkadi, B. Tyrosine kinase inhibitor resistance in cancer: role of ABC multidrug transporters. Drug Resist. Update 2005, 8, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Honjo, Y.; Hrycyna, C.A.; Yan, Q.W.; Medina-Perez, W.Y.; Robey, R.W.; van de Laar, A.; Litman, T.; Dean, M.; Bates, S.E. Acquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP-overexpressing cells. Cancer Res. 2001, 61, 6635–6639. [Google Scholar] [PubMed]
- Suvannasankha, A.; Minderman, H.; O'Loughlin, K.L.; Nakanishi, T.; Ford, L.A.; Greco, W.R.; Wetzler, M.; Ross, D.D.; Baer, M.R. Breast cancer resistance protein (BCRP/MXR/ABCG2) in adult acute lymphoblastic leukaemia: Frequent expression and possible correlation with shorter disease-free survival. Br. J. Haematol. 2004, 127, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Diestra, J.E.; Scheffer, G.L.; Catala, I.; Maliepaard, M.; Schellens, J.H.; Scheper, R.J.; Germa-Lluch, J.R.; Izquierdo, M.A. Frequent expression of the multi-drug resistance-associated protein BCRP/MXR/ABCP/ABCG2 in human tumours detected by the BXP-21 monoclonal antibody in paraffin-embedded material. J. Pathol. 2002, 198, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Saglam, A.; Hayran, M.; Uner, A.H. Immunohistochemical expression of multidrug resistance proteins in mature T/NK-cell lymphomas. APMIS 2008, 116, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Cervenak, J.; Andrikovics, H.; Ozvegy-Laczka, C.; Tordai, A.; Nemet, K.; Varadi, A.; Sarkadi, B. The role of the human ABCG2 multidrug transporter and its variants in cancer therapy and toxicology. Cancer Lett. 2006, 234, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Pawlowski, K.M.; Mucha, J.; Majchrzak, K.; Motyl, T.; Krol, M. Expression and role of PGP, BCRP, MRP1 and MRP3 in multidrug resistance of canine mammary cancer cells. BMC Vet. Res. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Zandvliet, M.; Teske, E.; Schrickx, J.A.; Mol, J.A. A longitudinal study of ABC transporter expression in canine multicentric lymphoma. Vet. J. 2015, 205, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.; Madej, J.A.; Dziegiel, P. Expression of Breast Cancer Resistance Protein (BCRP-1) in canine mammary adenocarcinomas and adenomas. In Vivo 2009, 23, 705–709. [Google Scholar] [PubMed]
- Scotto, K.W. Transcriptional regulation of ABC drug transporters. Oncogene 2003, 22, 7496–7511. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tang, Y.; Guo, C.; Wang, J.; Boral, D.; Nie, D. Nuclear receptors in the multidrug resistance through the regulation of drug-metabolizing enzymes and drug transporters. Biochem. Pharmacol. 2012, 83, 1112–1126. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Moore, J.T.; Wade, L.; Staudinger, J.L.; Watson, M.A.; Jones, S.A.; McKee, D.D.; Oliver, B.B.; Willson, T.M.; Zetterstrom, R.H.; Perlmann, T.; Lehmann, J.M. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell 1998, 92, 73–82. [Google Scholar] [CrossRef]
- Timsit, Y.E.; Negishi, M. CAR and PXR: The xenobiotic-sensing receptors. Steroids 2007, 72, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Harmsen, S.; Meijerman, I.; Febus, C.L.; Maas-Bakker, R.F.; Beijnen, J.H.; Schellens, J.H. PXR-mediated induction of P-glycoprotein by anticancer drugs in a human colon adenocarcinoma-derived cell line. Cancer Chemother. Pharmacol. 2010, 66, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Urquhart, B.L.; Tirona, R.G.; Kim, R.B. Nuclear receptors and the regulation of drug-metabolizing enzymes and drug transporters: Implications for interindividual variability in response to drugs. J. Clin. Pharmacol. 2007, 47, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Chen, T. Overcoming drug resistance by regulating nuclear receptors. Adv. Drug Deliv. Rev. 2010, 62, 1257–1264. [Google Scholar] [CrossRef] [PubMed]
- Piek, C.J.; Rutteman, G.R.; Teske, E. Evaluation of the results of a L-asparaginase-based continuous chemotherapy protocol versus a short doxorubicin-based induction chemotherapy protocol in dogs with malignant lymphoma. Vet. Q. 1999, 21, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Gavazza, A.; Lubas, G.; Valori, E.; Gugliucci, B. Retrospective survey of malignant lymphoma cases in the dog: Clinical, therapeutical and prognostic features. Vet. Res. Commun. 2008, 32 (Suppl. 1), S291–S293. [Google Scholar] [CrossRef] [PubMed]
- Price, G.S.; Page, R.L.; Fischer, B.M.; Levine, J.F.; Gerig, T.M. Efficacy and toxicity of doxorubicin/cyclophosphamide maintenance therapy in dogs with multicentric lymphosarcoma. J. Vet. Intern. Med. 1991, 5, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Marconato, L.; Stefanello, D.; Valenti, P.; Bonfanti, U.; Comazzi, S.; Roccabianca, P.; Caniatti, M.; Romanelli, G.; Massari, F.; Zini, E. Predictors of long-term survival in dogs with high-grade multicentric lymphoma. J. Am. Vet. Med. Assoc. 2011, 238, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Kneuer, C.; Honscha, W.; Gabel, G.; Honscha, K.U. Adaptive response to increased bile acids: Induction of MDR1 gene expression and P-glycoprotein activity in renal epithelial cells. Pflugers Arch. 2007, 454, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Allenspach, K.; Bergman, P.J.; Sauter, S.; Grone, A.; Doherr, M.G.; Gaschen, F. P-glycoprotein expression in lamina propria lymphocytes of duodenal biopsy samples in dogs with chronic idiopathic enteropathies. J. Comp. Pathol. 2006, 134, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Van der Heyden, S.; Croubels, S.; Gadeyne, C.; Ducatelle, R.; Daminet, S.; Murua Escobar, H.; Sterenczak, K.; Polis, I.; Schauvliege, S.; Hesta, M.; Chiers, K. Influence of P-glycoprotein modulation on plasma concentrations and pharmacokinetics of orally administered prednisolone in dogs. Am. J. Vet. Res. 2012, 73, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Pekcec, A.; Unkruer, B.; Stein, V.; Bankstahl, J.P.; Soerensen, J.; Tipold, A.; Baumgartner, W.; Potschka, H. Over-expression of P-glycoprotein in the canine brain following spontaneous status epilepticus. Epilepsy Res. 2009, 83, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.V.; Ueda, K.; Pastan, I.; Gottesman, M.M. Modulation of activity of the promoter of the human MDR1 gene by Ras and p53. Science 1992, 255, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Miltenberger, R.J.; Farnham, P.J.; Smith, D.E.; Stommel, J.M.; Cornwell, M.M. v-Raf activates transcription of growth-responsive promoters via GC-rich sequences that bind the transcription factor Sp1. Cell Growth Differ. 1995, 6, 549–556. [Google Scholar] [PubMed]
- Yamada, T.; Takaoka, A.S.; Naishiro, Y.; Hayashi, R.; Maruyama, K.; Maesawa, C.; Ochiai, A.; Hirohashi, S. Transactivation of the multidrug resistance 1 gene by T-cell factor 4/beta-catenin complex in early colorectal carcinogenesis. Cancer Res. 2000, 60, 4761–4766. [Google Scholar] [PubMed]
- Tomiyasu, H.; Watanabe, M.; Goto-Koshino, Y.; Fujino, Y.; Ohno, K.; Sugano, S.; Tsujimoto, H. Regulation of expression of ABCB1 and LRP genes by mitogen-activated protein kinase/extracellular signal-regulated kinase pathway and its role in generation of side population cells in canine lymphoma cell lines. Leuk. Lymphoma 2013, 54, 1309–1315. [Google Scholar] [CrossRef] [PubMed]
- Tomiyasu, H.; Goto-Koshino, Y.; Fujino, Y.; Ohno, K.; Tsujimoto, H. Antitumour effect and modulation of expression of the ABCB1 gene by perifosine in canine lymphoid tumour cell lines. Vet. J. 2014, 201, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Tomiyasu, H.; Goto-Koshino, Y.; Fujino, Y.; Ohno, K.; Tsujimoto, H. The regulation of the expression of ABCG2 gene through mitogen-activated protein kinase pathways in canine lymphoid tumor cell lines. J. Vet. Med. Sci. 2014, 76, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Tomiyasu, H.; Fujiwara-Igarashi, A.; Goto-Koshino, Y.; Fujino, Y.; Ohno, K.; Tsujimoto, H. Evaluation of DNA methylation profiles of the CpG island of the ABCB1 gene in dogs with lymphoma. Am. J. Vet. Res. 2014, 75, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Momoi, Y.; Okai, Y.; Watari, T.; Goitsuka, R.; Tsujimoto, H.; Hasegawa, A. Establishment and characterization of a canine T-lymphoblastoid cell line derived from malignant lymphoma. Vet. Immunol. Immunopathol. 1997, 59, 11–20. [Google Scholar] [CrossRef]
- Rutgen, B.C.; Hammer, S.E.; Gerner, W.; Christian, M.; de Arespacochaga, A.G.; Willmann, M.; Kleiter, M.; Schwendenwein, I.; Saalmuller, A. Establishment and characterization of a novel canine B-cell line derived from a spontaneously occurring diffuse large cell lymphoma. Leuk. Res. 2010, 34, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Nakaichi, M.; Taura, Y.; Kanki, M.; Mamba, K.; Momoi, Y.; Tsujimoto, H.; Nakama, S. Establishment and characterization of a new canine B-cell leukemia cell line. J. Vet. Med. Sci. 1996, 58, 469–471. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Hughes, C.S.; Fine, R.L.; Page, R.L. P-glycoprotein expression in canine lymphoma: A relevant, intermediate model of multidrug resistance. Cancer 1996, 77, 1892–1898. [Google Scholar] [CrossRef]
- Moore, A.S.; Leveille, C.R.; Reimann, K.A.; Shu, H.; Arias, I.M. The expression of P-glycoprotein in canine lymphoma and its association with multidrug resistance. Cancer Invest. 1995, 13, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Dhaliwal, R.S.; Kitchell, B.E.; Ehrhart, E.; Valli, V.E.; Dervisis, N.G. Clinicopathologic significance of histologic grade, pgp, and p53 expression in canine lymphoma. J. Am. Anim. Hosp. Assoc. 2013, 49, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Culmsee, K.; Gruber, A.D.; von Samson-Himmelstjerna, G.; Nolte, I. Quantification of MDR-1 gene expression in canine tissues by real-time reverse transcription quantitative polymerase chain reaction. Res. Vet. Sci. 2004, 77, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Rassnick, K.M.; Moore, A.S.; Collister, K.E.; Northrup, N.C.; Kristal, O.; Chretin, J.D.; Bailey, D.B. Efficacy of combination chemotherapy for treatment of gastrointestinal lymphoma in dogs. J. Vet. Intern. Med. 2009, 23, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Gramer, I.; Kessler, M.; Geyer, J. Determination of MDR1 gene expression for prediction of chemotherapy tolerance and treatment outcome in dogs with lymphoma. Vet. Comp. Oncol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Cidlowski, J.A. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 2013, 132, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.K.; Cidlowski, J.A. Glucocorticoid-induced apoptosis of healthy and malignant lymphocytes. Prog. Brain Res. 2010, 182, 1–30. [Google Scholar] [PubMed]
- Tissing, W.J.; den Boer, M.L.; Meijerink, J.P.; Menezes, R.X.; Swagemakers, S.; van der Spek, P.J.; Sallan, S.E.; Armstrong, S.A.; Pieters, R. Genomewide identification of prednisolone-responsive genes in acute lymphoblastic leukemia cells. Blood 2007, 109, 3929–3935. [Google Scholar] [CrossRef] [PubMed]
- Schlossmacher, G.; Stevens, A.; White, A. Glucocorticoid receptor-mediated apoptosis: Mechanisms of resistance in cancer cells. J. Endocrinol. 2011, 211, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Kfir-Erenfeld, S.; Sionov, R.V.; Spokoini, R.; Cohen, O.; Yefenof, E. Protein kinase networks regulating glucocorticoid-induced apoptosis of hematopoietic cancer cells: Fundamental aspects and practical considerations. Leuk. Lymphoma 2010, 51, 1968–2005. [Google Scholar] [CrossRef] [PubMed]
- Manceau, S.; Giraud, C.; Decleves, X.; Batteux, F.; Chereau, C.; Chouzenoux, S.; Scherrmann, J.M.; Weill, B.; Perrot, J.Y.; Treluyer, J.M. Expression and induction by dexamethasone of ABC transporters and nuclear receptors in a human T-lymphocyte cell line. J. Chemother. 2012, 24, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Krasil'nikov, M.A.; Shatskaya, V.A.; Stavrovskaya, A.A.; Erohina, M.; Gershtein, E.S.; Adler, V.V. The role of phosphatidylinositol 3-kinase in the regulation of cell response to steroid hormones. Biochim. Biophys. Acta 1999, 1450, 434–443. [Google Scholar] [CrossRef]
- Squire, R.A.; Bush, M.; Melby, E.C.; Neeley, L.M.; Yarbrough, B. Clinical and pathologic study of canine lymphoma: Clinical staging, cell classification, and therapy. J. Natl. Cancer Inst. 1973, 51, 565–574. [Google Scholar] [PubMed]
- Matsuda, A.; Tanaka, A.; Muto, S.; Ohmori, K.; Furusaka, T.; Jung, K.; Karasawa, K.; Okamoto, N.; Oida, K.; Itai, A.; Matsuda, H. A novel NF-kappaB inhibitor improves glucocorticoid sensitivity of canine neoplastic lymphoid cells by up-regulating expression of glucocorticoid receptors. Res. Vet. Sci. 2010, 89, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Zandvliet, M.; Rutteman, G.R.; Teske, E. Prednisolone inclusion in a first-line multidrug cytostatic protocol for the treatment of canine lymphoma does not affect therapy results. Vet. J. 2013, 197, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Angulo, A.M.; Morales-Vasquez, F.; Hortobagyi, G.N. Overview of resistance to systemic therapy in patients with breast cancer. Adv. Exp. Med. Biol. 2007, 608, 1–22. [Google Scholar] [PubMed]
- Leonard, R.C.; Lind, M.; Twelves, C.; Coleman, R.; van Belle, S.; Wilson, C.; Ledermann, J.; Kennedy, I.; Barrett-Lee, P.; Perren, T.; Verrill, M.; Cameron, D.; Foster, E.; Yellowlees, A.; Crown, J.; Anglo-Celtic Cooperative Oncology Group. Conventional adjuvant chemotherapy versus single-cycle, autograft-supported, high-dose, late-intensification chemotherapy in high-risk breast cancer patients: A randomized trial. J. Natl. Cancer Inst. 2004, 96, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Crown, J. Optimising treatment outcomes: A review of current management strategies in first-line chemotherapy of metastatic breast cancer. Eur. J. Cancer 1997, 33 (Suppl. 7), S15–S19. [Google Scholar] [CrossRef]
- Fisher, G.A.; Lum, B.L.; Hausdorff, J.; Sikic, B.I. Pharmacological considerations in the modulation of multidrug resistance. Eur. J. Cancer 1996, 32A, 1082–1088. [Google Scholar] [CrossRef]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Ward, K.W.; Azzarano, L.M. Preclinical pharmacokinetic properties of the P-glycoprotein inhibitor GF120918A (HCl salt of GF120918, 9,10-dihydro-5-methoxy-9-oxo-N-[4-[2-(1,2,3,4-tetrahydro-6,7-dimethoxy-2-i soquinolinyl)ethyl]phenyl]-4-acridine-carboxamide) in the mouse, rat, dog, and monkey. J. Pharmacol. Exp. Ther. 2004, 310, 703–709. [Google Scholar] [PubMed]
- Cagliero, E.; Ferracini, R.; Morello, E.; Scotlandi, K.; Manara, M.C.; Buracco, P.; Comandone, A.; Baroetto Parisi, R.; Baldini, N. Reversal of multidrug-resistance using Valspodar (PSC 833) and doxorubicin in osteosarcoma. Oncol. Rep. 2004, 12, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Ito, D.; Childress, M.; Mason, N.; Winter, A.; Leary, J.; Henson, M.; Borgatti, A.; Lewellen, M.; Krick, E.; Stewart, J.; Lahrman, S.; Leary, J.; Seelig, D.; Koopmeiners, J.; Ruetz, S.; Modiano, J.F.; Childress, M.O. A double blinded, placebo-controlled pilot study to examine reduction of CD34+/CD117+/CD133+ lymphoma progenitor cells and duration of remission induced by neoadjuvant valspodar in dogs with large B-cell lymphoma. F1000 Research 2015. Available online: http://f1000research.com/articles/4-42/v1 (accessed on 6 August 2015).
- Friedenberg, W.R.; Rue, M.; Blood, E.A.; Dalton, W.S.; Shustik, C.; Larson, R.A.; Sonneveld, P.; Greipp, P.R. Phase III study of PSC-833 (valspodar) in combination with vincristine, doxorubicin, and dexamethasone (valspodar/VAD) versus VAD alone in patients with recurring or refractory multiple myeloma (E1A95): A trial of the Eastern Cooperative Oncology Group. Cancer 2006, 106, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Tarasova, N.I.; Seth, R.; Tarasov, S.G.; Kosakowska-Cholody, T.; Hrycyna, C.A.; Gottesman, M.M.; Michejda, C.J. Transmembrane inhibitors of P-glycoprotein, an ABC transporter. J. Med. Chem. 2005, 48, 3768–3775. [Google Scholar] [CrossRef] [PubMed]
- Heike, Y.; Hamada, H.; Inamura, N.; Sone, S.; Ogura, T.; Tsuruo, T. Monoclonal anti-P-glycoprotein antibody-dependent killing of multidrug-resistant tumor cells by human mononuclear cells. JPN. J. Cancer Res. 1990, 81, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Morizono, K.; Xie, Y.; Ringpis, G.E.; Johnson, M.; Nassanian, H.; Lee, B.; Wu, L.; Chen, I.S. Lentiviral vector retargeting to P-glycoprotein on metastatic melanoma through intravenous injection. Nat. Med. 2005, 11, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Ye, D.; Fisher, M.; Juliano, R.L. Selective inhibition of P-glycoprotein expression in multidrug-resistant tumor cells by a designed transcriptional regulator. J. Pharmacol. Exp. Ther. 2002, 302, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, M.; Lavasanifar, A.; Uludag, H. Recent attempts at RNAi-mediated P-glycoprotein downregulation for reversal of multidrug resistance in cancer. Med. Res. Rev. 2013, 33, 33–53. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Wu, C.P.; Ambudkar, S.V. Development of inhibitors of ATP-binding cassette drug transporters: Present status and challenges. Expert Opin. Drug Metab. Toxicol. 2008, 4, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, C.; Ozvegy-Laczka, C.; Apati, A.; Magocsi, M.; Nemet, K.; Orfi, L.; Keri, G.; Katona, M.; Takats, Z.; Varadi, A.; Szakacs, G.; Sarkadi, B. Interaction of nilotinib, dasatinib and bosutinib with ABCB1 and ABCG2: Implications for altered anti-cancer effects and pharmacological properties. Br. J. Pharmacol. 2009, 158, 1153–1164. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Robey, R.W.; Bates, S.E.; Ambudkar, S.V. Sunitinib (Sutent, SU11248), a small-molecule receptor tyrosine kinase inhibitor, blocks function of the ATP-binding cassette (ABC) transporters P-glycoprotein (ABCB1) and ABCG2. Drug Metab. Dispos. 2009, 37, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Jovelet, C.; Benard, J.; Forestier, F.; Farinotti, R.; Bidart, J.M.; Gil, S. Inhibition of P-glycoprotein functionality by vandetanib may reverse cancer cell resistance to doxorubicin. Eur. J. Pharm. Sci. 2012, 46, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Thamm, D.H.; Rose, B.; Kow, K.; Humbert, M.; Mansfield, C.D.; Moussy, A.; Hermine, O.; Dubreuil, P. Masitinib as a chemosensitizer of canine tumor cell lines: A proof of concept study. Vet. J. 2012, 191, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Zandvliet, M.; Teske, E.; Chapuis, T.; Fink-Gremmels, J.; Schrickx, J.A. Masitinib reverses doxorubicin resistance in canine lymphoid cells by inhibiting the function of P-glycoprotein. J. Vet. Pharmacol. Ther. 2013, 36, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Huang, S.L.; Zhang, X.Q.; Zhang, B.; Zhu, H.; Yang, V.W.; Zou, X.P. Reversal effects of pantoprazole on multidrug resistance in human gastric adenocarcinoma cells by down-regulating the V-ATPases/mTOR/HIF-1alpha/P-gp and MRP1 signaling pathway in vitro and in vivo. J. Cell. Biochem. 2012, 113, 2474–2487. [Google Scholar] [CrossRef] [PubMed]
- Spugnini, E.P.; Baldi, A.; Buglioni, S.; Carocci, F.; de Bazzichini, G.M.; Betti, G.; Pantaleo, I.; Menicagli, F.; Citro, G.; Fais, S. Lansoprazole as a rescue agent in chemoresistant tumors: A phase I/II study in companion animals with spontaneously occurring tumors. J. Transl. Med. 2011, 9, 221. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, M.; Rothweiler, F.; Klassert, D.; von Deimling, A.; Weber, K.; Fehse, B.; Kammerer, B.; Doerr, H.W.; Cinatl, J., Jr. Reversal of P-glycoprotein-mediated multidrug resistance by the murine double minute 2 antagonist nutlin-3. Cancer Res. 2009, 69, 416–421. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zandvliet, M.; Teske, E. Mechanisms of Drug Resistance in Veterinary Oncology— A Review with an Emphasis on Canine Lymphoma. Vet. Sci. 2015, 2, 150-184. https://doi.org/10.3390/vetsci2030150
Zandvliet M, Teske E. Mechanisms of Drug Resistance in Veterinary Oncology— A Review with an Emphasis on Canine Lymphoma. Veterinary Sciences. 2015; 2(3):150-184. https://doi.org/10.3390/vetsci2030150
Chicago/Turabian StyleZandvliet, Maurice, and Erik Teske. 2015. "Mechanisms of Drug Resistance in Veterinary Oncology— A Review with an Emphasis on Canine Lymphoma" Veterinary Sciences 2, no. 3: 150-184. https://doi.org/10.3390/vetsci2030150
APA StyleZandvliet, M., & Teske, E. (2015). Mechanisms of Drug Resistance in Veterinary Oncology— A Review with an Emphasis on Canine Lymphoma. Veterinary Sciences, 2(3), 150-184. https://doi.org/10.3390/vetsci2030150